
Abstract.
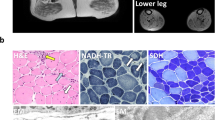
Fukuyama-type congenital muscular dystrophy (FCMD), one of the most common autosomal recessive disorders in the Japanese population, is characterized by congenital muscular dystrophy in combination with cortical dysgenesis (micropolygyria). Recently we identified on chromosome 9q31 the gene responsible for FCMD, which encodes a novel 461 amino acid protein that we have termed fukutin. Most FCMD-bearing chromosomes (87%) derive from a single ancestral founder, whose mutation consisted of a 3-kb retrotransposal insertion in the 3' noncoding region of the fukutin gene. Two independent point mutations causing premature termination confirmed that that this gene is responsible for FCMD. FCMD is the first human disease to be caused by an ancient retrotransposal integration. Fukutin contains an amino-terminal signal sequence, which together with results from transfection experiments suggests that it is an extracellular protein. Discovery of the FCMD gene represents an important step toward greater understanding of the pathogenesis of muscular dystrophies and also of normal brain development.
Similar content being viewed by others


Author information
Authors and Affiliations
Additional information
Electronic Publication
Rights and permissions
About this article
Cite this article
Toda, T., Kobayashi, K. Fukuyama-type congenital muscular dystrophy: the first human disease to be caused by an ancient retrotransposal integration. J Mol Med 77, 816–823 (1999). https://doi.org/10.1007/s001099900065
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s001099900065