
Abstract
Inflammatory bowel disease (IBD) is a prototypic complex disease in the gastrointestinal tract that has been increasing in incidence and prevalence in recent decades. Although the precise pathophysiology of IBD remains to be elucidated, a large body of evidence suggests the critical roles of mitochondria and intestinal microbiota in the pathogenesis of IBD. In addition to their contributions to the disease, both mitochondria and gut microbes may interact with each other and modulate disease-causing cell activities. Therefore, we hypothesize that dissecting this unique interaction may help to identify novel pathways involved in IBD, which will further contribute to discovering new therapeutic approaches to the disease. As poorly treated IBD significantly affects the quality of life of patients and is associated with risks and complications, successful treatment is crucial. In this review, we stratify previously reported experimental and clinical observations of the role of mitochondria and intestinal microbiota in IBD. Additionally, we review the intercommunication between mitochondria, and the intestinal microbiome in patients with IBD is reviewed along with the potential mediators for these interactions. We specifically focus on their roles in cellular metabolism in intestinal epithelial cells and immune cells. To this end, we propose a potential therapeutic intervention strategy for IBD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inflammatory bowel disease (IBD) is a common complex disease in the gut, and its global incidence and prevalence have been increasing worldwide in recent decades [1]. IBD primarily includes two subtypes: Crohn’s disease (CD) and ulcerative colitis (UC). The pathophysiology of IBD is complex, involving genetic and environmental factors [2] and impaired intestinal barrier function and disruption of innate and adaptive immune responses [3, 4]. However, the precise underlying disease mechanisms are not yet fully understood, despite many scientific efforts. Current therapies for IBD include untargeted therapies, such as glucocorticoids, and targeted biologic therapies, which are effective for some but not all patients [2]. Therefore, identifying pathways and mediators involved in disease pathology is the key to discovering therapeutic targets and developing novel therapeutic options for IBD.
This review summarizes current knowledge about the pathological roles of the microbiome and mitochondria in chronic inflammatory diseases in the gut, namely IBD. Furthermore, we discuss the potential interaction between mitochondria and microbiota and their contribution to IBD pathology, particularly cellular metabolism. This concept may contribute to elucidating novel pathways involved in IBD and to the discovery of new therapeutic options for IBD as well as other chronic inflammatory diseases in the gut. Finally, we propose a potential approach to modulate such a mitochondria-microbiota axis thereby controlling the disease.
Structure of the intestinal barrier at the cellular level
The gastrointestinal tract consists of four layers: mucosa, submucosa, muscular layer, and serosa [5]. The mucosa is further subdivided into three layers: a simple columnar epithelium, surface mucus layer, and underlying immune cell-containing lamina propria [6]. The epithelium consists of heterogenous cell types with its specialized functions. These include intestinal epithelial cells (enterocytes), goblet cells, Paneth cells, tuft cells, microfold (M) cells, enteroendocrine cells, and epithelial stem cells [6, 7]. Small and large intestine greatly differ in structure and cellular composition [8]. The small intestine comprises the duodenum, jejunum, and ileum, while the large intestine includes the cecum, colon, and rectum. The small intestinal epithelium forms crypts and has villi, and epithelial cell possesses striated microvilli. In contrast, the large intestine has crypts but lacks villi. Mucus covers the small and large intestine, and its layer is much thicker in the large intestine. The small intestine has Peyer’s patches, the main site of M cells. Intestinal epithelial cells serve as the intestinal barrier and are responsible for nutrient absorption, sensing pathogen-associated molecular patterns, secretion of antimicrobial peptides (AMPs), and modulation of immune cells. Goblet cells secrete mucus that forms an additional barrier of host protection and are more prevalent in the large intestine. Paneth cells secrete AMPs and growth factors for epithelial stem cells and are present in the small intestine [9]. M cells are specialized cells controlling the initiation of mucosal immune response by transporting antigens and microorganisms to the underlying lymphoid tissue [10]. Tuft cells monitor intestinal content, such as substances and intestinal pathogens using succinate, taste receptors, and respond to these by secreting cytokines and endocrine signaling molecules [11]. Enteroendocrine cells have multiple chemosensory receptors and able to detect intestinal microbes and microbe-derived metabolites. Upon these stimulations, enteroendocrine cells secrete peptide hormones and cytokines, thus modulate immune system [12]. As such, the intestinal epithelium plays an essential role in the selective absorption of nutrients and functions as a physical barrier separating mucosal tissues from the exterior environment, e.g., luminal commensal bacteria, pathogens, and dietary antigens.
Another important cell compartment in the intestinal mucosa is immune cells. These immune cells generally reside within one of three compartments: the epithelium (termed intraepithelial lymphocytes, which are primarily T cells with T-cell receptor γδ and T-cell receptor αβ lineages), the lamina propria (termed lamina propria lymphocytes, mainly CD4+ T cells, CD8+ T cells and plasma cells as well as dendritic cells, macrophages, mast cells, eosinophils, and innate lymphoid cells; ILCs), and specialized intestine-associated lymphoid structures in Peyer’s patches and mesenteric lymph nodes (including B cells, CD4+ T cells, CD8+ T cells, dendritic cells, and macrophages) [13]. These immune cells in the intestinal mucosa, together with intestinal epithelial cells, coordinate the mucosal homeostasis, which makes the host tolerant to foodborne antigens and commensal bacteria or develops a response against pathogenic bacteria. If this communication between immune cells and epithelial cells is dysregulated for any reasons, it will result in pathogenic mucosal inflammation. In fact, the disruption of the epithelial barrier and excessive immune response are hallmarks of IBD, as mentioned above. For example, intestinal T-cell infiltrate in IBD patients demonstrates higher levels of proinflammatory T cells and impaired activity of regulatory T cells compared with that in healthy controls and nonactive IBD tissue, and the subsets of monocytes and macrophages in the peripheral blood and colon tissue of IBD patients were significantly altered compared with those of healthy controls [14]. In the next sections, we will review factors that are thought to contribute to the dysregulation of these cells leading to IBD: gut microbiota and mitochondria.
Evidence for the important role of the gut microbiota in IBD
Association between dysbiosis and IBD
Due to the complex nature of pathological processes, multiple factors are thought to contribute to the disease pathology in complex diseases, including IBD. A number of genetic studies have been conducted and successfully identified over 200 genetic loci associated with IBD [15,16,17,18]. Moreover, over 70% of IBD cases cannot be explained only by genetics [15]. Another contributing factor to IBD is the environment, which includes diet lifestyle, air pollution such as particle matters and ozone, temperature (seasons), and climate changes [19, 20], a factor shared with other noncommunicable diseases [21, 22]. Considering the sharp increase in IBD incidence during recent decades, what has also largely changed is our lifestyle, including diet, antibiotic use, social life, and physical activities. In contrast, our genome has not been dramatically changed within the same time period. Importantly, such changes in our lifestyle can influence the composition of commensal bacteria in our gut [23].
Multiple studies have demonstrated that an imbalance of the gut microbial community, namely, dysbiosis, is associated with human diseases, including IBD [24]. Moreover, a direct causal effect of dysbiosis in IBD has not been proven in humans to date [25], while there are observations indicating the major role of intestinal microbiota in IBD. These observations include (1) clinical disease improvement after antibiotic treatment in CD patients [26, 27]; (2) the effectiveness of fecal transplantation in IBD patients, especially those suffering from UC [28,29,30]; and (3) the global increase in IBD associated with changes in lifestyle, such as diet and environment [31]. Nevertheless the outcome of antibiotics treatment efficacy was inconsistent between clinical trials [32, 33].
Classically, there is strong evidence of specific bacteria that are associated with pathological processes in IBD. These include adherent-invasive Escherichia coli, Fusobacterium nucleatum, and Clostridium difficile [34,35,36]. These bacterial species are known to be invasive or toxin-producing, thus have been implicated in pathogenesis of intestinal diseases [37]. In fact, IBD patients particularly under biologic and antibiotics treatment are at higher risk of Clostridium difficile infection, which was associated with relapse and higher mortality [38].
Bacterial metabolites as disease mediators: secondary bile acids, SCFAs
Dysbiosis changes not only the composition of the intestinal microbiota but also its metabolites, which are likely involved in the pathogenesis of IBD [37, 39,40,41]. Such examples include sphingolipids, bile acids, and short-chain fatty acids (SCFAs), among others [39, 42, 43].
In a recent study of untargeted metabolomic and metagenomic profiling in two IBD cohorts, the levels of bile acids (cholate and chenodeoxycholate, thus primary bile acids) and sphingolipids in IBD patients were significantly higher than those in healthy individuals [39]. In relation to the increased levels of primary bile acids, a complementary decrease in secondary bile acids was observed in IBD patients in the same study because primary bile acids support the digestion of lipids and are deconjugated by microbes to secondary bile acids. The effect of secondary bile acids on intestinal epithelial cells was demonstrated by several experimental studies. Secondary bile acids (lithocholic acid and deoxycholic acid) exhibited anti-inflammatory effects on intestinal mucosa by inhibiting proinflammatory cytokine IL-1beta and IL-8 secretion in the Caco-2 human colon adenocarcinoma cell line [44] and promoted intestinal epithelial regeneration in mice [45].
Similar to secondary bile acids, significantly lower levels of SCFAs were identified in IBD patients than in healthy individuals [46]. SCFAs are also involved in epithelial barrier function. SCFAs include acetate, propionate, and butyrate, and these are the most abundantly produced by anaerobic fermentation of dietary fibers in the gut [43]. SCFAs promoted cell proliferation [47], epithelial barrier integrity [48], and the production of antimicrobial peptides, which are a class of peptides with inhibitory effects against pathogenic microbes [49], in intestinal epithelial cells in experimental settings. This experimental evidence indicates that bacteria-derived metabolites play a critical role not only in maintaining epithelial barrier function but also as first-line defense effectors against pathogens. In addition to such effects of bacterially derived metabolites on intestinal epithelial cells, both secondary bile acids and SCFAs exert modulatory functions in intestinal immune cells, which are relevant to IBD pathology. Intestinal macrophages, T regulatory cells, and effector T cells are known to be regulated by secondary bile acid metabolites [42]. Similarly, SCFAs were reported to regulate colonic T regulatory cells via the receptor GPR43 [50] and to promote anti-inflammatory effects in colonic macrophages and dendritic cells via the receptor GPR109a, promoting the differentiation of T regulatory cells and IL-10-producing T cells [51]. Furthermore, enema treatment with butyrate in UC patients significantly decreased the disease activity index accompanied by a reduction in NF-kappa B activation in lamina propria macrophages [52]. On the other hand, butyrate enema showed no or only minor effect on disease activity index, clinical symptoms, endoscopic and histological scores, and inflammatory markers in IBD patients in other clinical studies [53,54,55]. Application of butyrate was exclusively via enema, which is a safe approach, in all of these clinical studies. Therefore, evaluation of other administration strategies, such as intestinally targeted drug delivery system using a nanocapsulated form, may be a potential option to improve the clinical outcome of butyrate.
In summary, dysbiosis is associated with IBD and may be involved in the disease pathology by changing not only the composition of the intestinal microbiota (e.g., increased levels of pathobionts) but also the profile of gut microbe-derived metabolites, which impact the activities of intestinal epithelial cells and immune cell populations that contribute to IBD pathology. However, therapeutic application of such intestinal microbial metabolites in IBD still requires further reliable data from randomized controlled trials, despite the sufficient experimental evidence that such mediators have beneficial effects at cellular levels.
Mitochondrial dysfunction is associated with IBD
Mitochondrial dysfunction as etiology of IBD
Bacterial involvement in pathophysiology of IBD was discussed in the previous sections, as one of the environmental factors associated with IBD. Next, mitochondrial dysfunction, as a genetic factor, is discussed. Mitochondrial dysfunction is involved in pathogenesis of a wide range of diseases in multiple organs, including IBD. For example, genome wide association studies have identified over 200 candidate loci to date [16, 56, 57]. These include mitochondrial functionally related genes. For example, ALDH2, LLRK2, and STAT3 were identified as genome-wide significantly associated genes with IBD [16, 58]; SLC22A5, C13orf31, GPX1, and GPX4 were those with CD [56], and PARK7 was that with UC [57]. These genes are relevant to cellular metabolism (ALDH2, SLC22A5, and C13orf31), apoptosis (LLRK2), redox sensing (PARK7), and redox balance (GPX1 and GPX4). In addition, independent unbiased analysis of the data [16] with specific focus of mitochondrial ontology reveled that additional 22 genes involved in mitochondrial function within the subset of 574 out of total 22,353 genes within 100 kb of a genome-wide significant IBD locus [59]. These genes include those related with mitochondrial iron transport (SLC25A28, VARS, and RNF5), mitochondrial unfolded protein responses (HSPA1-A, -B, and -L), and mitochondrial oxidative phosphorylation machinery (NDUFAF3, SDHC, and UQCR10) [59]. As such, mutations in genes responsible for mitochondrial function are partly involved in pathogenesis of IBD.
Mitochondrial function
Mitochondria are cellular organelles that process dietary nutrients into usable energy in the form of adenosine triphosphate (ATP) through oxidative phosphorylation (OxPhos) in serial enzyme complexes called the electron respiratory chain (ETC) in the inner membrane of the mitochondria. During the OxPhos reaction, mitochondria generate reactive oxygen species (ROS) as a byproduct [60]. Mitochondrial ROS control cell death [61] and are also major inflammatory mediators that activate the NLRP3 inflammasome [62]. In the mitochondrial matrix, there are enzymes that are involved in the tricarboxylic acid (TCA) cycle, fatty acid oxidation (FAO), amino acid oxidation, heme synthesis, and iron sulfur cluster formation [63]. Of note, the TCA cycle produces not only NADH+ and FADH2+, which fuel complex I and complex II in OxPhos but also generates intermediates such as acetyl coenzyme A (acetyl-CoA), alpha-ketoglutarate, and succinate. These metabolic intermediates function as signaling molecules and control cellular responses [63]. Therefore, mitochondria are called the cellular metabolic hub. In addition, mitochondria control Ca2+ homeostasis [64], epigenetic modification [65], and apoptosis [66]. The induction of mitochondria-driven apoptosis pathway is triggered by permeabilization of mitochondrial outer membrane. Upon diverse cellular stresses such as growth-factor deprivation and DNA damage, mitochondrial outer membrane permeabilization, caused by effector pro-apoptotic members of the B cell lymphoma 2 (BCL-2) family of proteins, such as BAX and BAK, mediates the leakage of soluble proteins (such as cytochrome c) from the mitochondrial intermembrane space to the cytosol. Cytochrome c released from the mitochondria subsequently binds to the cytosolic protein apoptotic protease-activating factor and forms apoptosome, which recruits and activates the initiator caspase 9, followed by cleavage and activation of caspase 3 and 7 resulting in cell death [66]. Similarly, the voltage-dependent anion channel 1 (VDAC1), a protein locating at the outer membrane of mitochondria, is also involved in the regulation of release of mitochondrial pro-apoptotic proteins, cytochrome c, and interacting with BCL-2 family proteins [67]. Since mitochondria are present in almost all cell types, functional deterioration of mitochondria affects the functions of any organ. In fact, mitochondrial dysfunctions are directly linked to common diseases, including neurodegenerative, metabolic, age-related, and chronic inflammatory disorders, including IBD [68,69,70].
Mitochondrial function in intestinal epithelial cells and its pathological relevance to IBD
Intestinal epithelial cells consist of distinct cell types, including enterocytes, goblet cells, Paneth cells, and Lgr5+ crypt base columnar stem cells, as mentioned earlier. All intestinal epithelial cell types facilitate distinct metabolic profiles according to specialized function, thereby maintaining homeostasis in the gut [71]. Dysregulation of cellular metabolism in such fine-tuned cellular compartments alters intestinal functions and consequently results in disease involvement. In fact, there is evidence showing mitochondrial functional relevance in IBD patients, which will be presented in this section.
Mitochondria isolated from colon mucosa obtained from Brazilian UC patients showed mitochondrial respiratory chain complexes II, II, and IV were significantly decreased by around 50 to 60% compared with the mitochondria isolated from control colon mucosa [72]. Similarly, tissue homogenates of colon mucosal biopsy samples from Indian UC patients showed significant reduction of complex II activities compared with controls [73]. These studies suggest an involvement of mitochondrial dysfunction in pathogenesis of UC. A recent multicenter study conducting a bulk RNA-sequencing study of UC patients revealed a striking downregulation of genes involved in mitochondrial metabolism–associated genes and pathways in UC [74]. A proteomics analysis of UC-diseased colon mucosa demonstrated a significant reduction in mitochondrial proteins involved in energy production [75]. For CD, there is a case report suggesting a potential involvement of mitochondrial dysfunction (in complex III and IV) in the pathogenesis of CD [76]. Energy deficiency caused by mitochondrial dysfunction in intestinal cells is thought to link to the pathological consequence in both UC and CD. More specifically, energy deficiency in enterocytes leads to compromised nutrient absorption in the small intestine and water and electrolytes absorption in the colon [77]. Goblet cells with energy deficits result in insufficient mucus production in the colon [78]. Energy deficiency in Paneth cells impacts stem cell homeostasis and production of AMPs, resulting in compromised turnover of epithelial cells and dysbiosis in the small intestine, respectively [79]. These contribute to the ileal and colonic pathogenesis in CD and UC, respectively.
As mentioned earlier, mitochondria also control apoptosis. To keep intestinal homeostasis, continuous division of stem cells leads to generation of progenitor cells, which rapidly proliferate and transform into mature intestinal epithelial cells. Excessive apoptosis of differentiated intestinal epithelial cells will accelerate proliferation of immature crypt cells, which can damage the intestinal epithelial barrier. This will result in hyperpermeability and invasion of commensal or environmental microbes into the tissue, which are involved in pathology of IBD [80]. In fact, upregulated expression of VDAC1, which mediates mitochondria-driven apoptosis, was observed in the colon tissue from chronic colitis and UC patients and in the ileocecal junction from CD patients [81]. In contrast, mucosal T cells (in the lamina propria) from CD patients showed lower expression of BAX, proapoptotic protein, which indicate their resistance to apoptotic signals [82, 83]. This phenomenon was not observed in mucosal T cells from UC patients. Further studies confirmed that the mitochondria-driven apoptosis defect of mucosal T cells, thus abnormal T-cell mediated immune reaction in CD [84, 85]. However, it is not clear why levels of apoptosis differ between different tissue/cell types in CD.
Furthermore, the mitochondrial stress response, antioxidant defense mechanisms, such as heat-shock proteins 90 and 60, H+-transporting two-sector ATPase, prohibitin (PHB), mitochondrial malate dehydrogenase, voltage-dependent anion-selective channel protein 1, thioredoxin peroxidase, and thiol-specific antioxidants were also shown to be involved in UC [75]. The protein expression of PHB was also significantly decreased in disease-involved area of colonic mucosal biopsy samples from CD patients [86]. In an experimental setting, the functional consequence of PHB1 was confirmed using mice with intestinal epithelial cell-specific deletion of the Phb1 gene. Both intestinal epithelial cell- and Paneth cell-specific deletion of the Phb1 gene caused spontaneous ileitis characterized by mitochondrial dysfunction in mice [87]. The same study showed that treatment with the mitochondria-targeted antioxidant Mito-Tempo ameliorated the ileitis, suggesting the pathogenic role of mitochondrial ROS in CD. Other experimental evidence suggested the pathological involvement of mitochondrial ROS using mice deficient in the Mdr1a (multidrug resistance protein 1a) gene [59]. In the same study, these researchers showed that the experimental induction of mitochondrial ROS in the colon by treatment with rotenone (mitochondrial complex I-specific inhibitor) or deletion of the Sod2 (superoxide dismutase 2, mitochondrial) gene exacerbated experimental colitis in mice and ameliorated colitis by treatment with a mitochondrial-specific antioxidant (e.g., MitoQ), confirming the role of mitochondrial ROS in colitis. As such, current knowledge of mitochondrial involvement in IBD is primarily mitochondrial ROS that are produced by intestinal cells and cause inflammation in the gut.
Mitochondrial function in immune cells: immunometabolism
Development and activation of immune cells, another important cell type residing in the intestinal mucosa, are also finely regulated by mitochondrial metabolism. To meet their energy demand to function, immune cells reprogram their metabolic pathways. These metabolic pathways include glycolysis, the pentose phosphate pathway, the TCA cycle, FAO, FA synthesis, and amino acid metabolism and are thus heavily involved in mitochondrial functions. The metabolic status of immune cells largely impacts on their phenotype, such as pro- and anti-inflammatory status [88]. For T cells, mitochondrial ATP production during the first 24 to 48 h after T-cell activation is critical for full effector T-cell activation and proliferation [89]. An integrated study of the proteome and phosphoproteome during T-cell activation demonstrated that many mitochondrial metabolic pathways (e.g., mTORC1-dependent mitoribosome biogenesis and COX10-mediated complex IV activity) were altered, suggesting the critical involvement of mitochondrial metabolism in the initiation of T-cell activation [90]. In this line, impairment in OxPhos complex IV or the complex III subunit was shown to impact T-cell activation in vitro and in vivo [91,92,93]. Macrophages, another player in IBD, are known to polarize to either inflammatory M1 macrophages or anti-inflammatory M2 macrophages, depending on the immune response [94], and indeed, many M1 proinflammatory macrophages were observed in the gut of IBD patients [95]. Interestingly, mitochondrial metabolites from the TCA cycle, itaconate, and α-ketoglutarate are known to inhibit secretion of the proinflammatory cytokines IL-1 beta and IL-6 secretion and to increase IL-10 production when stimulated with lipopolysaccharide [96]. Nevertheless, whether dysregulations of immunometabolism in the intestinal mucosal immune cells are involved in IBD pathogenesis or whether those in the peripheral blood are observed in IBD patients need to be determined.
Communication between gut microbiota and mitochondria in IBD
Both dysbiosis and mitochondrial dysfunction result in alteration in gut barrier function and immune cell activities, as discussed in the previous sections. In addition to their synergetic contribution to disease pathology, mitochondria and intestinal microbes are thought to interact via mediators, including endocrine, immune, and humoral links [97].
Signals from gut microbiota to mitochondria: intestinal microbial metabolites
The most studied gut microbe-derived metabolites, SCFAs, are predominantly metabolized by enterocytes and the liver and are the energy source for tissue metabolism [98]. In brief, SCFAs are converted into acetyl-CoA, which is transferred to the TCA cycle in mitochondria, and further metabolic intermediates are generated. During this process, NADH+ and FADH+ are generated, fueled into the ETC, and utilized in ATP production. Butyrate inhibits glycolysis and switches cell metabolism toward gluconeogenic conditions, thus promoting lactate utilization [99]. Positive effects of SCFAs on the intestinal barrier have been reported. Treatment of human colon mucosal epithelial cells (NCM460) with sodium butyrate (SB) increased mitochondrial respiration (i.e., increased maximal respiration, spare capacity, and OxPhos-dependent ATP production) and expression of genes involved in mitochondrial bioenergetics (TFAM), complex I (NDUFA1, NDUFA4, and NDUFA6), complex IV (COX6A1), and complex V (ATP5E and ATP8). Additionally, SB treatment promoted barrier integrity by increasing tight junction gene (zonulin-1 and occludin) expression. These effects of SB were more pronounced when NCM460 cells were treated with TNF, suggesting the potential therapeutic potential of SB for inflammation-induced barrier disfunction [100]. Another recent study reported that microbially produced butyrate promoted intestinal homeostasis and that butyrate treatment in mice protected against colitis by repressing hexokinase 2 expression, which promoted epithelial cell death and impaired mitochondrial respiration [101]. SCFAs are also known to control the differentiation of CD4+ T cells ex vivo and in vivo [102,103,104]. More specifically, differentiation to Th1 cells was promoted, while that to Th17 cells was inhibited. Interestingly, treatment of Th1 and Th17 cells with SCFAs stimulated the production of the anti-inflammatory cytokine IL-10. The mechanism of this effect of SCFAs on CD4+ T cells involves regulation of mTOR (mammalian target of rapamycin; a master regulator of cell growth and metabolism) pathway. The increased production of ATP and the depletion of AMP activate mTOR pathway because ATP is an inhibitor, and AMP is an activator of AMPK, which has repressive activity against mTOR [105]. Additional explanation of this mechanism is that increased levels of acetyl-CoA by SCFAs promote the inhibition of histone deacetylase, resulting the elevation in acetylation of ribosomal protein S6 kinase beta-1, which is the downstream target of the mTOR pathway, thus leading to the activation of the pathway [105]. Additionally, the modulatory effects of SCFAs (primarily butyrate) on mitochondrial metabolism have been reported in macrophages, B cells, and innate lymphoid cells [105]. Thus, these experimental observations support that SCFAs are microbially derived mediators that modulate cellular metabolism in intestinal epithelial cells and immune cells by controlling mitochondrial function.
Large amounts of hydrogen sulfide (H2S) are also produced by the intestinal microbes via anaerobic metabolism [106]. At lower concentrations, H2S is cytoprotective against oxidative damage, increases sensitivity to several antibiotics, and increases resistance to the host immune response, while at higher concentrations, it is toxic [107]. To counteract excess bacterially derived exogenous H2S, host cells have a detoxifying mechanism [108]. H2S is oxidized to thiosulfate and sulfate by the mitochondria at the level of coenzyme Q (CoQ) through sulfide quinone oxidoreductase (SQR). During this oxidation by SQR, two electrons are released and transferred by flavin adenine dinucleotide to CoQ and are thus donated to the ETC stimulating mitochondrial bioenergetics [108]. As mentioned earlier, SCFAs are oxidized to acetyl-CoA, which is further utilized to generate ATP. Of note, butyrate is a major energy source in colonocytes, and H2S is known to inhibit beta-oxidation of butyrate [109]. As such, mitochondria in colonocytes process a limited amount of sulfide, which mitochondria are capable to detoxify this molecule, while they are able to recover energy [106]. Exogenous H2S was shown to control the substrate utilization from fatty acid oxidation to glucose oxidation in cardiomyocytes in mice [110]. These observations clearly indicate that H2S impacts mitochondrial metabolism, which further controls cellular metabolism defining cellular activities. Therefore, it is not surprising that H2S modulates the adaptive immune system, where differential immune subpopulations (e.g., Th1, Th2, Th17, and T regulatory cells) exhibit a well-defined metabolic profile [111, 112]. In the context of IBD, a previous study showed that increased sulfate-reducing bacteria, thereby increasing bacterially derived H2S levels, increased Th17- and T regulatory-cell-type cytokine production and activation profiles in mesenteric lymph nodes in experimental colitis [113]. This mechanism also applies to intestinal epithelial cells, supported by a report describing that microbially derived (and endogenous) H2S modulates colonocyte energy metabolism and is thus involved in pathophysiology in the gut [106].
Other examples of microbial metabolites that are known to modulate mitochondrial function are p-cresol and ammonia. p-cresol belongs to phenolic compounds, which are produced from aromatic amino acid L-tyrosine primarily anaerobic microbes in the large intestine [114]. When p-cresol presents in excess in colonocytes, it inhibits mitochondrial oxygen consumption and consequently reduces cell proliferation [115]. The same study also showed that when colonocytes were treated with p-cresol, anion superoxide production and DNA-double strand break were increased; thus, p-cresol is genotoxic. Ammonia is also produced by intestinal microbes and present at relatively high concentration in the colon. A recent study demonstrated that ammonia treatment on Caco-2 cells induced oxidative stress and mitochondrial dysfunction (e.g., reduced mitochondrial gene expression, reduction of TCA cycle intermediates, reduced mitochondrial membrane potential) [116]. At the same time, reduction of membrane localization of tight junction protein (ZO-1, ZO-2, occludin, claudin-1, and -3) and increased permeability in colonocytes were induced by the ammonia treatment. This study suggested that ammonia treatment triggered mitochondrial dysfunction causing the increase of oxidative stress, which was causal for intestinal barrier dysfunction.
Signals from mitochondria to commensal bacteria
Mitochondria maintain homeostasis in the intestinal epithelial cells, and ROS are produced as byproduct of the OxPhos in the ETC, as mentioned earlier. The potential effect of mitochondrial ROS on gut microbiota is direct killing at higher concentrations [117]. This concept was supported by a study of mouse strains that produced differential levels of mitochondrial ROS, demonstrating the negative correlation between mitochondrial ROS levels and intestinal microbiota diversity [118].
Apart from exogenous bacterially derived H2S, host cells also endogenously produce H2S from the dietary amino acid methionine [108], and endogenous H2S controls cellular metabolism to maintain intestinal homeostasis together with exogenous H2S [106]. As discussed earlier, H2S is detoxified in the mitochondria. If any deficits in the mitochondrial H2S detoxification process exist, including genetic deficits in the SQR gene, H2S levels remain at toxic, which will cause tissue damage by modulating intestinal epithelial cellular metabolism, thus indirectly affecting the composition of the intestinal microbiota [119]. In fact, pathological relevance between mitochondrial H2S detoxification and IBD was reported: impaired H2S detoxification pathways were observed in colon biopsy samples obtained from CD patients. Moreover, the relative abundance of H2S-producing bacteria was increased in stool samples from CD patients [120]. In their study, whether mitochondrial dysfunction or intestinal dysbiosis is the primary cause of IBD was unclear.
Mitochondrial DNA as a common modulator of mitochondrial function and the composition of the intestinal microbiota
Mitochondria have their own genome, called mitochondrial DNA (mtDNA). This small (approximately 16 kb in mammals) circular DNA encodes 37 genes: 13 protein coding, 2 rRNA, and 22 tRNA genes. mtDNA is polymorphic, and mutations/variations in mtDNA are causal not only for primary mitochondrial diseases, such as MELAS and LHON, but also for common complex diseases, including type 2 diabetes, Parkinson’s disease, and cancers [121]. For IBD, two studies, one from Germany and another from the UK, showed an association between mtDNA variants and UC [122, 123]. While the former reported that one specific polymorphism, 11719A > G, in the MT-ND4 gene was associated with male patients affected with UC, the latter identified mtDNA variants with increased and/or decreased risks for UC. Interestingly, these UC candidate alleles are also risk factors or protective for other diseases, including schizophrenia, ankylosing spondylitis, ischemic stroke, Parkinson’s disease, and psoriasis [123]. This finding suggests that mtDNA variants modify the development of multiple late-onset diseases, including UC. There is experimental evidence reported that a mouse strain carrying mt-Co3 (m.9348G > A), mt-Tr (m.9821ins. AA), and mt-Nd3 (m.9461 T > C) (C57BL/6 J-mtNOD/LtJ) was significantly less susceptible to dextran sulfate sodium (DSS)–induced colitis, which is a murine experimental model for IBD, than wild-type C57BL/6 J mice [124]. In this study, the authors demonstrated that the functional consequence of these mtDNA variants was an increase in ATP production, thereby increasing the turnover of intestinal epithelial cells in the colon. Accordingly, this increased epithelial regeneration may be a protective effect against DSS–induced colitis by maintaining the intestinal barrier function. In contrast, another study demonstrated that the mucus layer of the colon in mice with a variant in the mt-Atp8 gene (C57BL/6 J-mtFVB/NJ) had significantly less defective goblet cell differentiation than that of wild-type mice at the steady state [78]. These findings of differential observations (protective/susceptible) in mice carrying distinct mtDNA variants are in line with the abovementioned human study describing that mtDNA variants are both risk factors and protective in common diseases, including UC [123]. Although this mouse strain (C57BL/6 J-mtFVB/NJ) was not evaluated for DSS–induced colitis, a differential disease susceptibility compared with wild-type is expected. The mucus layer has a critical role in the interaction with gut microbiota by providing nutrients and attachment sites [125]; thus, changes in the mucus layer would directly impact the microbes in the gut. In fact, we found that the composition of the gut microbiota in C57BL/6 J-mtFVB/NJ mice was significantly different from that in wild-type mice [126]. We also demonstrated that other mouse strains carrying distinct mtDNA variants (e.g., C57BL/6 J-mtNZB/BlnJ carrying multiple mtDNA variants and C57BL/6 J-mt129S1/SvlmJ carrying a variant in mt-Cytb) exhibited differential gut microbiota composition [126, 127]. With a differential gut microbiome, it is hypothesized that the profile of bacterial metabolites could be distinct between these mouse strains with differential mtDNA variants. Such changes may further result in the alteration of gut microbe-derived activation signals from the gut microbiota to intestinal immune cells and gut epithelial cells.
For the DSS-colitis-protective C57BL/6 J-mtNOD/LtJ mice, the histological evaluation of the colon demonstrated that the lower inflammatory score in the C57BL/6 J-mtNOD/LtJ mice with colitis compared with wild-type mice with colitis, and thus, evaluation of cellular metabolism in intestinal immune cells, including intestinal dendritic cells and CD4+ T cells, in these mouse strains carrying differential mtDNA variants at steady state and under stress is warranted. In fact, cellular metabolism, including the levels of OxPhos and glycolysis, was altered in quiescent CD4+ T cells from C57BL/6 J-mtFVB/NJ mice compared with those from wild-type C57BL/6 J mice [126, 128]. This finding suggests that the immune cell response toward immunological stress, including bacterial metabolites, will be variable between mice carrying different mtDNA variants. Thus, again, profiling cellular metabolism in intestinal immune cells would confirm this hypothesis.
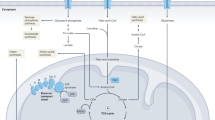
Therefore, we propose that mtDNA variants are common modulators of pathological players (e.g., gut epithelial cells and immune cells, gut microbiome, and mitochondria) in IBD and thus could be a potential therapeutic target (Fig. 1).

A schematic interaction between mitochondria and gut microbiota contributing to the disease pathology in IBD
Current studies of therapeutic approaches to modulate both gut microbiota and mitochondrial function in IBD
Currently, several clinical studies to evaluate the therapeutic potential of modulations of gut microbiota or mitochondrial function for IBD patients are ongoing. To modulate the composition of gut microbiota, several clinical trials have been conducted by nutritional intervention in IBD (e.g., exclusive enteral nutrition, the CD/UC exclusion diet, the low fermentable oligosaccharides, disaccharides, monosaccharides, and polyols; FODMAP diet and the gluten-free diet) [129] or fecal microbiota transplantation in CD patients [130]. Alteration of the gut microbial composition will further result in changes in the quality and quantities of their metabolites; thus, signals from gut microbiota to mitochondria are expected to be changed. The use of probiotics and prebiotics is also of interest as a potential therapy for IBD, although further studies involving larger well-designed clinical trials are required [131]. This hypothesis is supported by a recent study demonstrating that treatment with probiotic consortia (individual and a mixture of Lactobacillus spp. and Bifidobacterium spp.) and their metabolites altered gut microbial composition and successfully ameliorated DSS–induced colitis [132]. In this line, not only the natural probiotics but also a genetically engineered probiotic, E. coli Nissle 1917 overexpressing the antioxidant genes catalase and superoxide dismutase, showed therapeutic efficacy in a DSS–induced colitis model [133]. The uniqueness of this approach is that this genetically engineered probiotic was initially designed to eliminate ROS and reduce inflammation in the gut, and it indeed succeeded in doing so. This specific probiotic was also able to alter the composition of the gut microbiota by significantly reducing the abundance of Escherichia-Shigella, which promotes IBD pathology. Similarly, treatment with nanoparticles containing the antioxidant astaxanthin was able to relieve the disease severity in DSS–induced colitis by inhibiting ROS production and mitochondrial depolarization as well as altering the composition of the gut microbiota [134]. Although the precise mechanism of these findings was not discussed, it is hypothesized that the scavenging (mitochondrial) ROS can possess dual effects on both intestinal inflammation and the gut microbial composition, which may be the secondary effect. As a clinical trial for the use of mitochondrial antioxidant substance (MitoQ) to treat UC has been undertaken [135], it will be worth to evaluate the gut microbe composition in this trial.
As discussed earlier, mtDNA variants would be a potential common regulator of both mitochondrial function and the intestinal microbiota. Therefore, modulating pathologically relevant mtDNA variants may be a potential approach to control IBD. Currently, there are several approaches to manipulate mtDNA, e.g., RNA-free programmable nucleases, including restriction enzymes [136, 137], transcription activator-like effector (TALE) nuclease [138], and zinc finger nuclease [139, 140], fused to mitochondrial targeting signal sequences to induce double-strand breaks in mtDNA. These approaches utilize protein-restricted nucleolysis to achieve a heteroplasmy shift. More recently, a novel technology based on the TALE system using bacterial deaminase (dsDNA deaminase toxin A; DddA) to specifically convert cytosines to thymines (C to T) called the DddA–derived cytosine base editor (DdCBE) system was reported [141]. More recently, the technique has been further developed and generated TALE-linked deaminase, which enables the conversion of adenine to guanine as well [142]. These novel tools are now able to mutagenize mtDNA in mice [143, 144]. Notably, the load of pathogenic mtDNA variants may vary across cells, tissues, and organs [145], and mtDNA editing should be designed accordingly. Therefore, mitochondrial genome editing may be the future option if causal mtDNA variants are identified in IBD patients, although further basic studies are needed.
Conclusion and future prospects
Experimental and clinical studies conducted to date clearly indicate that mitochondrial functions in intestinal epithelial cells and immune cells together with gut microbiota and their metabolites contribute to the pathogenesis of IBD synergistically. Based on these findings, clinical trials and experimental studies (primarily using an experimental colitis mouse model) targeting either gut microbiota or mitochondria (primarily treatment with antioxidants to scavenge mitochondrial ROS) indeed lead to relief of the disease. These approaches (targeting either gut microbiota or mitochondrial ROS) were surprisingly able to modulate both the original therapeutic target and another, supporting the communication between gut microbiota and mitochondria. In addition to these therapeutic options under study, we propose that mtDNA variants relevant to IBD will be a novel target of treatment. One example to achieve this aim is the mitochondrial genome editing strategy, which has recently been rapidly advanced. Clinical application of such approaches is yet too early, requiring further basic studies including enabling organ- and cell-specific efficacy. This is a critical point because differential mucosal cell populations (e.g., epithelial cells, Paneth cells, intraepithelial lymphocytes, and macrophages) are the main players in the pathogenesis of IBD. Considering the increasing number of IBD patients and no availability of reliable treatment, such challenges and efforts to develop novel therapeutic options for IBD should be highly encouraged. In addition, such approaches will be further applied to many common complex diseases, which are not limited to gastroenterological disorders, but also to diseases including neurodegenerative diseases (e.g., Alzheimer’s disease) and metabolic diseases (obesity, type 2 diabetes, and cardiovascular disorders), as mtDNA variants are associated with such conditions, which affect majority of populations worldwide.
Availability of data and materials
Not applicable.
Abbreviations
- Acetyl-CoA:
-
Acetyl coenzyme A
- ATP:
-
Adenosine triphosphate
- IBD:
-
Inflammatory bowel disease
- CD:
-
Crohn’s disease
- ETC:
-
Electron transport chain
- H2S:
-
Hydrogen sulfide
- mtDNA:
-
Mitochondrial DNA
- OxPhos:
-
Oxidative phosphorylation
- ROS:
-
Reactive oxygen species
- SCFAs:
-
Short chain fatty acids
- UC:
-
Ulcerative colitis
References
GBD 2017 Inflammatory Bowel Disease Collaborators (2020) The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol 5:17–30. https://doi.org/10.1016/S2468-1253(19)30333-4
Chang JT (2020) Pathophysiology of inflammatory bowel diseases. N Engl J Med 383:2652–2664. https://doi.org/10.1056/NEJMra2002697
Kobayashi T, Siegmund B, Le Berre C et al (2020) Ulcerative colitis Nat Rev Dis Primers 6:74. https://doi.org/10.1038/s41572-020-0205-x
Roda G, Chien Ng S, Kotze PG et al (2020) Crohn’s disease Nat Rev Dis Primers 6:22. https://doi.org/10.1038/s41572-020-0156-2
Liao D-H, Zhao J-B, Gregersen H (2009) Gastrointestinal tract modelling in health and disease. World J Gastroenterol 15:169–176. https://doi.org/10.3748/wjg.15.169
Luissint A-C, Parkos CA, Nusrat A (2016) Inflammation and the intestinal barrier: leukocyte-epithelial cell interactions, cell junction remodeling, and mucosal repair. Gastroenterology 151:616–632. https://doi.org/10.1053/j.gastro.2016.07.008
Okumura R, Takeda K (2017) Roles of intestinal epithelial cells in the maintenance of gut homeostasis. Exp Mol Med 49:e338. https://doi.org/10.1038/emm.2017.20
Bowcutt R, Forman R, Glymenaki M et al (2014) Heterogeneity across the murine small and large intestine. World J Gastroenterol 20:15216–15232. https://doi.org/10.3748/wjg.v20.i41.15216
Sato T, van Es JH, Snippert HJ et al (2011) Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469:415–418. https://doi.org/10.1038/nature09637
Dillon A, Lo DD (2019) M cells: intelligent engineering of mucosal immune surveillance. Front Immunol 10:1499. https://doi.org/10.3389/fimmu.2019.01499
Hendel SK, Kellermann L, Hausmann A et al (2022) Tuft cells and their role in intestinal diseases. Front Immunol 13:822867. https://doi.org/10.3389/fimmu.2022.822867
Yu Y, Yang W, Li Y, Cong Y (2020) Enteroendocrine cells: sensing gut microbiota and regulating inflammatory bowel diseases. Inflamm Bowel Dis 26:11–20. https://doi.org/10.1093/ibd/izz217
Ahluwalia B, Magnusson MK, Öhman L (2017) Mucosal immune system of the gastrointestinal tract: maintaining balance between the good and the bad. Scand J Gastroenterol 52:1185–1193. https://doi.org/10.1080/00365521.2017.1349173
Zaiatz Bittencourt V, Jones F, Doherty G, Ryan EJ (2021) Targeting immune cell metabolism in the treatment of inflammatory bowel disease. Inflamm Bowel Dis 27:1684–1693. https://doi.org/10.1093/ibd/izab024
Lees CW, Barrett JC, Parkes M, Satsangi J (2011) New IBD genetics: common pathways with other diseases. Gut 60:1739–1753. https://doi.org/10.1136/gut.2009.199679
Liu JZ, van Sommeren S, Huang H et al (2015) Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 47:979–986. https://doi.org/10.1038/ng.3359
Luo Y, de Lange KM, Jostins L et al (2017) Exploring the genetic architecture of inflammatory bowel disease by whole-genome sequencing identifies association at ADCY7. Nat Genet 49:186–192. https://doi.org/10.1038/ng.3761
de Lange KM, Moutsianas L, Lee JC et al (2017) Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet 49:256–261. https://doi.org/10.1038/ng.3760
Agrawal M, Jess T (2022) Implications of the changing epidemiology of inflammatory bowel disease in a changing world. United European Gastroenterol J 10:1113–1120. https://doi.org/10.1002/ueg2.12317
Moon SJ, Lee YC, Kim TJ et al (2022) Effects of temperature, weather, seasons, atmosphere, and climate on the exacerbation of inflammatory bowel diseases: a systematic review and meta-analysis. PLoS ONE 17:e0279277. https://doi.org/10.1371/journal.pone.0279277
Frumkin H, Haines A (2019) Global environmental change and noncommunicable disease risks. Annu Rev Public Health 40:261–282. https://doi.org/10.1146/annurev-publhealth-040218-043706
Favé M-J, Lamaze FC, Soave D et al (2018) Gene-by-environment interactions in urban populations modulate risk phenotypes. Nat Commun 9:827. https://doi.org/10.1038/s41467-018-03202-2
Manichanh C, Borruel N, Casellas F, Guarner F (2012) The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol 9:599–608. https://doi.org/10.1038/nrgastro.2012.152
Carding S, Verbeke K, Vipond DT et al (2015) Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis 26:26191. https://doi.org/10.3402/mehd.v26.26191
Ni J, Wu GD, Albenberg L, Tomov VT (2017) Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol 14:573–584. https://doi.org/10.1038/nrgastro.2017.88
Prantera C, Lochs H, Grimaldi M et al (2012) Rifaximin-extended intestinal release induces remission in patients with moderately active Crohn’s disease. Gastroenterology 142:473-481.e4. https://doi.org/10.1053/j.gastro.2011.11.032
Thia KT, Mahadevan U, Feagan BG et al (2009) Ciprofloxacin or metronidazole for the treatment of perianal fistulas in patients with Crohn’s disease: a randomized, double-blind, placebo-controlled pilot study. Inflamm Bowel Dis 15:17–24. https://doi.org/10.1002/ibd.20608
Haifer C, Paramsothy S, Kaakoush NO et al (2022) Lyophilised oral faecal microbiota transplantation for ulcerative colitis (LOTUS): a randomised, double-blind, placebo-controlled trial. Lancet Gastroenterol Hepatol 7:141–151. https://doi.org/10.1016/S2468-1253(21)00400-3
Costello SP, Hughes PA, Waters O et al (2019) Effect of fecal microbiota transplantation on 8-week remission in patients with ulcerative colitis: a randomized clinical trial. JAMA 321:156–164. https://doi.org/10.1001/jama.2018.20046
Sokol H, Landman C, Seksik P et al (2020) Fecal microbiota transplantation to maintain remission in Crohn’s disease: a pilot randomized controlled study. Microbiome 8:12. https://doi.org/10.1186/s40168-020-0792-5
Kaplan GG, Ng SC (2017) Understanding and preventing the global increase of inflammatory bowel disease. Gastroenterology 152:313–321.e2. https://doi.org/10.1053/j.gastro.2016.10.020
Sutherland L, Singleton J, Sessions J et al (1991) Double blind, placebo controlled trial of metronidazole in Crohn’s disease. Gut 32:1071–1075. https://doi.org/10.1136/gut.32.9.1071
Selby W, Pavli P, Crotty B et al (2007) Two-year combination antibiotic therapy with clarithromycin, rifabutin, and clofazimine for Crohn’s disease. Gastroenterology 132:2313–2319. https://doi.org/10.1053/j.gastro.2007.03.031
Palmela C, Chevarin C, Xu Z et al (2018) Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 67:574–587. https://doi.org/10.1136/gutjnl-2017-314903
Issa M, Ananthakrishnan AN, Binion DG (2008) Clostridium difficile and inflammatory bowel disease. Inflamm Bowel Dis 14:1432–1442. https://doi.org/10.1002/ibd.20500
Strauss J, Kaplan GG, Beck PL et al (2011) Invasive potential of gut mucosa-derived Fusobacterium nucleatum positively correlates with IBD status of the host. Inflamm Bowel Dis 17:1971–1978. https://doi.org/10.1002/ibd.21606
Czepiel J, Dróżdż M, Pituch H et al (2019) Clostridium difficile infection: review. Eur J Clin Microbiol Infect Dis 38:1211–1221. https://doi.org/10.1007/s10096-019-03539-6
Balram B, Battat R, Al-Khoury A et al (2019) Risk factors associated with Clostridium difficile infection in inflammatory bowel disease: a systematic review and meta-analysis. J Crohns Colitis 13:27–38. https://doi.org/10.1093/ecco-jcc/jjy143
Franzosa EA, Sirota-Madi A, Avila-Pacheco J et al (2019) Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol 4:293–305. https://doi.org/10.1038/s41564-018-0306-4
Heinken A, Ravcheev DA, Baldini F et al (2019) Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome 7:75. https://doi.org/10.1186/s40168-019-0689-3
Heinken A, Hertel J, Thiele I (2021) Metabolic modelling reveals broad changes in gut microbial metabolism in inflammatory bowel disease patients with dysbiosis. NPJ Syst Biol Appl 7:19. https://doi.org/10.1038/s41540-021-00178-6
Thomas JP, Modos D, Rushbrook SM et al (2022) The emerging role of bile acids in the pathogenesis of inflammatory bowel disease. Front Immunol 13:829525. https://doi.org/10.3389/fimmu.2022.829525
Parada Venegas D, De la Fuente MK, Landskron G et al (2019) Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol 10:277. https://doi.org/10.3389/fimmu.2019.00277
Duboc H, Rajca S, Rainteau D et al (2013) Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 62:531–539. https://doi.org/10.1136/gutjnl-2012-302578
Sorrentino G, Perino A, Yildiz E et al (2020) Bile acids signal via TGR5 to activate intestinal stem cells and epithelial regeneration. Gastroenterology 159:956-968.e8. https://doi.org/10.1053/j.gastro.2020.05.067
Zhuang X, Li T, Li M et al (2019) Systematic review and meta-analysis: short-chain fatty acid characterization in patients with inflammatory bowel disease. Inflamm Bowel Dis 25:1751–1763. https://doi.org/10.1093/ibd/izz188
Park J-H, Kotani T, Konno T et al (2016) Promotion of intestinal epithelial cell turnover by commensal bacteria: role of short-chain fatty acids. PLoS ONE 11:e0156334. https://doi.org/10.1371/journal.pone.0156334
Kelly CJ, Zheng L, Campbell EL et al (2015) Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe 17:662–671. https://doi.org/10.1016/j.chom.2015.03.005
Zhao Y, Chen F, Wu W et al (2018) GPR43 mediates microbiota metabolite SCFA regulation of antimicrobial peptide expression in intestinal epithelial cells via activation of mTOR and STAT3. Mucosal Immunol 11:752–762. https://doi.org/10.1038/mi.2017.118
Smith PM, Howitt MR, Panikov N et al (2013) The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341:569–573. https://doi.org/10.1126/science.1241165
Singh N, Gurav A, Sivaprakasam S et al (2014) Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40:128–139. https://doi.org/10.1016/j.immuni.2013.12.007
Lührs H, Gerke T, Müller JG et al (2002) Butyrate inhibits NF-kappaB activation in lamina propria macrophages of patients with ulcerative colitis. Scand J Gastroenterol 37:458–466. https://doi.org/10.1080/003655202317316105
Hamer HM, Jonkers DMAE, Vanhoutvin SALW et al (2010) Effect of butyrate enemas on inflammation and antioxidant status in the colonic mucosa of patients with ulcerative colitis in remission. Clin Nutr 29:738–744. https://doi.org/10.1016/j.clnu.2010.04.002
Steinhart AH, Hiruki T, Brzezinski A, Baker JP (1996) Treatment of left-sided ulcerative colitis with butyrate enemas: a controlled trial. Aliment Pharmacol Ther 10:729–736. https://doi.org/10.1046/j.1365-2036.1996.d01-509.x
Scheppach W, Müller JG, Boxberger F et al (1997) Histological changes in the colonic mucosa following irrigation with short-chain fatty acids. Eur J Gastroenterol Hepatol 9:163–168. https://doi.org/10.1097/00042737-199702000-00010
Franke A, McGovern DPB, Barrett JC et al (2010) Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet 42:1118–1125. https://doi.org/10.1038/ng.717
Anderson CA, Boucher G, Lees CW et al (2011) Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet 43:246–252. https://doi.org/10.1038/ng.764
Huang H, Fang M, Jostins L et al (2017) Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 547:173–178. https://doi.org/10.1038/nature22969
Ho G-T, Aird RE, Liu B et al (2018) MDR1 deficiency impairs mitochondrial homeostasis and promotes intestinal inflammation. Mucosal Immunol 11:120–130. https://doi.org/10.1038/mi.2017.31
Wallace DC, Fan W, Procaccio V (2010) Mitochondrial energetics and therapeutics. Annu Rev Pathol 5:297–348. https://doi.org/10.1146/annurev.pathol.4.110807.092314
Orrenius S, Gogvadze V, Zhivotovsky B (2007) Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol 47:143–183. https://doi.org/10.1146/annurev.pharmtox.47.120505.105122
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–225. https://doi.org/10.1038/nature09663
Martínez-Reyes I, Chandel NS (2020) Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun 11:102. https://doi.org/10.1038/s41467-019-13668-3
Giorgi C, Marchi S, Pinton P (2018) The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol 19:713–730. https://doi.org/10.1038/s41580-018-0052-8
Zhu D, Li X, Tian Y (2022) Mitochondrial-to-nuclear communication in aging: an epigenetic perspective. Trends Biochem Sci 47:645–659. https://doi.org/10.1016/j.tibs.2022.03.008
Bock FJ, Tait SWG (2020) Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol 21:85–100. https://doi.org/10.1038/s41580-019-0173-8
Camara AKS, Zhou Y, Wen P-C et al (2017) Mitochondrial VDAC1: a key gatekeeper as potential therapeutic target. Front Physiol 8:460. https://doi.org/10.3389/fphys.2017.00460
Nunnari J, Suomalainen A (2012) Mitochondria: in sickness and in health. Cell 148:1145–1159. https://doi.org/10.1016/j.cell.2012.02.035
Sorrentino V, Menzies KJ, Auwerx J (2018) Repairing mitochondrial dysfunction in disease. Annu Rev Pharmacol Toxicol 58:353–389. https://doi.org/10.1146/annurev-pharmtox-010716-104908
Amorim JA, Coppotelli G, Rolo AP et al (2022) Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat Rev Endocrinol 18:243–258. https://doi.org/10.1038/s41574-021-00626-7
Ho G-T, Theiss AL (2022) Mitochondria and inflammatory bowel diseases: toward a stratified therapeutic intervention. Annu Rev Physiol 84:435–459. https://doi.org/10.1146/annurev-physiol-060821-083306
Sifroni KG, Damiani CR, Stoffel C et al (2010) Mitochondrial respiratory chain in the colonic mucosal of patients with ulcerative colitis. Mol Cell Biochem 342:111–115. https://doi.org/10.1007/s11010-010-0474-x
Santhanam S, Rajamanickam S, Motamarry A et al (2012) Mitochondrial electron transport chain complex dysfunction in the colonic mucosa in ulcerative colitis. Inflamm Bowel Dis 18:2158–2168. https://doi.org/10.1002/ibd.22926
Haberman Y, Karns R, Dexheimer PJ et al (2019) Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat Commun 10:38. https://doi.org/10.1038/s41467-018-07841-3
Hsieh S-Y, Shih T-C, Yeh C-Y et al (2006) Comparative proteomic studies on the pathogenesis of human ulcerative colitis. Proteomics 6:5322–5331. https://doi.org/10.1002/pmic.200500541
Restivo NL, Srivastava MD, Schafer IA, Hoppel CL (2004) Mitochondrial dysfunction in a patient with crohn disease: possible role in pathogenesis. J Pediatr Gastroenterol Nutr 38:534–538. https://doi.org/10.1097/00005176-200405000-00014
Guerbette T, Boudry G, Lan A (2022) Mitochondrial function in intestinal epithelium homeostasis and modulation in diet-induced obesity. Mol Metab 63:101546. https://doi.org/10.1016/j.molmet.2022.101546
Sünderhauf A, Hicken M, Schlichting H et al (2021) Loss of mucosal p32/gC1qR/HABP1 triggers energy deficiency and impairs goblet cell differentiation in ulcerative colitis. Cell Mol Gastroenterol Hepatol. https://doi.org/10.1016/j.jcmgh.2021.01.017
Khaloian S, Rath E, Hammoudi N et al (2020) Mitochondrial impairment drives intestinal stem cell transition into dysfunctional Paneth cells predicting Crohn’s disease recurrence. Gut 69:1939–1951. https://doi.org/10.1136/gutjnl-2019-319514
Wan Y, Yang L, Jiang S et al (2022) Excessive apoptosis in ulcerative colitis: crosstalk between apoptosis, ROS, ER stress, and intestinal homeostasis. Inflamm Bowel Dis 28:639–648. https://doi.org/10.1093/ibd/izab277
Verma A, Pittala S, Alhozeel B et al (2022) The role of the mitochondrial protein VDAC1 in inflammatory bowel disease: a potential therapeutic target. Mol Ther 30:726–744. https://doi.org/10.1016/j.ymthe.2021.06.024
Itoh J, de La Motte C, Strong SA et al (2001) Decreased Bax expression by mucosal T cells favours resistance to apoptosis in Crohn’s disease. Gut 49:35–41. https://doi.org/10.1136/gut.49.1.35
Ina K, Itoh J, Fukushima K et al (1999) Resistance of Crohn’s disease T cells to multiple apoptotic signals is associated with a Bcl-2/Bax mucosal imbalance. J Immunol 163:1081–1090
Sturm A, Leite AZA, Danese S et al (2004) Divergent cell cycle kinetics underlie the distinct functional capacity of mucosal T cells in Crohn’s disease and ulcerative colitis. Gut 53:1624–1631. https://doi.org/10.1136/gut.2003.033613
Doering J, Begue B, Lentze MJ et al (2004) Induction of T lymphocyte apoptosis by sulphasalazine in patients with Crohn’s disease. Gut 53:1632–1638. https://doi.org/10.1136/gut.2003.037911
Theiss AL, Idell RD, Srinivasan S et al (2007) Prohibitin protects against oxidative stress in intestinal epithelial cells. FASEB J 21:197–206. https://doi.org/10.1096/fj.06-6801com
Jackson DN, Panopoulos M, Neumann WL et al (2020) Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut 69:1928–1938. https://doi.org/10.1136/gutjnl-2019-319523
Pearce EL, Pearce EJ (2013) Metabolic pathways in immune cell activation and quiescence. Immunity 38:633–643. https://doi.org/10.1016/j.immuni.2013.04.005
Chang C-H, Curtis JD, Maggi LB Jr et al (2013) Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153:1239–1251. https://doi.org/10.1016/j.cell.2013.05.016
Tan H, Yang K, Li Y et al (2017) Integrative proteomics and phosphoproteomics profiling reveals dynamic signaling networks and bioenergetics pathways underlying T cell activation. Immunity 46:488–503. https://doi.org/10.1016/j.immuni.2017.02.010
Sena LA, Li S, Jairaman A et al (2013) Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38:225–236. https://doi.org/10.1016/j.immuni.2012.10.020
Tarasenko TN, Pacheco SE, Koenig MK et al (2017) Cytochrome c oxidase activity is a metabolic checkpoint that regulates cell fate decisions during T cell activation and differentiation. Cell Metab 25:1254-1268.e7. https://doi.org/10.1016/j.cmet.2017.05.007
Weinberg SE, Singer BD, Steinert EM et al (2019) Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 565:495–499. https://doi.org/10.1038/s41586-018-0846-z
Mills EL, Kelly B, O’Neill LAJ (2017) Mitochondria are the powerhouses of immunity. Nat Immunol 18:488–498. https://doi.org/10.1038/ni.3704
Lissner D, Schumann M, Batra A et al (2015) Monocyte and M1 macrophage-induced barrier defect contributes to chronic intestinal inflammation in IBD. Inflamm Bowel Dis 21:1297–1305. https://doi.org/10.1097/MIB.0000000000000384
Viola A, Munari F, Sánchez-Rodríguez R et al (2019) The metabolic signature of macrophage responses. Front Immunol 10:1462. https://doi.org/10.3389/fimmu.2019.01462
Clark A, Mach N (2017) The crosstalk between the gut microbiota and mitochondria during exercise. Front Physiol 8. https://doi.org/10.3389/fphys.2017.00319
Schönfeld P, Wojtczak L (2016) Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. J Lipid Res 57:943–954. https://doi.org/10.1194/jlr.R067629
Morand C, Besson C, Demigne C, Remesy C (1994) Importance of the modulation of glycolysis in the control of lactate metabolism by fatty acids in isolated hepatocytes from fed rats. Arch Biochem Biophys 309:254–260. https://doi.org/10.1006/abbi.1994.1110
Conder E, Shay HC, Vekaria H et al (2023) Butyrate-induced mitochondrial function improves barrier function in inflammatory bowel disease (IBD). Inflamm Bowel Dis 29:S71–S72. https://doi.org/10.1093/ibd/izac247.135
Hinrichsen F, Hamm J, Westermann M et al (2021) Microbial regulation of hexokinase 2 links mitochondrial metabolism and cell death in colitis. Cell Metab 33:2355-2366.e8. https://doi.org/10.1016/j.cmet.2021.11.004
Park J, Kim M, Kang SG et al (2015) Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR–S6K pathway. Mucosal Immunol 8:80–93. https://doi.org/10.1038/mi.2014.44
Chen L, Sun M, Wu W et al (2019) Microbiota metabolite butyrate differentially regulates Th1 and Th17 cells’ differentiation and function in induction of colitis. Inflamm Bowel Dis 25:1450–1461. https://doi.org/10.1093/ibd/izz046
Luu M, Pautz S, Kohl V et al (2019) The short-chain fatty acid pentanoate suppresses autoimmunity by modulating the metabolic-epigenetic crosstalk in lymphocytes. Nat Commun 10:760. https://doi.org/10.1038/s41467-019-08711-2
Michaudel C, Sokol H (2020) The gut microbiota at the service of immunometabolism. Cell Metab 32:514–523. https://doi.org/10.1016/j.cmet.2020.09.004
Blachier F, Andriamihaja M, Larraufie P et al (2021) Production of hydrogen sulfide by the intestinal microbiota and epithelial cells and consequences for the colonic and rectal mucosa. Am J Physiol Gastrointest Liver Physiol 320:G125–G135. https://doi.org/10.1152/ajpgi.00261.2020
Toliver-Kinsky T, Cui W, Törö G et al (2019) H2S, a bacterial defense mechanism against the host immune response. Infect Immun 87:e00272-e318. https://doi.org/10.1128/IAI.00272-18
Paul BD, Snyder SH, Kashfi K (2021) Effects of hydrogen sulfide on mitochondrial function and cellular bioenergetics. Redox Biol 38:101772. https://doi.org/10.1016/j.redox.2020.101772
Roediger WE, Duncan A, Kapaniris O, Millard S (1993) Reducing sulfur compounds of the colon impair colonocyte nutrition: implications for ulcerative colitis. Gastroenterology 104:802–809. https://doi.org/10.1016/0016-5085(93)91016-b
Sun Y, Tian Z, Liu N et al (2018) Exogenous H2S switches cardiac energy substrate metabolism by regulating SIRT3 expression in db/db mice. J Mol Med (Berl) 96:281–299. https://doi.org/10.1007/s00109-017-1616-3
Pozzi G, Gobbi G, Masselli E et al (2022) Buffering adaptive immunity by hydrogen sulfide. Cells 11:325. https://doi.org/10.3390/cells11030325
Geltink RIK, Kyle RL, Pearce EL (2018) Unraveling the complex interplay between T cell metabolism and function. Annu Rev Immunol 36:461–488. https://doi.org/10.1146/annurev-immunol-042617-053019
Figliuolo VR, Dos Santos LM, Abalo A et al (2017) Sulfate-reducing bacteria stimulate gut immune responses and contribute to inflammation in experimental colitis. Life Sci 189:29–38. https://doi.org/10.1016/j.lfs.2017.09.014
Tanaka H, Sirich TL, Meyer TW (2015) Uremic solutes produced by colon microbes. Blood Purif 40:306–311. https://doi.org/10.1159/000441578
Andriamihaja M, Lan A, Beaumont M et al (2015) The deleterious metabolic and genotoxic effects of the bacterial metabolite p-cresol on colonic epithelial cells. Free Radic Biol Med 85:219–227. https://doi.org/10.1016/j.freeradbiomed.2015.04.004
Yokoo K, Yamamoto Y, Suzuki T (2021) Ammonia impairs tight junction barriers by inducing mitochondrial dysfunction in Caco-2 cells. FASEB J 35:e21854. https://doi.org/10.1096/fj.202100758R
Ballard JWO, Towarnicki SG (2020) Mitochondria, the gut microbiome and ROS. Cell Signal 75:109737. https://doi.org/10.1016/j.cellsig.2020.109737
Yardeni T, Tanes CE, Bittinger K et al (2019) Host mitochondria influence gut microbiome diversity: a role for ROS. Sci Signal 12. https://doi.org/10.1126/scisignal.aaw3159
Rath E, Haller D (2022) Intestinal epithelial cell metabolism at the interface of microbial dysbiosis and tissue injury. Mucosal Immunol 15:595–604. https://doi.org/10.1038/s41385-022-00514-x
Mottawea W, Chiang C-K, Mühlbauer M et al (2016) Altered intestinal microbiota–host mitochondria crosstalk in new onset Crohn’s disease. Nat Commun 7:13419. https://doi.org/10.1038/ncomms13419
Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39:359–407. https://doi.org/10.1146/annurev.genet.39.110304.095751
Dankowski T, Schröder T, Möller S et al (2016) Male-specific association between MT-ND4 11719 A/G polymorphism and ulcerative colitis: a mitochondria-wide genetic association study. BMC Gastroenterol 16:118. https://doi.org/10.1186/s12876-016-0509-1
Hudson G, Gomez-Duran A, Wilson IJ, Chinnery PF (2014) Recent mitochondrial DNA mutations increase the risk of developing common late-onset human diseases. PLoS Genet 10:e1004369. https://doi.org/10.1371/journal.pgen.1004369
Bär F, Bochmann W, Widok A et al (2013) Mitochondrial gene polymorphisms that protect mice from colitis. Gastroenterology 145:1055-1063.e3. https://doi.org/10.1053/j.gastro.2013.07.015
Paone P, Cani PD (2020) Mucus barrier, mucins and gut microbiota: the expected slimy partners? Gut 69:2232–2243. https://doi.org/10.1136/gutjnl-2020-322260
Hirose M, Künstner A, Schilf P et al (2017) Mitochondrial gene polymorphism is associated with gut microbial communities in mice. Sci Rep 7:15293. https://doi.org/10.1038/s41598-017-15377-7
Hirose M, Künstner A, Schilf P et al (2019) A natural mtDNA polymorphism in complex III is a modifier of healthspan in mice. Int J Mol Sci 20. https://doi.org/10.3390/ijms20092359
Schilf P, Künstner A, Olbrich M et al (2021) A mitochondrial polymorphism alters immune cell metabolism and protects mice from skin inflammation. Int J Mol Sci 22. https://doi.org/10.3390/ijms22031006
Adolph TE, Zhang J (2022) Diet fuelling inflammatory bowel diseases: preclinical and clinical concepts. Gut 71:2574–2586. https://doi.org/10.1136/gutjnl-2021-326575
de Caldeira L, F, Borba HH, Tonin FS et al (2020) Fecal microbiota transplantation in inflammatory bowel disease patients: a systematic review and meta-analysis. PLoS ONE 15:e0238910. https://doi.org/10.1371/journal.pone.0238910
Darb Emamie A, Rajabpour M, Ghanavati R et al (2021) The effects of probiotics, prebiotics and synbiotics on the reduction of IBD complications, a periodic review during 2009–2020. J Appl Microbiol 130:1823–1838. https://doi.org/10.1111/jam.14907
Xu L, Liu B, Huang L et al (2022) Probiotic consortia and their metabolites ameliorate the symptoms of inflammatory bowel diseases in a colitis mouse model. Microbiol Spectr 10:e0065722. https://doi.org/10.1128/spectrum.00657-22
Zhou J, Li M, Chen Q et al (2022) Programmable probiotics modulate inflammation and gut microbiota for inflammatory bowel disease treatment after effective oral delivery. Nat Commun 13:3432. https://doi.org/10.1038/s41467-022-31171-0
Zhang X, Zhao X, Hua Z et al (2023) ROS-triggered self-disintegrating and pH-responsive astaxanthin nanoparticles for regulating the intestinal barrier and colitis. Biomaterials 292:121937. https://doi.org/10.1016/j.biomaterials.2022.121937
Gwyer Findlay E, Sutton G, Ho G-T (2021) The MARVEL trial: a phase 2b randomised placebo-controlled trial of oral MitoQ in moderate ulcerative colitis. Immunother Adv 1:ltaa002. https://doi.org/10.1093/immadv/ltaa002
Tanaka M, Borgeld H-J, Zhang J et al (2002) Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J Biomed Sci 9:534–541. https://doi.org/10.1159/000064726
Bacman SR, Williams SL, Hernandez D, Moraes CT (2007) Modulating mtDNA heteroplasmy by mitochondria-targeted restriction endonucleases in a “differential multiple cleavage-site” model. Gene Ther 14:1309–1318. https://doi.org/10.1038/sj.gt.3302981
Bacman SR, Williams SL, Pinto M et al (2013) Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat Med 19:1111–1113. https://doi.org/10.1038/nm.3261
Gammage PA, Rorbach J, Vincent AI et al (2014) Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol Med 6:458–466. https://doi.org/10.1002/emmm.201303672
Gammage PA, Viscomi C, Simard M-L et al (2018) Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat Med 24:1691–1695. https://doi.org/10.1038/s41591-018-0165-9
Mok BY, de Moraes MH, Zeng J et al (2020) A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 583:631–637. https://doi.org/10.1038/s41586-020-2477-4
Cho S-I, Lee S, Mok YG et al (2022) Targeted A-to-G base editing in human mitochondrial DNA with programmable deaminases. Cell 185:1764-1776.e12. https://doi.org/10.1016/j.cell.2022.03.039
Lee H, Lee S, Baek G et al (2021) Mitochondrial DNA editing in mice with DddA-TALE fusion deaminases. Nat Commun 12:1190. https://doi.org/10.1038/s41467-021-21464-1
Silva-Pinheiro P, Nash PA, Van Haute L et al (2022) In vivo mitochondrial base editing via adeno-associated viral delivery to mouse post-mitotic tissue. Nat Commun 13:750. https://doi.org/10.1038/s41467-022-28358-w
Stewart JB, Chinnery PF (2021) Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat Rev Genet 22:106–118. https://doi.org/10.1038/s41576-020-00284-x
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the University of Sharjah and the German Research Foundation (DFG) - Project-ID 454193335 - SFB 1526. Deutsche Forschungsgemeinschaft, 454193335, Misa Hirose, 454193335, Saleh Mohamed Ibrahim.
Author information
Authors and Affiliations
Contributions
MH, PS, and MW wrote the manuscript and the figure design, with the support of MB, MR, and SMI.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hirose, M., Sekar, P., Eladham, M.W.A. et al. Interaction between mitochondria and microbiota modulating cellular metabolism in inflammatory bowel disease. J Mol Med 101, 1513–1526 (2023). https://doi.org/10.1007/s00109-023-02381-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-023-02381-w