
Abstract
Bayesian and pharmacophore modeling approaches were utilized to identify the fragments and critical chemical features of small molecules that enhance sirtuin1 (SIRT1) activity. Initially, 48 Bayesian models (BMs) were developed by exploring 12 different fingerprints (ECFC, ECFP, EPFC, EPFP, FPFC, FPFP, FCFC, FCFP, LCFC, LCFP, LPFC, and LPLP) with diameters of 4, 6, 8, and 10. Among them the BM1 model was selected as the best model based on its good statistical parameters including total accuracy: 0.98 and positive recalls: 0.95. Additionally, BM1 showed good predictive power for the test set (total accuracy: 0.87 and positive recall: 0.87). In addition, 10 qualitative pharmacophore models were generated using 6 well-known SIRT1 activators. Hypothesis2 (Hypo2) was selected as best hypothesis, among 10 Hypos, based on its discriminant ability between the highly active and least/moderately active SIRT1 activators. The best models, BM1 and Hypo2 were used as a query in virtual screens of a drug-like database and the hit molecules were sorted based on Bayesian score and fit value, respectively. In addition, the highest occupied molecular orbital, lowest unoccupied molecular orbital, and energy gap values were calculated for the selected virtual screening hits using density functional theory. Finally, 16 compounds were selected as leads based on their energy gap values, which represent the high reactivity of molecules. Thus, our results indicated that the combination of two-dimensional (2D) and 3D approaches are useful for the discovery and development of specific and potent SIRT1 activators, and will benefit medicinal chemists focused on designing novel lead compounds that activate SIRT1.
Similar content being viewed by others
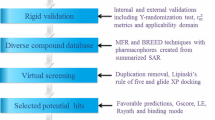

Introduction
Sirtuins (SIRTs), known as silent information regulator 2 (Sir2) proteins, are conserved from E. coli to mammals and involved in regulating cellular responses to a variety of stresses (Sakkiah et al., 2009). SIRTs belong to a family of histone deacetylases (HDACs), which counteract histone acetyltransferase enzymes (Frye, 2000; Mai et al., 2005). The HDAC family of proteins is divided into four different classes based on their cofactors, sequence homology and functions (Witt et al., 2009): Class I, HDAC1–3 and 8; Class II, HDAC4–7 and 9, 10; Class III, Sir2 also known as SIRTs and Class IV, HDAC11. Classes I, II and IV HDACs are similar in their catalytic domain and utilize a zinc ion to deacetylate histone and non-histone proteins. In contrast, SIRTs require NAD+ to deacetylate histone and non-histone proteins (Metoyer and Pruitt, 2008).
SIRTs are critical to cellular homeostasis and play an important role in a myriad of biological process, including life span regulation (Cohen et al., 2004; Howitz et al., 2003; Kaeberlein et al., 1999; Tissenbaum and Guarente, 2001), fat metabolism (Picard et al., 2004), insulin secretion (Moynihan et al., 2005), cellular stress responses (Anderson et al., 2003; Brunet et al., 2004; Cohen et al., 2004), axonal degeneration (Araki et al., 2004), basal transcription factor activity, regulation of enzyme activities, rDNA recombination, and apoptosis (Gertz et al., 2012). Thus, there has been much interest in uncovering the chemical and structural details of these fascinating enzymes (Sakkiah et al., 2013a). Until now, seven human sirtuins (SIRT1–7) have been identified (Haigis and Guarente, 2006; Leonard, 2007) and all isoforms share a similar catalytic domain which consists of ~275 amino acids. With the exception of SIRT1 all other sirtuins (SIRT2–7) consist of approximately 400 residues, whereas SIRT1 consists of 744 residues. However, SIRT size does not appear to dictate sirtuin function, as small (SIRT6) and large (SIRT1) sirtuins have overlapping functions in processes like aging and stress response (Gertz et al., 2012).
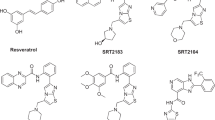
Among the seven sirtuins, SIRT1 is the most characterized member of the Sir2 family. SIRT1 contains two major domains: a large domain containing a Rossmann fold and a small domain, which splits into a zinc-binding domain and a helical domain (Sakkiah et al., 2009). The NAD+ cofactor binds within the cleft formed by the large and small domains adjacent to the acetylated protein (substrate) binding tunnel. The N-terminal residues (G183–I225) play a critical role in the enhancement of SIRT1 activity (Milne et al., 2007). The tumor suppressor protein p53 is a transcription factor that regulates the expression of genes, including p21 and Bax, to initiate cell-cycle arrest and senescence or apoptosis. Interestingly, SIRT1 deacetylates p53 at lysine 382 and attenuates its transcriptional activity in response to DNA damage (Brunet et al., 2004; Dryden et al., 2003; Motta et al., 2004; Rodgers et al., 2005; Vaziri et al., 2001; Yeung et al., 2004). SIRT1 has also been implicated as a mediator of caloric restriction, a low-calorie dietary regimen without malnutrition that extends the life span of yeast, worms, flies, and mammals and decreases the incidence of age-associated disorders like cardiovascular disease, diabetes, and cancer. Therefore, SIRT1 is emerging as a crucial drug target for designing activators and inhibitors for the treatment of various diseases (Taylor et al., 2008). There are two putative conceptual modes for activating SIRT1, the direct and allosteric methods (Westphal et al., 2007). In this work, we focus on the allosteric method to design activators for SIRT1 based on the reported activators including resveratrol (a natural substrate found in red wine which enhances SIRT1 activity, and thereby extends the lifespan of mammals Pillarisetti, 2008), SRT1720, SRT2183, and SRT1460 (Szczepankiewicz and Ng, 2008).
Currently there is no SIRT1 (allosteric site) three-dimensional (3D) structure available, hence to further our understanding of SIRT1 activators, we developed a Bayesian model (BM) based on the reported SIRT1 activators. An integrated Bayesian and pharmacophore modeling approach was utilized to identify the molecular fragments and critical chemical features of SIRT1 activators, respectively (Rogers et al., 2005; Sakkiah et al., 2009, 2010, 2011a, b). The generated 2D and 3D models were used as a query for virtual screening of a drug-like database. The combined 2D and 3D approaches enabled the elucidation of the molecular fragments and chemical features of small molecules that are important to enhance the SIRT1 activity. Orbital energies like highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) were calculated for the 44 known activators and were used as criteria for identifying hit leads from virtual screens.
Materials and methods
Molecular simulations
The molecular simulation studies were performed using the Discovery Studio v 3.1. (DS, Accelrys, Inc., San Diego, USA). Three different approaches, Bayesian modeling, pharmacophore modeling, and density functional theory (DFT), were applied to the discovery of chemical features important for the potency of SIRT1 activators.
BM generation
In chemoinformatics, the Bayesian network (an emerging new technique in drug discovery) acts as the best alternative tool for similarity-based virtual screening approaches. To generate and validate the BMs, a set of 360 SIRT1 activators were collected from various literatures and patents (Bemis et al., 2009; Mai et al., 2009; Vu et al., 2009) with a reported biological activity value (EC50). The collected SIRT1 activators were sketched with the help of ACD ChemSketch v12 and transformed to 3D using DS. Based on the EC50 values the collected 360 activators were divided into 184 highly active activators (EC50 ≤5 μM) and 176 least active activators (EC50 >5 μM). The force field of Chemistry at HARvard Macromolecular Mechanics, CHARMM (Brooks et al., 1983), a flexible and comprehensive empirical energy function that is a summation of many individual energy terms, was applied to all SIRT1 activators. The quality of the BM was directly proportional to the preeminent of training set compounds, descriptors/fingerprints, and statistical method (Prathipati et al., 2008).
Preparation of training set based on diversity
The molecular diversity in the training set plays a major role in finding an important fragment in the generation of BM. Hence, the Diverse Molecules/Library Analysis was applied to select the training and test sets compounds based on the subset of structure diversity with respect to functional class fingerprints of maximum diameter 4, FCFP_4 (Prathipati et al., 2008). The curated activators (360 activators) were divided into training set (20 highly active and 20 least active) and test set (164 highly active and 156 least active) based on a maximum dissimilarity approach. DS provides three types of conformational analysis: FAST, BEST, and CAESAR quality analysis (Sakkiah et al., 2011b). BEST conformation analysis ensures the best coverage of the conformational space, hence it was used to generate the conformations for the training and test set activators. A maximum number of 255 diverse conformations were generated for each compound in training and test sets using Monte-Carlo-like algorithm with an energy range of 20 kcal mol−1 together with the poling algorithm (Smellie et al., 1995).
Selection of molecular descriptors
Descriptors are classified based on the molecular representation such as 1D (molecular formula, volume, and LogP), 2D (molecular connectivity/topology), 3D (molecular geometry/stereochemistry/pharmacophore), and 4D/5D (conformational ensembles). Here, we focused on the 2D descriptors including ALogP, molecular properties counts and element counts, surface and volume using 2D estimation and Estate Keys to generate the BMs. The main aim in calculating the descriptors is to check how well these descriptors are correlated with the reported biological activity of compounds.
Fingerprints selection
Laplacian-modified Bayesian analysis combined with fingerprints is especially useful for high-throughput data analysis because it is fast, easily automated and scales linearly with the number of samples (Rogers et al., 2005). Hence, we selected 12 different fingerprints including (i) atom type extended connectivity fingerprints counts (ECFC), (ii) atom type extended connectivity fingerprints (ECFP), (iii) atom type connectivity fingerprints counts (EPFC), (iv) atom type daylight path-based fingerprints (EPFP), (v) functional class daylight path-based fingerprint counts (FPFC), (vi) functional class daylight path-based fingerprints (FPFP), (vii) functional class extended connectivity fingerprints counts (FCFC), (viii) functional class extended connectivity fingerprints (FCFP), (ix) ALogP types extended connectivity fingerprint counts (LCFC), (x) ALogP extended connectivity fingerprint (LCFP), (xi) ALogP types daylight path-based fingerprint counts (LPFC), and (xii) ALogP types daylight path-based fingerprints (LPLP), with a diameters of 4, 6, 8, and 10 to design BMs. Extended connectivity fingerprints generate higher-order features with each feature representing the presence of a structural unit. Atom environment counts generate higher-order features (Bender et al., 2004) and hashed atom environment counts use a hashing algorithm to create an integer representation of the atom environment counts. The only difference in the generation of a functional class or atom type is the assignment of the initial atom code for each heavy, non-hydrogen atom of the molecule. The initial code assigned to an atom type is based on the number of connections to the atom, element type, charge, atom mass, and valence. For the functional class, initial atom code is based on quick estimate of the functional role the atom plays, this role indicates that the atom must be a combination of hydrogen-bond acceptor (HBA), hydrogen-bond donor (HBD), positively ionized or positively ionizable (PI), aromatic, and halogen. Hence, these different fingerprints were chosen to find which one was more appropriate to differentiate the highly active from least active SIRT1 activators.
Laplace modified naïve Bayesian classifier
In recent studies, Bayesian inference network was introduced as a promising similarity search approach. In this work, Laplacian-corrected Bayesian classifier algorithm (Prathipati et al., 2008) was used to generate BMs for SIRT1 activators. Multiple reference structures were used or more weights assigned to some fragments in molecular structure. This implementation of Bayesian statistics used information from both highly active and least active SIRT1 activators in the training set and removed features from the model which were deemed to be less important. The following steps were taken for BM generation: (i) features of the sample were generated for each compound, (ii) the weight was calculated for each feature using a Laplacian-adjusted probability estimate, (iii) weights were summed to provide a probability estimate which is a relative predictor of the likelihood of that sample being from the good subset, and (iv) Laplacian-corrected estimator was used to adjust the uncorrected probability estimate of a feature to account for different sampling frequencies of different features. The value of “1” was assigned to highly active activators and “0” for least active activators. The BM was built based on Bayes’ theorem:
where h denotes the model. D is the observed data, P(h) indicates the prior belief (probability of pharmacophore model h before observing any data), P(d) is the data evidence (marginal probability of the data), P(d/h) is the likelihood (probability of data d if pharmacophore model h is true), and P(h/d) is the posterior probability (probability of pharmacophore model h being true given the observed data d; Sakkiah et al., 2013b). The modeling process creates a predictive model from the training set that can then be applied to score samples in the test set, and the score can be used to prioritize samples for screening.
Qualitative pharmacophore model generation
Pharmacophore modeling is one the most potent techniques used to identify the critical chemical features from known inhibitors/activators of a particular target. The ligand-based approach is one of the most powerful tools in the rational drug design process. Qualitative hypothesis (Hypo) using the Hip-Hop algorithm was utilized to generate a Hypo for SIRT1 activators.
Training set preparation
Initially 45 good SIRT1 activators were collected from the literature. The diversity of the training set was directly proportional to the quality of the generated Hypo. Hence Cluster Ligands protocol was used to select 6 highly diverse SIRT1 activators from the 45 activators. Cluster Ligands protocol recognized the common pattern or chemical features present in the SIRT1 activators and assigned a set of molecules into subsets or clusters based on the root mean square difference in the Tanimoto distance for fingerprints, such that molecules with similar properties clustered. The cluster analysis was performed by a relocation method based on maximal dissimilarity portioning. Initially, it randomly chose the data set as the first cluster center and based on the distance record from the first center, it selected the next cluster center. The process was repeated until a sufficient number of cluster centers were achieved. Six clusters were obtained through this process and finally six SIRT2 activators were selected as a training set (one SIRT1 activator from each cluster).
Pharmacophore model generation
DS provides a dictionary of chemical features important in drug-enzyme/receptor interactions such as HBA, HBD, hydrophobic (Hy), Hy aliphatic (HAli), Hy aromatic (HAro), ring aromatic (RA), and PI and negative ionizable (NI) chemical groups. Sakkiah et al. (2009) reported that the HBA, HBD, RA, PI, and Hy chemical features were important for a molecule to enhance SIRT1 activity. Hence these five chemical features were selected to generate a qualitative Hypo models.
The qualitative pharmacophore generation for SIRT1 activators was performed in three-steps (Sakkiah et al., 2011a; Ferreira da Silva et al., 2004): (i) generation of the conformation for each molecule in the training set, (ii) each conformer is examined for the presence of certain chemical features, and (iii) a 3D configuration of chemical features common to the input molecules is determined. In the Hypo generation methodology, the highest weight value of “2” was assigned for all compounds which ensures that all the chemical features present in the compound will be considered in building Hypo space and “0” for the principal and maximum omitting features columns (Arooj et al., 2013). The ranking score for each individual Hypo was calculated based on a ranking formula and the default definition of the “FIT” of a molecule to the Hypo, in order to determine the probability that a selected Hypo mapped with the training set molecule by a chance correlation. The top 10 common feature Hypos were generated with the best ranking scores. The quality of the Hypo was predicted by calculating the “fit-value” and this value was defined as the weight (f) × [1 − SSE(f)], where f is the mapping features, SSE(f) is the sum over location constraints c on f of [D(c)/T(c)] 2, D is the displacement of the feature from the center of the location constraint, and T (tolerance) is the radius of the location constraint sphere for the feature.
Pharmacophore model validation
The test set was prepared to identify the best pharmacophore model, as well as to check how accurately it was able to differentiate the highly active from moderately/least active SIRT1 activators. The test set contained 130 activators collected from the literature and classified into three sets based on their activity values: highly active (EC50 <5 μM), moderately active (5 ≤ EC50 ≤ 50 μM), and least active (EC50 >50 μM). All the activators were energy minimized by applying CHARMM force field and 255 conformations were generated for each compound based on the energy values. To select a best Hypo, Ligand Pharmacophore mapping module was used to screen the test set to find which Hypo was able to pick a reliable number of highly active SIRT1 activators.
Preparation of drug-like database
Chemists typically prioritize screening hits on the basis of “drug-like properties,” synthetic accessibility, intellectual property potential, and potency. Hence, a drug-like database was generated by removing the non-drug-like compounds with a high probability of having undesirable molecular features that would hamper their development. Initially, a simple Rule of 5 was applied to filter Maybridge (60,000), Chembridge (50,000), NCI (~200,000), and ChemDiv (~0.7 million) databases on the basis of their likelihood to be orally availability. Rule of 5 (Lipinski, 2000) states that the molecular weight should be less than or equal to 500 kDa, LogP less than or equal to 5, HBA (O and N) less than or equal to 10, and HBD (OH and NH) less than or equal to 5. Furthermore, absorption, distribution, metabolism, excretion, and toxicity, ADMET (Sakkiah et al., 2010, 2011b) was applied to select the compounds having less toxicity, should not cross the blood–brain barrier (BBB), good solubility, and absorption. The ADMET functionality estimates the values of BBB penetration, solubility, cytochrome P450 (CYP450) 2D6 inhibition, hepatotoxicity, human intestinal adsorption (HIA), plasma protein binding (PPB) and access a broad range of ligand toxicity measures. This approach is based on the assumption that compounds resembling known drugs are more likely to possess desirable biological properties such as low toxicity, high oral absorption and permeability, resistance to metabolic degradation, and the absence of rapid excretion. Finally, Prepare ligand module was used to eliminate the duplicate structures, as well as to generate the possible tautomer’s and isomers.
Virtual screening using BM and pharmacophore models
Initially, the best BM was used as an input for virtual screening to select compounds from the drug-like database. BM screening retrieved compounds from the drug-like database with similar molecular fragments present in highly active SIRT1 activators. The screened compounds which had the similar molecular fragment were further validated by mapping to best qualitative pharmacophore model to identify whether these compounds had the important chemical features found in highly active SIRT1 activators.
Density functional theory
There is growing evidence that DFT provides an accurate description of the electronic and structural properties of small molecules by computing the electronic structure of matter. The energy of SIRT1 activators was calculated using Calculate Energy module by combining the quantum mechanics (QM) and molecular mechanics (MM) force field (Sherwood et al., 2003). DFT calculated the QM–MM single point energies and geometry optimization minimizations using Dmol3 as the quantum server with CHARMM force field (Momany and Rone, 1992). This protocol simulated the systems by dividing the input into two regions, central and outer regions, which were treated by QM and MM methods. It also calculated the electronic orbital properties for a molecule including the highest occupied atomic orbital (HOMO) and lowest occupied atomic orbital (LUMO). Bayesian training set molecules were optimized by applying the QM–MM at Becke exchange plus Lee–Yang–Parr correlation (BLYP) hybrid DFT. The optimized molecules were used to calculate the HOMO and LUMO energy values.
Results and discussion
The major aims of this study were to derive a BM and a pharmacophore model that would be able to select the beneficial chemical fragments and chemical features, with suitable geometric constraints to discriminate between good and weak SIRT1 activators.
Bayesian classifier model
A BM is a type of classification model that captures a simple two-class relationship (active and inactive) to distinguish between “good” and “bad” fragments. The main advantages of Bayesian categorization are: (i) it can handle large amounts of data, (ii) it learns fast, (iii) it is tolerant of random noise, and (iv) it is not prone to over fitting.
Development and validation of BMs
The training set contained 40 (20 active and 20 inactive) SIRT1 activators, which were selected based on molecular diversity. A total of 48 BMs were generated from 12 different fingerprints with diameters of 4, 6, 8, and 10 and validated using the 10-fold cross validation (Table S1). In this validation process, one tenth of the samples were left out and the remaining samples were used to build the models. The generated models were used to predict the scores for the left out samples. This process was repeated until all samples had prediction values. The samples, sorted out by decreasing the score value, were used to estimate the predictive power of the resultant BMs based on several statistical parameters like receiver operating characteristic (ROC) value, total accuracy, and enrichment values. The ROC value indicates that the classification power of the models by representing how often the model correctly identifies the true positive (TP) and true negative (TN) in the dataset. In addition, ROC values between 0.50–0.60, 0.60–0.70, 0.70–0.80, 0.80–0.90, and 0.90–1 indicate the accuracy of the model as fail, poor, fair, good, and excellent, respectively. In our study, out of 48, 9 models showed a ROC value between 0.9 and 1 which indicates that these models are excellent when compared with the remaining models.
The nine excellent BMs were obtained from three fingerprints (EPFC, FPFC, and LPFC) with a diameter of 10, 8, and 6. Table S2 summarizes the key statistical discriminative parameters of these models in comparison with the performance of nine derived models. The EPFC_10 Bayesian model (BM1) was chosen as a best model, among the nine derived models, based on the highest average prediction accuracy for highly active activators (positive recalls 19) and the highest total prediction accuracy (total accuracy 0.98; Table S3). The ROC plot revealed that the BM1 had an excellent prediction accuracy for training set (ROC 0.96; Fig. 1), as well as for the test set (ROC 0.94).

ROC plot between true positive (TP) and false positive (FP) for Bayesian BM1
Enrichment plots display the percentage of true category members captured at a particular percentage cutoff. The BM1 enrichment plot indicated very high and reasonable enrichment values for the training and test set, respectively. The best split is a statistical value used to evaluate the quality of the BM. The spilt minimized the sum of percent misclassified for category members and nonmembers, using the cross-validated score for each sample. Based on the split, a contingency table was constructed containing the number of TP, false negative (FN), false positive (FP) and TN (Alcendor et al., 2004; Yang et al., 2013). The total accuracy, negative recall, precision, EF, Mathew’s correlation coefficient (MCC), sensitivity (positive recall) and specificity were calculated based on TP, TN, FP and FN (Table S2). Finally, a distribution curve for the training set suggested that the selected BM1 model can segregate the highly active from least active activators. While the Laplacian-modified Bayesian score for the highly active activators was mostly in the range of 0.598–0.551, the least active activators were predominantly between −2.129 and −1.655. In summary, all statistical parameters suggested that the BM1 model had a higher predictive ability than other BMs. The list of good and bad substructures preferentially present in highly and least active activators can serve as a look-up guide for chemists/computational chemists during hit-to-lead or lead optimization campaigns (Figs. 2, 3).

Fingerprints predicted to be crucial for enhancing SIRT1 activity keyed out from Bayesian theory

Unfavorable fingerprints identified by the Bayesian theory
Qualitative pharmacophore model generation
Pharmacophore approaches have attempted to create 3D models with the necessary key chemical features that interact with the protein by common substructure features present in known activators or inhibitors. Pharmacophore models are used to identify structure–activity relationships (SARs) as well as to develop new lead compounds which can enhance the activity and selectivity for a particular target (Sakkiah et al., 2011b). A total of 45 SIRT1 activators were collected from the literatures and 6 chemical clusters were produced based on the FCFP_6 fingerprints using Cluster Ligand module. One molecule from each cluster (to achieve the maximum diversity of the activators) with good structure diversity was used to generate the top 10 qualitative Hypos, which were selected as a training set (Fig. 4). The top 10 Hypos were generated with their ranking score values between 83.40 and 74.43 bits. Direct hit, partial hit mask value of “1” and “0” for all Hypos indicated that all the molecules of the dataset mapped all features of the Hypo and there was no partial mapping or missing features in the training set molecules, respectively (Table S4). Among these 10 hypotheses, Hypo1 and Hypo2 had similar chemical features and showed the highest-ranking score. The high-ranking scores indicated that these Hypos could select compounds with similar chemical features from any database. However, an external validation test set was used to validate these Hypos; how well they were able to pick the active activators from moderately or least active activators other than the training set compounds.

Training set compounds used for generating quantitative hypotheses. Their EC50 values in nM are indicated in brackets
Qualitative pharmacophore models validation
The test set, containing 130 compounds that were not present in the training set, was used to evaluate the effectiveness of the top 10 Hypos. These top 10 Hypos were divided into two groups contingent on the number of chemical features: Group I, contained four hypotheses (Hypo1: 2-RA, 2-Hy, 2-HBA; Hypo2: 2-RA, 2-Hy, 2-HBA; Hypo3: 1-RA, 3-Hy, 2-HBA and Hypo4: 1-RA, 3-Hy, 2-HBA) with six pharmacophore features and Group II, consisted of six hypotheses (Hypo5: 2-RA, 1-Hy, 2-HBA; Hypo6: 2-RA, 1-Hy, 2-HBA; Hypo7: 2-RA, 1-Hy, 2-HBA; Hypo8: 2-RA, 1-Hy, 2-HBA; Hypo9: 2-RA, 1-Hy, 2-HBA and Hypo10: 2-RA, 1-Hy, 2-HBA) with five pharmacophore features. All of the pharmacophore models in Group I were able to retrieve a good number of active compounds (greater than 90 % from the test set), whereas Group II Hypos selected less than 60 % of active compounds from the test set (Table S5). Thus, we focused on Group I Hypos, among the four hypotheses, Hypo1 was able to screen only 94 % of the active compounds, which was less compared to the other remaining hypotheses (Hypo2: 100 %, Hypo3: 96 % and Hypo4: 96 %). Hypo1 and Hypo2 have similar chemical features, however, due to the geometric constraints of the chemical features Hypo1 retrieved a lower percentage of activators from the test set. On the other hand, Hypo2 picked the least number of compounds from the moderately and least active activators compared with Hypo3 and Hypo4 (Table S6). Hypo2, which shows a good ranking score and predictive ability, consists of 2-RA, 2-Hy, and 2-HBA and its corresponding geometric constrains are summarized in Table S4. Thus, Hypo2 was selected as the best Hypo from the qualitative Hypos (Fig. 5). The best and least fit compounds by Hypo2 are shown in Fig. 6. Therefore, we suggest that the chemical features of Hypo2 reflect the characteristics of the atoms present in SIRT1 activators.

Hypo2 chemical features and spatial arrangement. HBA hydrogen-bond acceptor (green), RA ring aromatic (brown), and Hy hydrophobic (cyan) (Color figure online)

Best (a) and least (b) fit training set molecules in Hypo2. HBA hydrogen-bond acceptor (green), RA ring aromatic (brown), and Hy hydrophobic (cyan) (Color figure online)
Virtual screening using best BM and pharmacophore models
Many virtual screening techniques such as substructure and similarity searches, molecular docking, and quantitative SARs (QSAR) have been the focus of medicinal chemists to reduce the time, cost, and man power of identifying potent activators and inhibitors. Virtual screening of chemical databases can serve two main purposes: (i) to validate the quality of a generated Hypo by selective detection of compounds with known inhibitory activity and (ii) to find novel potential leads which are suitable for further characterization.
The best BM and quantitative pharmacophore models were used to screen a generated drug-like database. First, BM1, best BM, was used to check whether the important chemical fragments were present in the drugs-like molecules or not. The BM1 picked nearly 40,000 compounds from the drug-like database, and these compounds were subsequently screened using Hypo2 to select the ones with suitable chemical features and with geometrical constrains to enhance the activity of SIRT1. Interestingly, 196 compounds contained all the chemical features in Hypo2 with a suitable geometric constrain. The reactivity of 196 compounds was predicted by calculating the energy gap value using DFT.
Density functional theory
DFT is a popular and successful tool for computing the electronic structure of matter. DFT has been used to predict the stability of charge transfers and to analyze the reaction mechanisms responsible for antioxidant activity (Estévez and Mosquera, 2008; Ferreira da Silva et al., 2004; Leopoldini et al., 2010). Thus, we utilized the DFT approach to theoretically characterize the electronic properties and structure properties that correlate with SIRT1 activators. DFT predicts a great variety of molecular properties such as molecular structures, vibration frequencies, atomization energies, ionization energies, electric and magnetic properties, reaction paths, etc. Initially the orbital energies, HOMO and LUMO, were calculated for 44 SIRT1 activators by applying the BLYP, which are responsible for the charge transfer in a chemical reaction (Sakkiah et al., 2009). The geometry obtained from BLYP was used to calculate the orbital energy values. The reaction occurs when the HOMO of the nucleophile overlaps with the LUMO of the electrophilic species. The orbital energies of frontier orbitals are small, ranging between −0.20 and −0.12 au (−5.297 to −2.212 eV) for HOMO and −0.11 to −0.06 au (−2.598 to −1.413 eV) for LUMO (Table S7). Along the reaction coordinates, the HOMO–LUMO gap becomes narrower and favors reactivity. The small values of orbital energies indicate that the molecules are more reactive and rapid electron transfer and exchanges are equally possible. The molecules that have small energy gaps, signifies stability, are likely to undergo changes in the charge distribution through rapid electron transfer between HOMO and LUMO. The higher HOMO value implies that the molecule has good electron donating ability. On the other hand, lower HOMO values imply a weak electron donating ability. In most of the reported SIRT1 activators, the peptide bond was perfectly mapped by HBA and was also confirmed by the HOMO and LUMO values (Fig. 7). The electron density of the HOMO at an atom is a measure of relative reactivity of the HOMO at the atom within a single molecule, while the energy level of the HOMO reflects the reactivity of different molecule, thus molecules with smaller ionization (-HOMO) are expected to be more reactive as nucleophiles.

Identification of SIRT1 activators. (a) Chemical structures of representative SIRT1 activators. (b) Molecules that fit in Hypo2 hypothesis. (c) Highest occupied molecular orbital (HOMO) distribution. (d) Lowest unoccupied molecular orbital (LUMO) distribution. HBA hydrogen-bond acceptor (green), RA ring aromatic (brown), and Hy hydrophobic (cyan) (Color figure online)
The smaller energy gap between the LUMO and HOMO energies illustrates the high reactivity of the molecules. The wide gap is unfavorable for the electron to be excited from HOMO to LUMO, which consequently leads to the weak affinity of the SIRT1 activators. The orbital energy gap values for all the reported activators are in the range of 0.02–0.13 au. Considering the above energy gap value (0.13 au) as a reference, the orbital energy values were calculated for the 196 hit leads from the virtual screening process. Among the 196 compounds only 16 compounds showed an energy gap value less than 0.13 au. Finally, these 16 molecules were selected as the best leads, which can rapidly transfer electrons, by applying the cutoff value of less than 1 (Table S8). Thus, these protocols are useful for improving the activity of the molecules, as well as to identify the novel structures that share the similar chemical features and may thus elicit the desired biological response.
Application of the BM and pharmacophore models
The BM and pharmacophore models were well defined for virtual screening. As an illustration, the selected Bayesian BM1 model was used as a 3D query to screen the drug-like database. In addition, the results of the model will be useful for designing and optimizing compounds with SIRT1 activity, either by replacing the inactive substructures with active ones, removing inactive substructures altogether or by adding active substructures to small fragments with promising cellular activity. Such strategies are especially useful in lead optimization campaigns with compounds identified in cellular screens, where SARs are often difficult to interpret and maintaining cellular activity can be a challenge. The 3D QSAR pharmacophore model, Hypo2, could be useful to medical chemists to determine whether lead compounds contain the suitable chemical features. The identified compounds were subjected to a novelty study using PubChem structure search tools and Scifinder Scholar. This study confirmed that the identified hits were not previously reported elsewhere for the activation of SIRT1. These compounds remain virtually identified and the experimental verification of these compounds will be required to confirm their activation profiles.
Conclusions and future directions
SIRT1 is an emerging chemotherapeutic drug target. To date, there is lack of 3D structural detail; hence ligand-based approaches have been applied to define the critical chemical fragments and pharmacophoric features for SIRT1 activators. BMs, derived based on the 2D molecular properties and fingerprints, were found to effectively discriminate highly active from least active activators. Since the training set design and choice of fingerprints critically affects the predictive ability of QSAR models, model development and validation involved the exploration of 12 different fingerprint types with diameters of 4, 6, 8, and 10 which resulted in a total of 48 BMs. The best Bayesian model BM1, prioritized using average total accuracy and positive recall, was derived using ECFC_10 and a few global descriptors on a training set. This model demonstrated good discriminant ability in general; with excellent discriminant statistics for the training set (total accuracy and positive recall) and a good predictive ability of test set (total accuracy and positive recall). With such encouraging results, the model was used to screen a drug-like database and several compounds were highlighted as potentially potent SIRT1 activators. To confirm these 2D conformations present in the BM, a 3D qualitative pharmacophore model was used to confirm the existence of critical chemical features that would enhance the SIRT1 activator activity of lead compounds. Ten quantitative Hypos were generated based on six known activators, and Hypo2 was selected as the best Hypo based on its ability to discriminate between active from moderately/least active activators of SIRT1. The BM1 (Bayesian model) and the Hypo2 (qualitative pharmacophore model) were used as query in virtual screening of a drug-like database. A total of 196 compounds from drug-like databases contained similar chemical fragments and features present in BM1 and Hypo2. These 196 compounds were subjected to a DFT study in order to determine the reactivity of the compounds. Finally 16 compounds were selected as lead activators of SIRT1. The knowledge of 2D and 3D structural relationships has the potential to accelerate the discovery of SIRT1 activators. The approach described in this study can be used to determine the functional characteristics of SIRT1 activators. Finally, we suggest that the generated BM and pharmacophore models offer an alternative way to screen the various chemical databases and to design novel scaffolds for SIRT1 activators.
Associated content
Supporting information
Supporting tables with ROC scores and the best split values of the 48 BMs, the statistical values for the 9 best BMs, the statistical values of Bayesian BM1 model, the details of the top 10 Hypos generated using Hip-Hop, the percentage comparison for the number of molecules screened from each Hypo, the distance between every two pharmacophore features present in Hypo2, the orbital energy values of 44 SIRT1 activators and the orbital energy values of 16 hit lead compounds.
Abbreviations
- ADMET:
-
Absorption, distribution, metabolism, excretion and toxicity
- BLYP:
-
Becke exchange plus Lee–Yang–Parr correlation
- BM:
-
Bayesian model
- BBB:
-
Blood–brain barrier
- CHARMM:
-
Chemistry at HARvard Macromolecular Mechanics
- CYP450:
-
Cytochrome P450
- DFT:
-
Density functional theory
- DS:
-
Discovery Studio v3.1
- ECFC:
-
Atom type extended connectivity fingerprints counts
- ECFP:
-
Atom type connectivity fingerprints counts
- ECFP:
-
Atom type extended connectivity fingerprints
- EPFC:
-
Functional class daylight path-based fingerprints
- FCFC:
-
Functional class extended connectivity fingerprints counts
- FCFP:
-
Functional class extended connectivity fingerprints
- FN:
-
False negative
- FP:
-
False positive
- FPFC:
-
Functional class daylight path-based fingerprint counts
- FPFP:
-
Functional class daylight path-based fingerprints
- HAli:
-
Hydrophobic aliphatic
- HAro:
-
Hydrophobic aromatic
- HBA:
-
Hydrogen-bond acceptor
- HBD:
-
Hydrogen-bond donor
- HDAC:
-
Histone deacetylase
- HIA:
-
Human intestinal adsorption
- HOMO:
-
Highest occupied molecular orbital
- Hy:
-
Hydrophobic
- Hypo:
-
Hypothesis
- LCFC:
-
ALogP types extended connectivity fingerprint counts
- LCFP:
-
ALogP extended connectivity fingerprint
- LPFC:
-
ALogP types daylight path-based fingerprint counts
- LPLP:
-
ALogP types daylight path-based fingerprints
- LUMO:
-
Lowest unoccupied molecular orbital
- MCC:
-
Mathew’s correlation coefficient
- MM:
-
Molecular mechanics
- NI:
-
Negative ionizable
- PI:
-
Positively ionized or positively ionizable
- PPB:
-
Plasma protein binding
- QM:
-
Quantum mechanics
- QSAR:
-
Quantitative structure–activity relationships
- RA:
-
Ring aromatic
- ROC:
-
Receiver operating characteristic
- SIRT1:
-
Sirtuin1
- TN:
-
True negatives
- TP:
-
True positives
References
Alcendor RR, Kirshenbaum LA, Imai S-I, Vatner SF, Sadoshima J (2004) Silent information regulator 2α, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ Res 95:971–980
Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA (2003) Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423:181–185
Araki T, Sasaki Y, Milbrandt J (2004) Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305:1010–1013
Arooj M, Sakkiah S, Cao Gp, Lee KW (2013) An innovative strategy for dual inhibitor design and its application in dual inhibition of human thymidylate synthase and dihydrofolate reductase enzymes. PLoS ONE 8:e60470
Bemis JE, Vu CB, Xie R, Nunes JJ, Ng PY, Disch JS, Milne JC, Carney DP, Lynch AV, Jin L et al (2009) Discovery of oxazolo[4,5-b]pyridines and related heterocyclic analogs as novel SIRT1 activators. Bioorg Med Chem Lett 19:2350–2353
Bender A, Mussa HY, Glen RC, Reiling S (2004) Similarity searching of chemical databases using atom environment descriptors (MOLPRINT 2D): evaluation of performance. J Chem Inf Comput Sci 44:1708–1718
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 4:187–217
Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY et al (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303:2011–2015
Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA (2004) Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305:390–392
Dryden SC, Nahhas FA, Nowak JE, Goustin A-S, Tainsky MA (2003) Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol Cell Biol 23:3173–3185
Estévez L, Mosquera RA (2008) Molecular structure and antioxidant properties of delphinidin. J Phys Chem A 112:10614–10623
Ferreira da Silva P, Lima JC, Quina FH, Maçanita AL (2004) Excited-state electron transfer in anthocyanins and related flavylium salts. J Phys Chem A 108:10133–10140
Frye RA (2000) Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun 273:793–798
Gertz M, Nguyen GTT, Fischer F, Suenkel B, Schlicker C, Fränzel B, Tomaschewski J, Aladini F, Becker C, Wolters D et al (2012) A molecular mechanism for direct sirtuin activation by resveratrol. PLoS ONE 7:e49761
Haigis MC, Guarente LP (2006) Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction. Genes Dev 20:2913–2921
Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang L-L et al (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425:191–196
Kaeberlein M, McVey M, Guarente L (1999) The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13:2570–2580
Leonard G (2007) Sirtuins in aging and disease. Cold Spring Harb Symp Quant Biol 72:483–488
Leopoldini M, Rondinelli F, Russo N, Toscano M (2010) Pyranoanthocyanins: a theoretical investigation on their antioxidant activity. J Agric Food Chem 58:8862–8871
Lipinski CA (2000) Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods 44:235–249
Mai A, Massa S, Lavu S, Pezzi R, Simeoni S, Ragno R, Mariotti FR, Chiani F, Camilloni G, Sinclair DA (2005) Design, synthesis, and biological evaluation of sirtinol analogues as class III histone/protein deacetylase (sirtuin) inhibitors. J Med Chem 48:7789–7795
Mai A, Valente S, Meade S, Carafa V, Tardugno M, Nebbioso A, Galmozzi A, Mitro N, De Fabiani E, Altucci L et al (2009) Study of 1,4-dihydropyridine structural scaffold: discovery of novel sirtuin activators and inhibitors. J Med Chem 52:5496–5504
Metoyer CF, Pruitt K (2008) The role of sirtuin proteins in obesity. Pathophysiology 15:103–108
Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB et al (2007) Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450:712–716
Momany FA, Rone R (1992) Validation of the general purpose QUANTA ®3.2/CHARMm® force field. J Comput Chem 13:888–900
Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L (2004) Mammalian SIRT1 represses forkhead transcription factors. Cell 116:551–563
Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Méneur C, Permutt MA, Imai S-I (2005) Increased dosage of mammalian Sir2 in pancreatic β cells enhances glucose-stimulated insulin secretion in mice. Cell Metab 2:105–117
Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado de Oliveira R, Leid M, McBurney MW, Guarente L (2004) Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-[gamma]. Nature 429:771–776
Pillarisetti S (2008) A review of Sirt1 and Sirt1 modulators in cardiovascular and metabolic diseases. Recent Pat Cardiovasc Drug Discov 3:156–164
Prathipati P, Ma NL, Keller TH (2008) Global Bayesian models for the prioritization of antitubercular agents. J Chem Inf Model 48:2362–2370
Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P (2005) Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434:113–118
Rogers D, Brown RD, Hahn M (2005) Using extended-connectivity fingerprints with Laplacian-modified Bayesian analysis in high-throughput screening follow-up. J Biomol Screen 10:682–686
Sakkiah S, Krishnamoorthy N, Gajendrarao P, Thangapandian S, Lee Y-O, Kim S-M, Suh J-K, Kim H-H, Lee K-W (2009) Pharmacophore mapping and virtual screening for SIRT1 activators. Bull Korean Chem Soc 30:1152–1156
Sakkiah S, Thangapandian S, John S, Kwon YJ, Lee KW (2010) 3D QSAR pharmacophore based virtual screening and molecular docking for identification of potential HSP90 inhibitors. Eur J Med Chem 45:2132–2140
Sakkiah S, Thangapandian S, John S, Lee KW (2011a) Identification of critical chemical features for Aurora kinase-B inhibitors using Hip-Hop, virtual screening and molecular docking. J Mol Struct 985:14–26
Sakkiah S, Thangapandian S, John S, Lee KW (2011b) Pharmacophore based virtual screening, molecular docking studies to design potent heat shock protein 90 inhibitors. Eur J Med Chem 46:2937–2947
Sakkiah S, Arooj M, Kumar MR, Eom SH, Lee KW (2013a) Identification of inhibitor binding site in human sirtuin 2 using molecular docking and dynamics simulations. PLoS ONE 8:e51429
Sakkiah S, Arullaperumal V, Hwang S, Lee KW (2013b) Ligand-based pharmacophore modeling and Bayesian approaches to identify c-Src inhibitors. J Enzym Inhib Med Chem 29:69–80
Sherwood P, de Vries AH, Guest MF, Schreckenbach G, Catlow CRA, French SA, Sokol AA, Bromley ST, Thiel W, Turner AJ et al (2003) QUASI: a general purpose implementation of the QM/MM approach and its application to problems in catalysis. J Mol Struct THEOCHEM 632:1–28
Smellie A, Teig SL, Towbin P (1995) Poling: promoting conformational variation. J Comput Chem 16:171–187
Szczepankiewicz BG, Ng PY (2008) Sirtuin modulators: targets for metabolic diseases and beyond. Curr Top Med Chem 8:1533–1544
Taylor DM, Maxwell MM, Luthi-Carter R, Kazantsev AG (2008) Biological and potential therapeutic roles of sirtuin deacetylases. Cell Mol Life Sci 65:4000–4018
Tissenbaum HA, Guarente L (2001) Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 410:227–230
Vaziri H, Dessain SK, Eaton EN, Imai S-I, Frye RA, Pandita TK, Guarente L, Weinberg RA (2001) hSIR2SIRT1 functions as an NAD-dependent p53 deacetylase. Cell 107:149–159
Vu CB, Bemis JE, Disch JS, Ng PY, Nunes JJ, Milne JC, Carney DP, Lynch AV, Smith JJ, Lavu S et al (2009) Discovery of imidazo[1,2-b]thiazole derivatives as novel SIRT1 activators. J Med Chem 52:1275–1283
Westphal CH, Dipp MA, Guarente L (2007) A therapeutic role for sirtuins in diseases of aging? Trends Biochem Sci 32:555–560
Witt O, Deubzer HE, Milde T, Oehme I (2009) HDAC family: what are the cancer relevant targets? Cancer Lett 277:8–21
Yang Y, Zhang W, Cheng J, Tang Y, Peng Y, Li Z (2013) Pharmacophore, 3D-QSAR, and Bayesian model analysis for ligands binding at the benzodiazepine site of GABAA receptors: the key roles of amino group and hydrophobic sites. Chem Biol Drug Des 81:583–590
Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW (2004) Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 23:2369–2380
Acknowledgments
This work was supported by the Basic Science Research Program (2012R1A1A4A01013657), Pioneer Research Center Program (2009-0081539), the Management of Climate Change Program (2010-0029084) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, and the Next Generation BioGreen 21 Program (PJ009486) from Rural Development Administration (RDA) of Republic of Korea.
Conflict of interest
The authors declare no competing financial interests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Sakkiah, S., Arooj, M., Lee, K.W. et al. Theoretical approaches to identify the potent scaffold for human sirtuin1 activator: Bayesian modeling and density functional theory. Med Chem Res 23, 3998–4010 (2014). https://doi.org/10.1007/s00044-014-0983-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-014-0983-3