
Abstract
Wakefulness and consciousness depend on perturbation of the cortical soliloquy. Ascending activation of the cerebral cortex is characteristic for both waking and paradoxical (REM) sleep. These evolutionary conserved activating systems build a network in the brainstem, midbrain, and diencephalon that contains the neurotransmitters and neuromodulators glutamate, histamine, acetylcholine, the catecholamines, serotonin, and some neuropeptides orchestrating the different behavioral states. Inhibition of these waking systems by GABAergic neurons allows sleep. Over the past decades, a prominent role became evident for the histaminergic and the orexinergic neurons as a hypothalamic waking center.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Activation of the cerebral cortex
The cerebral cortex is active day and night, but we are not always aware of its activity. During slow wave sleep, the electroencephalogram (EEG) is dominated by high-voltage δ-waves (0.5–3 Hz) indicating a high degree of cortical inactivation or synchronization. Consciousness depends on external perturbation that causes a radical change in the cortical mode of function visible in the EEG as cortical activation or desynchronization, with low voltage and fast frequency (mainly β and γ, 20 and 60 Hz). This is achieved by ascending afferents leading to cortical activation during waking or paradoxical sleep (synonym REM sleep). Both of these behavioral states are conscious though in different ways.
Moruzzi and Magoun [1] demonstrated cortical arousal in the cat by stimulating and lesioning the brain stem reticular formation and formulated the concept of the ascending reticular activating system (ARAS), that reaches the cortex through the non-specific thalamus, the medial and intralaminar nuclei, as well as through extrathalamic pathways. During the following decades, stimulations, lesions, and brain transsections in combination with electrophysiological recordings (from EEG to single cells) have been used to determine the structures involved in the regulation of sleep and waking. Acute preparations of high brainstem transsection [2] or isolated forebrain display continuous slow synchronous high-amplitude activity, similar to that seen during deep slow wave sleep. These studies led to the conclusion that the cerebral cortex does not possess an intrinsic mechanism for its own activation and have identified four brain regions that can activate the cortex: (1) the thalamus, medial and intralaminar nuclei; (2) the basal forebrain (substantia innominata and adjacent areas); (3) the monoaminergic nuclei of the brainstem; (4) the posterior hypothalamus.
The hypothalamus, though long suspected to play a role in sleep-waking regulation, has been relatively neglected in the past and will be treated with preference here in the network formed by the four regions listed above. The thalamus, basal forebrain, and brainstem have been extensively reviewed in this context [3–11].
The ascending reticular activating system
The ARAS-concept [1] (Fig. 1) has been supported and complemented, especially at the cellular and electrophysiological levels, mainly by Steriade and co-workers [5, 6, 9, 12]. The excitatory inputs to the thalamus and other subcortical relay structures include cholinergic neurons of the mesopontine tegmentum, aminergic neurons in the brainstem and hypothalamus, and glutamatergic neurons located in the large brainstem reticular core [4, 8, 13, 14]. The reticulothalamocortical pathway is not the only system involved, however, as cortical EEG desynchronization can reappear following extensive destruction of the mesencephalic reticular formation [15] or its thalamic relay [16–18] indicating the existence of extrathalamic systems, capable of activating the cortex, that have drawn more attention recently: the magnocellular substantia innominata, the adjacent basal forebrain and the cholinergic and GABAergic corticopetal neurons [6–8], as well as the posterior hypothalamus with the histamine and orexin systems [19, 20].

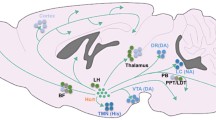
Ascending activation of the cortex. The ascending reticular activation system (ARAS) reaches the cortex through a ventral pathway (hypothalamus, basal forebrain), through the aminergic nuclei (containing catecholamines, acetylcholine, and serotonin) and a dorsal pathway, the thalamic relay. Switching between paradoxical sleep (REM-sleep) and slow wave sleep occurs in the reticular formation, whereas the switch between sleep and waking lies in the hypothalamus
The basal forebrain
Cholinergic neurons of the basal forebrain discharge tonically during both wakefulness and paradoxical sleep [6, 18]. They can excite cortical neurons directly and suppress the thalamic reticular nucleus oscillation generating the cortical spindles and drowsiness or light slow wave sleep [21]. In keeping with this, electrical stimulation of certain basal forebrain sites elicits cortical acetylcholine release and cortical desynchronization, while chemical inactivation or unilateral lesion in the basal forebrain cholinergic zone decrease cortical fast rhythms and increase slow activity [18, 22, 23]. Like thalamocortical neurons, basal forebrain cholinergic neurons can relay excitation (e.g., from glutamatergic, noradrenergic, and histaminergic neurons) from the lower brain reticular structures to the cortex [7, 10, 24]. GABAergic ascending neurons in the basal forebrain also project to the cortex [7] and might act in synergy with the cholinergic neurons in cortical activation, likely by ascending disinhibition, since they largely innervate inhibitory cortical neurons [25]. Thus, there is little doubt that the substantia innominata and the adjacent basal forebrain as a whole, including cholinergic, GABAergic, and perhaps further, non-identified neurons, play an important role in cortical activation both during waking and paradoxical sleep and in the modulation of different cortical rhythmic activities. However, the basal forebrain is not indispensable for the long-term maintenance of fast, low-voltage cortical activity, since, in the cat, extensive destruction of the basal forebrain, including the adjacent lateral preoptic areas, does not abolish cortical activation [26]. Ibotenic acid lesioning of the cholinergic zone within the basal forebrain results in a transitory reduction in waking lasting 1–2 days, after which the sleep-wake cycle returns to the pre-lesioning level [27].
Monoaminergic systems
An intense interest in the diffuse ascending projections from the brainstem monoaminergic neurons arose in the 1960s from the histochemical demonstration of their locations and projections [28] and the pharmacological intervention on monoaminergic transmission in major psychiatric disorders, schizophrenia, and depression. These diseases include disturbed sleep-waking regulation. Inhibition of catecholamine synthesis results in decreased waking and behavioral somnolence. Moreover, psychostimulants, such as amphetamine or cocaine, lead to an accumulation of catecholamines, causing a waking state and behavioral excitation.
Noradrenergic and serotonergic (but not most of the dopaminergic) neurons discharge tonically during waking, decrease their activity during slow wave sleep, and cease firing during paradoxical sleep [5, 9, 29–31]. In mice, locus coeruleus noradrenergic neurons show the earliest activation at wake onset among the known waking systems (137). In the cat, lesioning of the ventral tegmental area and the substantia nigra, containing dopaminergic ascending neurons, induces behavioral unresponsiveness and akinesia, but is not associated with loss of cortical activation [32].
A group of diffusely projecting dopaminergic cells in the ventral periaqueductal grey matter expresses c-fos after natural or forced wakefulness and their lesioning results in increased sleep. These cells may provide the long-sought ascending dopaminergic activation [33]. Large lesions of the mediopontine tegmentum, including the locus coeruleus and the ascending noradrenergic pathway, impair waking [11], while electrolytic or chemical lesioning confined to the locus coeruleus region [34] or selective DSP-4 lesioning of locus coeruleus ascending projections to the cortex [35] does not cause a major deficit of waking. Ascending dopaminergic neurons, as a whole, can thus play an important role in waking, notably locomotion, motivation and cognitive activities, whereas the brainstem noradrenergic neurons (groups A1–A7, including neurons in the locus coeruleus) are permissive in paradoxical sleep, and modulate behavioral and cortical arousal, as well as qualitative and cognitive aspects of waking, such as perception [5, 8, 11, 14, 29, 31, 36] or the expression of immediate early genes [37]. Although dorsal raphe serotonergic neurons possess widespread ascending projections to the cortex and discharge tonically during waking [5, 30], and although serotonin may participate in cortical activation when associated with acetylcholine [38], pharmacological depletion of serotonin using para-chlorophenylalanine or lesioning/inactivation of the dorsal raphe nucleus results in insomnia [39]. The postsynaptic action of serotonin on many neuronal targets is dominated by the strong inhibition through 5-HT 1A receptors, while the waking amines, such as noradrenaline or histamine display predominantly excitatory or excitation potentiating actions [7, 8, 10, 14, 40, 41]. A modulation of the sleep-wake cycle by serotonin is evident but it seems not directly and centrally involved in cortical activation and waking (Fig. 2).

The waking brain. Schematic localization of the brain’s waking systems and their inhibition. NA (noradrenaline, locus coeruleus); ACh (acetylcholine, pedunculo-pontine nucleus, PPN; lateral dorsal tegmentum, LDT; basal forebrain, BF); DA (dopamine, periaquaeductal grey, PAG), 5-HT (serotonin, dorsal raphe, DR); HA (histamine, tuberomamillary nucleus, TMN), OX (orexins, periventricular nucleus, PVN); POA: preoptic area, GABAergic neurons which inhibit all the waking systems, black lines indicate inhibitory pathways
The posterior hypothalamus is a waking center
The posterior hypothalamus is a heterogeneous structure made up of different neuronal populations containing diverse neurotransmitters (histamine, dopamine, glutamate, GABA) and neuropeptides [orexins, melanin concentrating hormone (MCH), galanin, enkephalins, substance P, thyreotropin releasing hormone (TRH)]. This region has extensive reciprocal anatomical connections with many brain regions, notably those involved in sleep-wake control such as the cortex, thalamus, preoptic-anterior hypothalamus and other forebrain structures, the brainstem cholinergic, and monoaminergic nuclei [42–45].
The posterior hypothalamus has only recently been recognized as a major waking center in spite of early indications (Fig. 2). After the influenza epidemic of 1918, von Economo identified hypothalamic lesions in the anterior or in the posterior hypothalamus correlating with insomnia and hypersomnia (encephalitis lethargica [46]). Subsequent studies in cats, monkeys, and rats have confirmed that electrolytic lesioning of the posterior hypothalamus causes somnolence, hypersomnia, or coma [4]. Nauta defined a waking center in the posterior hypothalamus and a sleep center in the preoptic/anterior hypothalamus on the basis of lesion studies in the rat. He suggested a reciprocal interaction between these two hypothalamic centers in the alternation of sleep and wakefulness [47]. The posterior hypothalamus as a waking center is also supported by the fact that electrical stimulation of this region in the normal [48] or mesencephalic transsectioned [22] cat causes EEG desynchronization.
This role of the posterior hypothalamus has recently received increasing interest with the identification of widespread hypothalamo-cortical projection systems [49–51] and electrophysiological studies revealing several types of neurons, discharging with neocortical activation [42, 52–54], suggesting a source for driving cortical arousal. Sakai et al. [55] have identified three types of tonic unitary activity in the cat: type-I neurons, discharging during waking and paradoxical sleep, and type-II neurons with a significantly higher discharge rate during paradoxical sleep than during waking and slow wave sleep. Both patterns are encountered diffusely in the posterior hypothalamus. Type-III neurons displaying paradoxical sleep-off or waking-specific discharge have been identified in the tuberomamillary nucleus and the ventrolateral area of the posterior hypothalamus. Thus, the posterior hypothalamus, like the thalamus and the basal forebrain, represents a major component of the ascending activating system.
As electrical lesions [4] destroy not only cellular somata but also fibers en passage, more recent studies [15, 56] have used chemical agents such as excitatory amino acids (kainic or ibotenic acid) to induce selective cell death following over-excitation of neurons. Cellular destruction, under anesthesia, of large areas in the cat posterior hypothalamus including the most caudal part and the hypothalamo-mesencephalic junction produces hypersomnia including both paradoxical sleep and slow wave sleep lasting 1–2 days, accompanied by narcoleptic episodes, i.e., direct onsets of paradoxical sleep from waking (sleep onset REM); while lesions restricted to the rostral part of the posterior hypothalamus, sparing the hypothalamo-mesencephalic junction produce a significant decrease in waking and an increase in slow wave sleep lasting for 1–3 weeks.
Muscimol (GABAA-receptor agonist) injections can acutely inactivate different hypothalamic loci and deliver functional information on their role in sleep-wake states. In normal freely moving animals, muscimol microinjection into the preoptic/anterior hypothalamus or the hypothalamo-mesencephalic junction provokes increased waking and hyperactivity. In sharp contrast, the same injection in the rostral and middle parts of the posterior hypothalamus induces a pronounced and long-lasting increase in deep slow wave sleep, accompanied by a reduction in, or suppression of, paradoxical sleep. When the injection is performed in the caudal part, the increase in deep slow wave sleep is followed by an increase either in waking or paradoxical sleep, depending upon the exact injection site. In the latter case, paradoxical sleep can even occur directly from waking as narcolepsy (sleep onset REM) [57].
The rostral and middle parts of the posterior hypothalamus, so far the sole brain region associated with such a pronounced hypersomnia after inactivation by muscimol, are therefore the main hypothalamic waking territory. Under physiological conditions, this region must be inactivated to allow the appearance and maintenance of sleep likely by the local release of GABA that inhibits the wake on neurons. A selective increase in GABA during slow wave sleep is indeed seen in the cat posterior hypothalamus [58].
Further support for the central role of the posterior hypothalamus in the maintenance of waking comes from a number of observations in insomniac cats: insomnia caused by inhibiting the synthesis of serotonin by para-chlorophenylalanine is reversed by muscimol injection in the TM and adjacent areas with restoration of slow wave sleep and paradoxical sleep with short latency [57]. Similarly, lesioning of the preoptic and anterior hypothalamus results in long-lasting insomnia and hyperthermia, both effects being reversed by muscimol microinjection into the TM and adjacent areas with restoration of both slow wave sleep and paradoxical sleep [59].
During the long-lasting and total waking state following the enhancement of dopaminergic transmission by amphetamine, slow wave sleep (but not paradoxical sleep) is restored at short latency by microinjection of muscimol into the TM area. The wake-promoting drug modafinil, which causes long-lasting quiet waking without behavioral activation, acts through dopamine D2 receptors in the ventral tegmental area [60] and other arousal systems but not in TM histamine neurons. This waking state is reversed by local injection of muscimol [19, 61, 62]. The histamine neurons display an unusually low sensitivity to D1 and D2R agonists but are highly sensitive to l-Dopa, which they can take up and convert to dopamine [63].
Thus, inactivation of the posterior hypothalamus induces hypersomnia in normal cats and restores sleep in various models of insomnia, suggesting a key role of this region in the maintenance of cortical activation and the waking state. Posterior hypothalamic neurons are likely in a state of hyperactivity during insomnia and offer themselves as targets for medication. Nelson et al. [64] reproduced the muscimol microinjection and also injected gabazine in the rat posterior hypothalamus TMN which antagonized propofol anesthesia and loss of the righting reflex. They attribute a key role to this region for the action of GABAergic anesthetics. The posterior hypothalamus also controls sympathetic and behavioral functions, such as thermoregulation, cardiovascular and respiratory regulation, locomotion, emotional reactions, and feeding behaviors [4, 65–68]. It therefore seems likely that, during waking, cortical EEG activity and the concomitant behavioral signs of arousal are coordinated at the level of the posterior hypothalamus, thus organizing an integral functional activation of the brain.
Neuronal substrates involved in arousal in the posterior hypothalamus
The posterior hypothalamus contains different categories of neurons: those involved in the control of cortical activation and waking display arborizing projections, allowing the modulation of large brain areas. The dopaminergic A11 group has massive hypothalamic projections and sends fibers to the mesopontine tegmentum [69], which plays an important role in the cortical activation during waking and paradoxical sleep [5, 9, 21, 70]. Muscimol microinjection in the dorsolateral and perifornical regions, which, in the cat, contain both type-I and type-II tonic neurons [55] induces, with a certain latency, continuous deep slow wave sleep accompanied by suppression of paradoxical sleep [57]. These populations, including orexin and MCH neurons, send out widespread ascending and descending projections [51].
Experimental data obtained from our laboratories as well as the results from other groups suggest a major role of the histaminergic neurons located in the tuberomamillary nucleus and ventrolateral part of the posterior hypothalamus in waking. The sedation caused by classical antihistamines (H1-receptor antagonists) has long been known as an undesirable side-effect in the treatment of allergy [71]. Only after histamine was recognized as a transmitter in the brain [72–77] a block of histaminergic transmission was made responsible for the drowsiness caused by antihistamines [78] and many drugs used in the treatment of neuropsychiatric diseases that bind to the H1-receptors.
The histaminergic system in brain
Histamine is synthesized from histidine by histidine-decarboxylase; its levels in the brain measured by microdialysis display a circadian rhythmicity in accordance with the firing of histamine neurons during waking [79]. Extracellular histamine levels in the preoptic/anterior hypothalamus follow the oscillations of different sleep stages (wakefulness > non-REM sleep > REM sleep). Sleep deprivation does not affect histamine levels, suggesting the relay of circadian rather than homoeostatic sleep drive [80]. Philippu and Prast [81] have demonstrated a direct correlation between histamine levels in the hypothalamus and behavioral state by electroencephalography. Synthesis and release of histamine are controlled by feedback through H3-autoreceptors located on somata and axonal varicosities [82]. Inactivation of histamine in the extracellular space of the CNS is achieved solely by methylation through neuronal histamine N-methyltransferase [83].
The tuberomamillary nucleus contains ca. 3,000 neurons in the rat and about 64,000 in man, and is the only source of neuronal histamine in the adult vertebrate brain and histamine is its main transmitter. Further transmitters (or their synthetic enzymes) expressed within tuberomamillary nucleus neurons include GABA, galanin, enkephalins, TRH, and substance P. Histamine neurons have widespread projections to the whole brain.
Histaminergic neurons present morphological and electrophysiological properties similar to those seen in other aminergic neuron populations [84]. They fire slow and regular at a membrane potential of about −50 mV with 1–4 action potentials per second [85]. The action potentials have a significant contribution from Ca2+ channels triggering a strong afterhyperpolarization. The excitatory arm of the pacemaker cycle includes dendritic Ca2+ potentials and a non-inactivating Na+ current. The Ca2+-currents are likely instrumental for histamine release from dendrites and axons; they are blocked by H3-autoreceptor activation. In behaving cats, rats and mice, the firing is more variable during waking and absent upon drowsiness and during sleep [52, 55, 86, 87]. This is the most wake-selective firing pattern identified in the brain to date.
Tuberomamillary neurons are influenced by many transmitters and other humoral signals. Excitatory and inhibitory synaptic potentials (EPSPs and IPSPs) evoked by stimulations of several locations are mediated by glutamate and GABA. Monoaminergic, cholinergic, and peptidergic fibers innervate the tuberomamillary neurons. Many peptides function as signaling molecules in the hypothalamus where they are involved in endocrine and homoeostatic functions. They can be co-expressed and differentially released with other neurotransmitters; in many neurons, however, they represent the main transmitter or hormone.
Aminergic and some peptidergic neurons are mutually connected, mostly through excitation, occasionally also inhibition, forming an orchestra that is to a certain extent self-organizing. The orexin neurons give the signals for sleep-waking architecture and the histaminergic neurons are the dominant cell group with respect to cortical arousal and wake quality. Multifold arborizing histaminergic axons reach the entire central nervous system through two ascending and one descending bundle [72, 88–92]. The highest density of histaminergic fibers is seen in the hypothalamus. In the posterior part, the fibers often make close contact to the brain surface. The septal nuclei and those of the diagonal band receive a very strong innervation.
Four metabotropic histamine receptor types (H1R–H4R) have been cloned so far. H1R, H2R, and H3R are expressed in abundance in the brain. All histamine receptors display constitutive activity. The sedative effects of antihistamines (H1-antagonists) have prompted early suggestions of histamine as a waking substance [78]. Neuronal excitation is achieved by activation of H1R, Gq/11-proteins and phospholipase C, the formation of the two second messengers DAG and IP3, as well as intracellular Ca2+ release, which can trigger: (1) opening of cation channels, causing depolarization; (2) activation of an electrogenic Na–Ca-exchanger (NCX), causing depolarization; (3) formation of NO and cyclic GMP; (4) opening of Ca2+-dependent potassium channels, resulting in a hyperpolarization [93]. Blocking a potassium leak conductance through direct G-protein action can shift the thalamic relay mode towards an open state and cortical activation [94]; or directly excite cortical neurons [95]. Activation of a tetrodotoxin insensitive Na-current is proposed for the excitation of cholinergic septal neurons [96] and a mixed cation channel for the excitation of dorsal raphe serotonergic neurons [97]. Firing is also increased in the suprachiasmatic nucleus [98] and cholinergic basal forebrain neurons [24].
The histamine H2 receptors, β-adrenergic receptors, serotonin 5-HT2 receptors, among others, are coupled to Gs-protein, adenylyl cyclase and PKA, which phosphorylates proteins and activates the transcription factor CREB. The direct action on neurons is usually excitatory or excitation potentiating. Through this signaling pathway these transmitters block a Ca2+-dependent potassium conductance, which is responsible for long-lasting afterhyperpolarizations and the accommodation of firing. This effect modulates the response of target neurons (e.g., in cerebral cortex and hippocampus): an identical stimulus can thus elicit a response consisting of few or many action potentials depending on the aminergic activation. Such a potentiation of excitation is perfectly suited to raise attention.
H3 receptors function as autoreceptors on histaminergic cell somata, dendrites, and axons (varicosities) where they provide a negative feedback to restrict histamine synthesis and release. Importantly, as heteroreceptors, they are also located on many non-histaminergic axons where they modulate the release of glutamate, GABA, noradrenaline, and acetylcholine. H3 receptors are coupled to Gq and high-voltage activated Ca channels, a typical mechanism for the regulation of transmitter release (Fig. 3). A high degree of molecular and functional heterogeneity through different transcriptional and post-transcriptional processing (splice variants) is prototypic for the H3R [82, 99]. H3R-related drugs are being developed largely for the treatment of sleep disorders [100].

The histamine H3R as an autoreceptor mediating negative feedback on histamine synthesis and release, and, importantly, as a heteroreceptor suppressing the release of many other transmitters from their varicosities. H3R antagonists and partial agonists are widely developed for several neuropsychiatric indications including sleep disorders
Histamine and waking
The above data strongly indicate that histaminergic neurons activate or facilitate large brain areas through postsynaptic H1- and H2-receptors, thus contributing to cortical activation. Indeed, treatments that impair histamine-mediated neurotransmission enhance cortical slow activity and increase sleep. For instance, the blockade of histamine synthesis with α-fluoromethylhistidine markedly reduces histamine levels, decreases waking, and increases slow wave sleep in the cat [19] and rodents [101–103]. In contrast, enhancement of histaminergic neurotransmission by inhibiting histamine degradation promotes waking, reviewed in [19, 75, 102]. The absence of histamine synthesis in histidine decarboxylase knockout mice impairs the cortical EEG and has deleterious effects on both sleep and wake quality, causing permanent somnolence and behavioral deficits. Consequently, mice that lack brain histamine are unable to remain awake when high vigilance is required, at lights off, or when they are placed in a new environment [103] (Fig. 4). Like orexin neurons [104], histamine neurons may also be involved in CO2-mediated arousal; they are activated by short-term hypoxia [105] and are excited by mild acidification (Sergeeva, unpublished observations). Taken together, histaminergic neurons have a key role in maintaining the brain awake. They promote wakefulness through their direct widespread projections to the cerebral cortex and indirectly via their subcortical targets in the thalamus, basal forebrain, and brainstem [14, 75, 92].

Effects of an environmental change on the cortical EEG and behavioral states in wild-type (upper) and knockout-mice lacking histamine synthesis (lower). The environmental change (middle) consists of transferring the mice from their habitual home cage (A) to a new cage (B). Upper A wild-type mouse with an intact brain histaminergic system. This mouse placed in the new environment remains highly awake, as indicated by the alert behavior and waking EEG. Lower A knockout mouse without histamine. This mouse falls asleep a few minutes after being placed in the new environment, as shown by the sleeping behavior and EEG signs of slow wave sleep. Histaminergic neurons play a key role in maintaining the brain in an awake state in the presence of behavioral challenges. Modified from Parmentier et al. (2002) Journal of Neuroscience 22:7695–7711
Orexinergic/hypocretinergic neurons
Orexins/hypocretins are two peptides (Ox-A, OxB/HCrt1, HCrt2) derived from proteolytic cleavage of a precursor peptide encoded by the prepro-orexin(hypocretin) gene. The orexin-containing neurons are almost exclusively located in the perifornical area of the dorsolateral hypothalamus, therefore, just dorsorostral to the histaminergic tuberomamillary nucleus. Like histamine neurons, they project all over the brain [106]. They bind to two G-protein-coupled receptors (OX1/2-R, HCrt1/2-R) [107–111]. Orexin neurons also contain excitatory glutamate and inhibitory dynorphin [112]. Orexin receptors are expressed in numerous targets throughout and even outside the nervous system. The name orexins, indicating a function in food intake, was first envisaged [113]; however, it soon became apparent that these peptides fulfil important roles in the regulation of behavioral state and sleep architecture [114, 115] and serve many physiological functions [108]. Deficiency of the orexins is the cause of narcolepsy-cataplexy whereas their hyperactivity, for instance after sleep deprivation or metabolic challenges [116, 117], predisposes to addiction and compulsion (see [118]).
Orexin neurons display a wake-active discharge pattern, clearly correlated to muscle tone and posture change, with a significant decrease from active waking to quiet waking and from quiet waking to slow wave sleep [119–121]. In the rat, the discharge rate of orexin neurons during active waking is more than 4.5 times that of quiet waking, indicating that their main activity is to promote behavioral activation during waking [119, 120]. Cerebrospinal fluid Ox-A level [122] or c-fos expression in orexin neurons [123] increase after forced waking or behavioral activation. Finally, central application of orexins elicits active arousal and hyperactivity in rats, an effect prevented by SB-334867 [124, 125]. Taken together, we suggest that Ox-neurons promote locomotion and behavioral arousal and thus contribute to the maintenance of waking by enhancing locomotion [126].
Orexins are also involved in higher brain functions; they can facilitate memory performance and synaptic plasticity [127, 128]. An OX2R-dependent increase of GABAergic transmission in septo-hippocampal pathways may promote arousal via hippocampal disinhibition and theta rhythm [129] and a behavioral-state-dependent large-scale oscillatory brain activity associated with heightened synaptic plasticity and memory processing during REM-sleep, exploratory behavior, and stress [130]. Long-term potentiation of synaptic transmission (LTP) in the hippocampus [131], a cellular correlate of learning and memory is enhanced by orexin infusion into the rat dentate gyrus or locus coeruleus in vivo, while stimulus-induced LTP of Schaffer collateral-CA1 synapses in dorsal hippocampal slices as well as spatial memory in a water maze task is inhibited by orexins. OX-A induces an endogenous form of LTP at excitatory Schaffer collateral-CA1 synapses (LTPOX), relying on co-activation of metabotropic amino acid and biogenic amine receptors [132].
The respective roles of orexin and histamine-systems for waking
The histaminergic neurons are currently regarded as a downstream system driven by the orexin neurons through their dense axon arborizations in the tuberomamillary nucleus. However, recent studies show that the behavioral and sleep-wake phenotypes of histidine-decarboxylase (HDC, histamine-synthesizing enzyme)−/− mice are distinct from those of orexin knockout(−/−) mice [126, 133]. While both mouse strains display sleep fragmentation and increased paradoxical sleep, they present a number of marked differences:
(1) The paradoxical sleep-increase in HDC−/−mice is seen during lightness, whereas that in Ox−/−mice occurs during darkness; (2) Contrary to HDC−/−, Ox−/−mice have neither waking deficiency around lights-off, nor an abnormal EEG and respond to a new environment with increased waking; (3) Only Ox−/−, but not HDC−/−mice, display narcolepsy and deficient waking when faced with a motor challenge. Wild-type, but not littermate Ox−/−mice, when placed on a wheel, voluntarily spend their time in turning it, and as a result, remain highly awake (Fig. 5); this is accompanied by dense c-fos expression in many areas of their brains, including Ox-neurons in the dorsolateral hypothalamus. The waking and motor deficiency of Ox−/−mice is due to the absence of Ox as intraventricular dosing of Ox-A restores their waking amount and motor performance. SB-334867 (Ox1-receptor antagonist, i.p.) impairs waking and locomotion of wild-type mice during the test.

Different behavioral performance and ability to maintain waking between wild-type and orexin knockout mice when faced with a motor challenge demonstrated using simultaneous electroencephalogram and elecromyogram monitoring (upper). When wild-type mice (middle left) were placed on a wheel, they voluntarily spent their time in turning it and, as a result, remained highly awake. In contrast, orexin knockout mice (middle right) usually tried to adapt a position to stay immobile, thus falling asleep. Note the absence of orexin neurons in the knockout mice (lower). Modified from Anaclet et al. (2009) Journal of Neuroscience 29:14423–14438
Thus, histamine- and orexin-neurons, with their reciprocal interactions, exert a synergistic and complementary control over waking, the histaminergic system being mainly responsible for cortical activation (EEG) and cognitive activities and the orexinergic system being more involved in the behavioral arousal during waking, including muscle tone, posture, locomotion, food intake, and emotional reactions. Orexin deficiency is in most cases the direct cause of narcoleptic episodes in humans (DREMs, direct onsets of REMs from wake or SOREMs, sleep onsets REMs) and cataplexy [134], whereas decreased histaminergic transmission likely accounts for the somnolence and excessive daytime sleepiness seen in this disease and other sleep disorders [103, 126, 135, 136]. The advent of optogenetic stimulation has opened new ways to study the impact of defined neuronal populations on behavior. Carter et al. [137] reported recently such activation of orexin/hypocretin neurons causing increased waking and c-Fos expression in locus coeruleus and the tuberomamillary nucleus, but this effect was lost after sleep deprivation, presumably being overwhelmed by homoeostatic mechanisms (see below). Interestingly, the increase in waking was unchanged in HDC-KO mice lacking histamine: the histaminergic system is an important, but only one of many targets of the widely arborizing orexin/hypocretin axons. There clearly is redundancy within the wake-active systems. They act together like an orchestra and are able to compensate for the failure of some of their players. For instance, 3 weeks after triple saporin-induced lesions of the cholinergic forebrain, the tuberomamillary nucleus and the locus coeruleus in the rat, the remaining phenotype is a wake deficit during the light to dark period [138], similar to that identified in HDC-KO mice [133]. The acute effects of such lesions, notably those on the neocortical EEG, remain to be investigated.
The search for substances increasing alertness led to the discovery of the wake-promoting action of montirelin (a non-hydrolyzable TRH analogue, which showed beneficial action in canine narcolepsy [139]). TMN neurons express two known TRH receptors, are excited by TRH and montirelin, and the wake-promoting action of montirelin is missing in histamine-deficient mice [133]. Thus, the histaminergic system represents an attractive target for wake-promoting medication in narcolepsy [136] and e.g., in Parkinson’s disease [140], where most arousal centers undergo degeneration while the histaminergic system remains intact and an H3-receptor inverse agonist increases alertness [100, 140]. Interestingly, modafinil exerts only a minor action in PD [140] in accordance with the degeneration of dopaminergic neurons and subsequent down-regulation of D2R.
Other posterior hypothalamic neurons regulating sleep-wake alternation
The posterior hypothalamus is a heterogeneous structure also from a functional point of view. In addition to its well-recognized role in wake, it has long been suggested that this region exerts hypothalamic control over the brainstem paradoxical sleep-generating mechanisms and may play an important role in cortical activation not only during wakefulness but also during paradoxical sleep, reviewed in [3, 11, 55].
In addition to the histaminergic wake-specific and orexinergic wake-active discharge patterns, neurons firing selectively and tonically during paradoxical sleep were also identified in the cat and more recently in the rat posterior hypothalamus [55, 141]. This pattern is driven, at least in part, by neurons containing MCH in the rat [141]. Although MCH-containing cells are found in the same area where orexin cells are located, the perifornical area of the dorsolateral hypothalamus [49, 142], the fact that they discharge in a reciprocal manner to orexin or histamine neurons [141] suggests that they might play a different role than orexin or histamine neurons in sleep-wake regulation. However, intracerebroventricular supply of MCH increases paradoxical sleep supporting a role in promoting this behavioral state, whereas mice lacking MCH-R1 also show enhanced paradoxical sleep [143]. Thus, the hypothalamic mechanisms involved in cortical activation during paradoxical sleep remain to be clarified, but it seems important to determine the possible interactions between MCH-containing cells and orexin and histamine neurons to further understand the hypothalamic control on sleep-wake alternation via the three widespread projecting systems.
Interaction between the waking systems and sleep-generating mechanisms
The waking systems are inactivated to allow sleep. Sleep-wake alternation results from an interaction between the waking systems and the brain’s sleep-generating mechanisms. The best defined brain structure for sleep generation is the preoptic-anterior hypothalamus, which contains dense populations of sleep-active neurons discharging at a high rate during slow wave sleep [26, 144]. A lesion of this region causes severe insomnia [59]; reviewed in Szymusiak [145]. Later on, it was proposed that the ventrolateral [146], median [147] or dorsolateral [148] preoptic area and adjacent regions generate sleep mainly through GABAergic inhibition of the aminergic and peptidergic (orexins) waking systems.
Early studies also suggest that waking occurs by direct or indirect inhibition of the preoptic area by the waking systems. Microinjection of histamine in this area enhances waking in the cat [149]. In vitro studies demonstrated direct and indirect inhibition of sleep-active preoptic neurons by the ascending activating neurotransmitters noradrenaline, serotonin, acetylcholine [150], and histamine [151].
The classical view on the orchestration of sleep-wake alteration by interactions between sleep-generating and wake-promoting structures is currently challenged or modified: sleep-active neurons are also identified in wake-promoting structures like the tuberomamillary nucleus and the adjacent posterior hypothalamus whereas wake-specific neurons are also found in the preoptic and the adjacent basal forebrain. Moreover, at the transition from wake to slow wave sleep, sleep-active neurons discharge not before, but after, cessation of activity of the wake-specific neurons, indicating that release of the inhibition by wake-promoting systems plays a major role in sleep generation [144, 152, 153].
Circadian and homoeostatic regulation of sleep and waking
A two-process model of sleep-waking regulation proposes the factors C (circadian pacemaker) and S (sleep propensity increasing with the duration of waking, homoeostasis) [154, 155]. A search for endogenous substances interacting with this regulation continues for decades, in particular for promoting sleep [156]. Adenosine has been identified as a candidate that accumulates in the brain during strong nervous activity, during prolonged waking. It causes sedation [157] and likely induces sleep [158, 159]. Adenosine A1 receptors are positively coupled to various potassium channels, negatively to Ca2+-channels and cyclic AMP, exerting post- and presynaptic inhibition at many sites in the brain, specifically in the cholinergic basal forebrain [160]. Interestingly, the histaminergic neuron’s firing is unaffected by adenosine [161]. Adenosine A2A receptors are more localized, and they mediate excitation of sleep-active neurons in the preoptic area. Both these adenosine receptors are blocked by caffeine, resulting in arousal, especially at times when endogenous adenosine has accumulated during sleep deprivation [159, 162]. Whereas adenosine levels correlate with low energy reserve [163], high levels of the energy-rich adenosine-triphosphate (ATP) can mediate an increased excitability through closure of KATP-channels and, after release to the extracellular space, to direct excitation through ionotropic and metabotropic purine receptors of the P2 type, e.g., in histaminergic neurons [161].
In summary, multiple waking systems operate together to ensure the complex vital function wakefulness. The different activating and inhibiting systems form a complex distributed network. Disturbances of sleep and waking are frequent, socially and economically relevant. Understanding the regulation and the neural mechanisms opens the way for successful intervention: recent efforts in drug discovery concern the waking center in the posterior hypothalamus.
References
Moruzzi G, Magoun HW (1949) Brain stem reticular formation and activation of the EEG. Electroencephalogr Clin Neurophysiol 1:455–473
Bremer F (1935) Cerveau isolé et physiologie du sommeil. C R Soc Biol (Paris) 118:1235–1242
Jouvet M (1993) From amines to sleep—a citation-classic commentary on the role of monoamines and acetylcholine-containing neurons in the regulation of the sleep-waking cycle by Jouvet, M. Curr Contents Life Sci 8
Moruzzi G (1972) The sleep-waking cycle. Ergebnisse der Physiologie Biologischen Chemie und Experimentellen Pharmakologie 64:1
Steriade M, McCarley RW (1990) Brainstem control of wakefulness and sleep. Plenum Press, New York
Steriade M, Buzsáki G (1990) Parallel activation of thalamic and cortical neurons by brainstem and basal forebrain cholinergic system. In: Steriade M, Biesold D (eds) Brain cholinergic systems. Oxford University Press, Oxford, pp 3–64
Jones B, Muhlethaler M (1999) Cholinergic and GABAergic neurons of the basal forebrain: role in cortical activation. In: Lydic R, Baghdoyan HA (eds) Handbook of behavioral state control: cellular and molecular mechanisms. CRC Press, Florida, pp 213–233
Jones BE (1999) Basic mechanisms of sleep-wake states. In: Kryger MH, Roth T, Dement WC (eds) Principles and practice of sleep medicine. Saunders, Philadelphia, pp 145–162
Steriade M, McCarley R (2005) Brainstem control of wakefulness and sleep. Plenum, New York
Jones BE (2005) From waking to sleeping: neuronal and chemical substrates. Trends Pharmacol Sci 26:578–586
Jouvet M (1972) Role of monoamines and acetylcholine-containing neurons in regulation of sleep-waking cycle. Ergebnisse der Physiologie Biologischen Chemie und Experimentellen Pharmakologie 64:166
Steriade M (2003) The corticothalamic system in sleep. Front Biosci 8:d878–d899
Steriade M, McCormick DA, Sejnowski TJ (1993) Thalamocortical oscillations in the sleeping and aroused brain. Science 262:679–685
McCormick DA (1992) Neurotransmitter actions in the thalamus and cerebral-cortex and their role in neuromodulation of thalamocortical activity. Prog Neurobiol 39:337–388
Denoyer M, Sallanon M, Buda C, Kitahama K, Jouvet M (1991) Neurotoxic lesion of the mesencephalic reticular-formation and or the posterior hypothalamus does not alter waking in the cat. Brain Res 539:287–303
Angeleri F, Marchesi GF, Quattrini.A (1969) Effects of chronic thalamic lesions on electrical activity of neocortex and on sleep. Archives Italiennes de Biologie 107:633
Vanderwolf CH, Stewart DJ (1988) Thalamic control of neocortical activation: a critical re-evaluation. Brain Res Bull 20:529–538
Buzsaki G, Bickford RG, Ponomareff G, Thal LJ, Mandel R, Gage FH (1988) Nucleus basalis and thalamic control of neocortical activity in the freely moving rat. J Neurosci 8:4007–4026
Lin JS (2000) Brain structures and mechanisms involved in the control of cortical activation and wakefulness, with emphasis on the posterior hypothalamus and histaminergic neurons. Sleep Med Rev 4:471–503
Eriksson KS, Sergeeva OA, Haas HL, Selbach O (2010) Orexins/hypocretins and aminergic systems. Acta Physiol (Oxf) 198:263–275
Steriade M (1991) Alertness, quiet sleep, dreaming. In: Peters A (ed) Cerebral cortex. Plenum Press, New York, pp 279–357
Belardetti F, Borgia R, Mancia M (1977) Prosencephalic mechanisms of ecog desynchronization in cerveau-isole cats. Electroencephalogr Clin Neurophysiol 42:213–225
Casamenti F, Deffenu G, Abbamondi AL, Pepeu G (1986) Changes in cortical acetylcholine output induced by modulation of the nucleus basalis. Brain Res Bull 16:689–695
Khateb A, Fort P, Pegna A, Jones BE, Muhlethaler M (1995) Cholinergic nucleus basalis neurons are excited by histamine in vitro. Neuroscience 69:495–506
Freund TF, Meskenaite V (1992) Gamma-aminobutyric acid-containing basal forebrain neurons innervate inhibitory interneurons in the neocortex. Proc Natl Acad Sci USA 89:738–742
Szymusiak R, McGinty D (1986) Sleep-related neuronal discharge in the basal forebrain of cats. Brain Res 370:82–92
Lin JS (1994) Systeme histaminergique central et les etats de vigilance chez le chat. Universite Claude Bernard, Lyon, pp 1–238 (thesis/dissertation)
Fuxe K, Dahlstrom A, Hoistad M, Marcellino D, Jansson A, Rivera A, Diaz-Cabiale Z, Jacobsen K, Tinner-Staines B, Hagman B, Leo G, Staines W, Guidolin D, Kehr J, Genedani S, Belluardo N, Agnati LF (2007) From the Golgi-Cajal mapping to the transmitter-based characterization of the neuronal networks leading to two modes of brain communication: wiring and volume transmission. Brain Res Rev 55:17–54
Hobson JA, McCarley RW, Wyzinski PW (1975) Sleep cycle oscillation—reciprocal discharge by 2 brain-stem neuronal groups. Science 189:55–58
McGinty DJ, Harper RM (1976) Dorsal raphe neurons—depression of firing during sleep in cats. Brain Res 101:569–575
Steriade M, Hobson J (1976) Neuronal activity during the sleep-waking cycle. Prog Neurobiol 6:155–376
Jones BE, Bobillier P, Pin C, Jouvet M (1973) Effect of lesions of catecholamine-containing neurons upon monoamine content of brain and EEG and behavioral waking in cat. Brain Res 58:157–177
Lu J, Jhou TC, Saper CB (2006) Identification of wake-active dopaminergic neurons in the ventral periaqueductal gray matter. J Neurosci 26:193–202
Jones BE, Harper ST, Halaris AE (1977) Effects of locus coeruleus lesions upon cerebral monoamine content, sleep-wakefulness states and response to amphetamine in cat. Brain Res 124:473–496
Gonzalez MMD, Debilly G, Valatx JL (1998) Noradrenaline neurotoxin DSP-4 effects on sleep and brain temperature in the rat. Neurosci Lett 248:93–96
Foote SL, Bloom FE, Astonjones G (1983) Nucleus locus ceruleus—new evidence of anatomical and physiological specificity. Physiol Rev 63:844–914
Cirelli C, Pompeiano M, Tononi G (1996) Neuronal gene expression in the waking state: a role for the locus coeruleus. Science 274:1211–1215
Dringenberg HC, Vanderwolf CH (1998) Involvement of direct and indirect pathways in electrocorticographic activation. Neurosci Biobehav Rev 22:243–257
Petitjean F, Buda C, Janin M, Sallanon M, Jouvet M (1985) Insomnia due to the administration of parachlorophenylalanine—reversibility by peripheral or central injection of 5-hydroxytryptophane and serotonin. Sleep 8:56–67
Haas HL, Konnerth A (1983) Histamine and noradrenaline decrease calcium-activated potassium conductance in hippocampal pyramidal cells. Nature 302:432–434
Madison DV, Nicoll RA (1982) Noradrenaline blocks accommodation of pyramidal cell discharge in the hippocampus. Nature 299:636–638
Sakai K, Yoshimoto Y, Luppi PH, Fort P, Elmansari M, Salvert D, Jouvet M (1990) Lower brain-stem afferents to the cat posterior hypothalamus—a double-labeling study. Brain Res Bull 24:437–455
Yoshimoto Y, Sakai K, Luppi PH, Fort P, Salvert D, Jouvet M (1989) Forebrain afferents to the cat posterior hypothalamus—a double labeling study. Brain Res Bull 23:83–104
Sherin JE, Elmquist JK, Torrealba F, Saper CB (1998) Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci 18:4705–4721
Ford B, Holmes CJ, Mainville L, Jones BE (1995) Gabaergic neurons in the rat pontomesencephalic tegmentum—codistribution with cholinergic and other tegmental neurons projecting to the posterior lateral hypothalamus. J Comp Neurol 363:177–196
von Economo C (1926) Die Pathologie des Schlafes. In: von Bethe A, Bergmann GV, Embden G, Ellinger A (eds) Handbuch der Normalen und Pathologischen Physiologie. Springer, Berlin, pp 591–610
Nauta WHJ (1946) Hypothalamic regulation of sleep in rats. Experimental study. J Neurophysiol 9:285–316
Wilson CL, Motter BC, Lindsley DB (1976) Influences of hypothalamic-stimulation upon septal and hippocampal electrical-activity in cat. Brain Res 107:55–68
Saper CB, Akil H, Watson SJ (1986) Lateral hypothalamic innervation of the cerebral-cortex—immunoreactive staining for a peptide resembling but immunochemically distinct from pituitary arcuate alpha-melanocyte stimulating hormone. Brain Res Bull 16:107–120
Sakai K, Salvert D, Kitahama K, Kimura H, Maeda T, Jouvet M (1983) Ascending and descending projections of caudal hypothalamic neurons stained by serotonin immunohistochemistry after administration of 5-hydroxytryptophan in the cat. Comptes Rendus de l Academie des Sciences Serie Iii-Sciences de la Vie-Life Sciences 296:1013–1018
Saper CB (1985) Organization of cerebral cortical afferent systems in the rat. 2. Hypothalamocortical projections. J Comp Neurol 237:21–46
Vanni-Mercier G, Gigout S, Debilly G, Lin JS (2003) Waking selective neurons in the posterior hypothalamus and their response to histamine H3-receptor ligands: an electrophysiological study in freely moving cats. Behav Brain Res 144:227–241
Krilowicz BL, Szymusiak R, McGinty D (1994) Regulation of posterior lateral hypothalamic arousal related neuronal discharge by preoptic-anterior hypothalamic warming. Brain Res 668:30–38
Steininger TL, Alam MN, Gong H, Szymusiak R, McGinty D (1999) Sleep-waking discharge of neurons in the posterior lateral hypothalamus of the albino rat. Brain Res 840:138–147
Sakai K, El Mansari M, Lin JS, Zhang JG, Vanni-Mercier G (1990) The posterior hypothalamus in the regulation of wakefulness and paradoxical sleep. In: Mancia M, Marini G (eds) The diencephalon and sleep. Raven Press, New York, pp 171–198
Sallanon M, Sakai K, Buda C, Puymartin M, Jouvet M (1988) Increase of paradoxical sleep induced by microinjections of ibotenic acid into the ventrolateral part of the posterior hypothalamus in the cat. Arch Ital Biol 126:87–97
Lin JS, Sakai K, Vanni MG, Jouvet M (1989) A critical role of the posterior hypothalamus in the mechanisms of wakefulness determined by microinjection of muscimol in freely moving cats. Brain Res 479:225–240
Nitz D, Siegel JM (1996) GABA release in posterior hypothalamus across sleep-wake cycle. Am J Physiol 271:R1707–R1712
Sallanon M, Denoyer M, Kitahama K, Aubert C, Gay N, Jouvet M (1989) Long-lasting insomnia induced by preoptic neuron lesions and its transient reversal by muscimol injection into the posterior hypothalamus in the cat. Neuroscience 32:669–683
Korotkova TM, Klyuch BP, Ponomarenko AA, Lin JS, Haas HL, Sergeeva OA (2007) Modafinil inhibits rat midbrain dopaminergic neurons through D2-like receptors. Neuropharmacology 52:626–633
Lin JS, Hou YP, Jouvet M (1996) Potential brain neuronal targets for amphetamine-, methylphenidate-, and modafinil-induced wakefulness, evidenced by c-fos immunocytochemistry in the cat. Proc Natl Acad Sci USA 93:14128–14133
Lin JS, Roussel B, Akaoka H, Fort P, Debilly G, Jouvet M (1992) Role of catecholamines in the modafinil and amphetamine induced wakefulness, a comparative pharmacological study in the cat. Brain Res 591:319–326
Yanovsky Y, Li S, Klyuch BP, Yao Q, Blandina P, Passani MB, Lin JS, Haas HL, Sergeeva OA (2011) L-Dopa activates histaminergic neurons. J Physiol [Epub ahead of print]
Nelson LE, Guo TZ, Lu J, Saper CB, Franks NP, Maze M (2002) The sedative component of anesthesia is mediated by GABA(A) receptors in an endogenous sleep pathway. Nat Neurosci 5:979–984
Shekhar A, Dimicco JA (1987) Defense reaction elicited by injection of GABA antagonists and synthesis inhibitors into the posterior hypothalamus in rats. Neuropharmacology 26:407–417
Waldrop TG, Bauer RM, Iwamoto GA (1988) Microinjection of GABA antagonists into the posterior hypothalamus elicits locomotor-activity and a cardiorespiratory activation. Brain Res 444:84–94
Bauer RM, Vela MB, Simon T, Waldrop TG (1988) A gabaergic mechanism in the posterior hypothalamus modulates baroreflex bradycardia. Brain Res Bull 20:633–641
Tsujino N, Sakurai T (2009) Orexin/hypocretin: a neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacol Rev 61:162–176
Sakai K (1991) Physiological properties and afferent connections of the locus coeruleus and adjacent tegmental neurons involved in the generation of paradoxical sleep in the cat. In: Barnes CD, Pompeiano O (eds) Progress in brain research. Elsevier, Amsterdam, pp 31–45
Jones BE (1993) The organization of central cholinergic systems and their functional importance in sleep-waking states. In: Cuello AC (ed) Progress in brain research. Elsevier, Amsterdam, pp 61–71
Douglas WW (1985) Histamine and serotonin and their antagonists. In: Gilman AG, Goodman LS, Rall TW, Murad F (eds) The pharmacological basis of therapeutics. Macmillan, New York, pp 605–635
Panula P, Yang HY, Costa E (1984) Histamine-containing neurons in the rat hypothalamus. Proc Natl Acad Sci USA 81:2572–2576
Prell GD, Green JP (1986) Histamine as a neuroregulator. Annu Rev Neurosci 9:209–254
Schwartz JC, Arrang JM, Garbarg M, Pollard H, Ruat M (1991) Histaminergic transmission in the mammalian brain. Physiol Rev 71:1–51
Haas HL, Sergeeva OA, Selbach O (2008) Histamine in the nervous system. Physiol Rev 88:1183–1241
Watanabe T, Taguchi Y, Shiosaka S, Tanaka J, Kubota H, Terano Y, Tohyama M, Wada H (1984) Distribution of the histaminergic neuron system in the central nervous system of rats; a fluorescent immunohistochemical analysis with histidine decarboxylase as a marker. Brain Res 295:13–25
Lin JS, Luppi PH, Salvert D, Sakai K, Jouvet M (1986) Histamine-containing neurons in the cat hypothalamus. Comptes Rendus de l Academie des Sciences Serie Iii-Sciences de la Vie-Life Sciences 303:371–376
Monnier M, Fallert M, Battacharya IC (1967) Waking action of histamine. Experientia 23:21
Mochizuki T, Yamatodani A, Okakura K, Horii A, Inagaki N, Wada H (1992) Circadian-rhythm of histamine-release from the hypothalamus of freely moving rats. Physiol Behav 51:391–394
Strecker RE, Nalwalk J, Dauphin LJ, Thakkar MM, Chen Y, Ramesh V, Hough LB, McCarley RW (2002) Extracellular histamine levels in the feline preoptic/anterior hypothalamic area during natural sleep-wakefulness and prolonged wakefulness: an in vivo microdialysis study. Neuroscience 113:663–670
Philippu A, Prast H (2001) Importance of histamine in modulatory processes, locomotion and memory. Behav Brain Res 124:151–159
Arrang JM, Garbarg M, Schwartz JC (1983) Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature 302:832–837
Barnes WG, Hough LB (2002) Membrane-bound histamine N-methyltransferase in mouse brain: possible role in the synaptic inactivation of neuronal histamine. J Neurochem 82:1262–1271
Grace AA, Onn SP (1989) Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded invitro. J Neurosci 9:3463–3481
Haas HL, Reiner PB (1988) Membrane properties of histaminergic tuberomammillary neurones of the rat hypothalamus in vitro. J Physiol Lond 399:633–646
John J, Wu MF, Boehmer LN, Siegel JM (2004) Cataplexy-active neurons in the hypothalamus: Implications for the role of histamine in sleep and waking behavior. Neuron 42:619–634
Takahashi K, Lin JS, Sakai K (2006) Neuronal activity of histaminergic tuberomammillary neurons during wake-sleep states in the mouse. J Neurosci 26:10292–10298
Kohler C, Swanson LW, Haglund L, Wu JY (1985) The cytoarchitecture, histochemistry and projections of the tuberomammillary nucleus in the rat. Neuroscience 16:85–110
Takeda N, Inagaki S, Taguchi Y, Tohyama M, Watanabe T, Wada H (1984) Origins of histamine-containing fibers in the cerebral cortex of rats studied by immunohistochemistry with histidine decarboxylase as a marker and transection. Brain Res 323:55–63
Wouterlood FG, Sauren YMHF, Steinbusch HWM (1986) Histaminergic neurons in the rat-brain—correlative immunocytochemistry, Golgi impregnation, and electron-microscopy. J Comp Neurol 252:227–244
Lin JS, Kitahama K, Fort P, Panula P, Denney RM, Jouvet M (1993) Histaminergic system in the cat hypothalamus with reference to type-B monoamine-oxidase. J Comp Neurol 330:405–420
Lin JS, Hou YP, Sakai K, Jouvet M (1996) Histaminergic descending inputs to the mesopontine tegmentum and their role in the control of cortical activation and wakefulness in the cat. J Neurosci 16:1523–1537
Selbach O, Brown RE, Haas HL (1997) Long-term increase of hippocampal excitability by histamine and cyclic AMP. Neuropharmacology 36:1539–1548
McCormick DA, Williamson A (1991) Modulation of neuronal firing mode in cat and guinea-pig LGNd by histamine—possible cellular mechanisms of histaminergic control of arousal. J Neurosci 11:3188–3199
Reiner PB, Kamondi A (1994) Mechanisms of antihistamine-induced sedation in the human brain—H-1 receptor activation reduces a background leakage potassium current. Neuroscience 59:579–588
Gorelova N, Reiner PB (1996) Histamine depolarizes cholinergic septal neurons. J Neurophysiol 75:707–714
Brown RE, Sergeeva OA, Eriksson KS, Haas HL (2002) Convergent excitation of dorsal raphe serotonin neurons by multiple arousal systems (orexin/hypocretin, histamine and noradrenaline). J Neurosci 22:8850–8859
Stehle J (1991) Effects of histamine on spontaneous electrical activity of neurons in rat suprachiasmatic nucleus. Neurosci Lett 130:217–220
Leurs R, Bakker RA, Timmerman H, De Esch IJP (2005) The histamine H-3 receptor: From gene cloning to H-3 receptor drugs. Nat Rev Drug Discov 4:107–120
Lin JS, Sergeeva OA, Haas HL (2010) Histamine H3-receptors and sleep-wake regulation. J Pharmacol Exp Ther
Kiyono S, Seo ML, Shibagaki M, Watanabe T, Maeyama K, Wada H (1985) Effects of alpha-fluoromethylhistidine on sleep-waking parameters in rats. Physiol Behav 34:615–617
Monti JM (1993) Involvement of histamine in the control of the waking state. Life Sci 53:1331–1338
Parmentier R, Ohtsu H, Djebbara-Hannas Z, Valatx JL, Watanabe T, Lin JS (2002) Anatomical, physiological, and pharmacological characteristics of histidine decarboxylase knock-out mice: Evidence for the role of brain histamine in behavioral and sleep-wake control. J Neurosci 22:7695–7711
Williams RH, Jensen LT, Verkhratsky A, Fugger L, Burdakov D (2007) Control of hypothalamic orexin neurons by acid and CO2. Proc Natl Acad Sci USA 104:10685–10690
Ohshima Y, Iwase M, Izumizaki M, Ishiguro T, Kanamaru M, Nakayama H, Gejyo F, Homma I (2007) Hypoxic ventilatory response during light and dark periods and the involvement of histamine H1 receptor in mice. Am J Physiol Regul Integr Comp Physiol 293:R1350–R1356
Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS (1998) Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci 18:9996–10015
de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG (1998) The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA 95:322–327
Date Y, Ueta Y, Yamashita H, Yamaguchi H, Matsukura S, Kangawa K, Sakurai T, Yanagisawa M, Nakazato M (1999) Orexins, orexigenic hypothalamic peptides, interact with autonomic, neuroendocrine and neuroregulatory systems. Proc Natl Acad Sci USA 96:748–753
Marcus JN, Aschkenasi CJ, Lee CE, Chemelli RM, Saper CB, Yanagisawa M, Elmquist JK (2001) Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol 435:6–25
Kukkonen JP, Holmqvist T, Ammoun S, Akerman KEO (2002) Functions of the orexinergic/hypocretinergic system. Am J Physiol Cell Physiol 283:C1567–C1591
Sakurai T (2002) Roles of orexins in regulation of feeding and wakefulness. Neuroreport 13:987–995
Chou TC, Lee CE, Lu J, Elmquist JK, Hara J, Willie JT, Beuckmann CT, Chemelli RM, Sakurai T, Yanagisawa M, Saper CB, Scammell TE (2001) Orexin (hypocretin) neurons contain dynorphin. J Neurosci 21:RC168
Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M (1998) Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior [see comments]. Cell 92:573–585
Siegel JM, Boehmer LN (2006) Narcolepsy and the hypocretin system—where motion meets emotion. Nat Clin Pract Neurol 2:548–556
Zeitzer JM, Nishino S (2006) Mignot E: the neurobiology of hypocretins (orexins), narcolepsy and related therapeutic interventions. Trends Pharmacol Sci 27:368–374
Willie JT, Chemelli RM, Sinton CM, Yanagisawa M (2001) To eat or to sleep? Orexin in the regulation of feeding and wakefulness. Annu Rev Neurosci 24:429–458
Sakurai T (2007) The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat Rev Neurosci 8:171–181
Aston-Jones G, Smith RJ, Moorman DE, Richardson KA (2009) Role of lateral hypothalamic orexin neurons in reward processing and addiction. Neuropharmacology 56:112–121
Lee MG, Hassani OK, Jones BE (2005) Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci 25:6716–6720
Mileykovskiy BY, Kiyashchenko LI, Siegel JM (2005) Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron 46:787–798
Takahashi K, Lin JS, Sakai K (2008) Neuronal activity of orexin and non-orexin waking-active neurons during wake-sleep states in the mouse. Neuroscience 153:860–870
Martins PJ, D’Almeida V, Pedrazzoli M, Lin L, Mignot E, Tufik S (2004) Increased hypocretin-1 (orexin-a) levels in cerebrospinal fluid of rats after short-term forced activity. Regul Pept 117:155–158
Valdes JL, Farias P, Ocampo-Garces A, Cortes N, Seron-Ferre M, Torrealba F (2005) Arousal and differential Fos expression in histaminergic neurons of the ascending arousal system during a feeding-related motivated behaviour. Eur J Neurosci 21:1931–1942
Hagan JJ, Leslie RA, Patel S, Evans ML, Wattam TA, Holmes S, Benham CD, Taylor SG, Routledge C, Hemmati P, Munton RP, Ashmeade TE, Shah AS, Hatcher JP, Hatcher PD, Jones DN, Smith MI, Piper DC, Hunter AJ, Porter RA, Upton N (1999) Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc Natl Acad Sci USA 96:10911–10916
Jones DN, Gartlon J, Parker F, Taylor SG, Routledge C, Hemmati P, Munton RP, Ashmeade TE, Hatcher JP, Johns A, Porter RA, Hagan JJ, Hunter AJ, Upton N (2001) Effects of centrally administered orexin-B and orexin-A: a role for orexin-1 receptors in orexin-B-induced hyperactivity. Psychopharmacology (Berl) 153:210–218
Anaclet C, Parmentier R, Ouk K, Guidon G, Buda C, Sastre JP, Akaoka H, Sergeeva OA, Yanagisawa M, Ohtsu H, Franco P, Haas HL, Lin JS (2009) Orexin/hypocretin and histamine: distinct roles in the control of wakefulness demonstrated using knock-out mouse models. J Neurosci 29:14423–14438
Jaeger LB, Farr SA, Banks WA, Morley JE (2002) Effects of orexin-A on memory processing. Peptides 23:1683–1688
Bayer L, Serafin M, Eggermann E, Saint-Mleux B, Marchard D, Jones BE, Muhlethaler M (2004) Exclusive postsynaptic action of hypocretin-orexin on sublayer 6b cortical neurons. J Neurosci 24:6760–6764
Gerashchenko D, Salin-Pascual R, Shiromani PJ (2001) Effects of hypocretin-saporin injections into the medial septum on sleep and hippocampal theta. Brain Res 913:106–115
Selbach O, Bohla C, Barbara A, Doreulee N, Eriksson KS, Sergeeva OA, Haas HL (2010) Orexins/hypocretins control bistability of hippocampal long-term synaptic plasticity through co-activation of multiple kinases. Acta Physiol (Oxf) (in press)
Reymann KG, Frey JU (2007) The late maintenance of hippocampal LTP: requirements, phases, ‘synaptic tagging’, ‘late-associativity’ and implications. Neuropharmacology 52:24–40
Selbach O, Doreulee N, Bohla C, Eriksson KS, Sergeeva OA, Poelchen W, Brown RE, Haas HL (2004) Orexins/hypocretins cause sharp wave- and theta-related synaptic plasticity in the hippocampus via glutamatergic, gabaergic, noradrenergic, and cholinergic signaling. Neuroscience 127:519–528
Parmentier R, Kolbaev S, Klyuch BP, Vandael D, Lin JS, Selbach O, Haas HL, Sergeeva OA (2009) Excitation of histaminergic tuberomamillary neurons by thyrotropin-releasing hormone. J Neurosci 29:4471–4483
Nishino S, Ripley B, Overeem S, Nevsimalova S, Lammers GJ, Vankova J, Okun M, Rogers W, Brooks S, Mignot E (2001) Low cerebrospinal fluid hypocretin (Orexin) and altered energy homeostasis in human narcolepsy. Ann Neurol 50:381–388
Parmentier R, Anaclet C, Guhennec C, Brousseau E, Bricout D, Giboulot T, Bozyczko-Coyne D, Spiegel K, Ohtsu H, Williams M, Lin JS (2007) The brain H-3-receptor as a novel therapeutic target for vigilance and sleep-wake disorders. Biochem Pharmacol 73:1157–1171
Lin JS, Dauvilliers Y, Arnulf I, Bastuji H, Anaclet C, Parmentier R, Kocher L, Yanagisawa M, Lehert P, Ligneau X, Perrin D, Robert P, Roux M, Lecomte JM, Schwartz JC (2008) An inverse agonist of the histamine H-3 receptor improves wakefulness in narcolepsy: Studies in orexin(−/−) mice and patients. Neurobiol Dis 30:74–83
Carter ME, Adamantidis A, Ohtsu H, Deisseroth K, De LL (2009) Sleep homeostasis modulates hypocretin-mediated sleep-to-wake transitions. J Neurosci 29:10939–10949
Blanco-Centurion C, Gerashchenko D, Shiromani PJ (2007) Effects of saporin-induced lesions of three arousal populations on daily levels of sleep and wake. J Neurosci 27:14041–14048
Nishino S, Arrigoni J, Shelton J, Kanbayashi T, Dement WC, Mignot E (1997) Effects of thyrotropin-releasing hormone and its analogs on daytime sleepiness and cataplexy in canine narcolepsy. J Neurosci 17:6401–6408
Arnulf I, Leu-Semenescu S (2009) Sleepiness in Parkinson’s disease. Parkinsonism Relat Disord 15(Suppl 3):S101–S104
Hassani OK, Lee MG, Jones BE (2009) Melanin-concentrating hormone neurons discharge in a reciprocal manner to orexin neurons across the sleep-wake cycle. Proc Natl Acad Sci USA 106:2418–2422
Bittencourt JC, Presse F, Arias C, Peto C, Vaughan J, Nahon JL, Vale W, Sawchenko PE (1992) The melanin-concentrating hormone system of the rat-brain—an immunization and hybridization histochemical characterization. J Comp Neurol 319:218–245
Adamantidis A, Salvert D, Goutagny R, Lakaye B, Gervasoni D, Grisar T, Luppi PH, Fort P (2008) Sleep architecture of the melanin-concentrating hormone receptor 1-knockout mice. Eur J Neurosci 27:1793–1800
Takahashi K, Lin JS, Sakai K (2009) Characterization and mapping of sleep-waking specific neurons in the basal forebrain and preoptic hypothalamus in mice. Neuroscience 161:269–292
Szymusiak R, Gvilia I, McGinty D (2007) Hypothalamic control of sleep. Sleep Med 8:291–301
Saper CB, Scammell TE, Lu J (2005) Hypothalamic regulation of sleep and circadian rhythms. Nature 437:1257–1263
Suntsova N, Guzman-Marin R, Kumar S, Alam MN, Szymusiak R, McGinty D (2007) The median preoptic nucleus reciprocally modulates activity of arousal-related and sleep-related neurons in the perifornical lateral hypothalamus. J Neurosci 27:1616–1630
Schmidt MH, Valatx JL, Sakai K, Fort P, Jouvet M (2000) Role of the lateral preoptic area in sleep-related erectile mechanisms and sleep generation in the rat. J Neurosci 20:6640–6647
Lin JS, Sakai K, Jouvet M (1994) Hypothalamo-preoptic histaminergic projections in sleep-wake control in the cat. Eur J Neurosci 6:618–625
Gallopin T, Fort P, Eggermann E, Cauli B, Luppi PH, Rossier J, Audinat E, Muhlethaler M, Serafin M (2000) Identification of sleep-promoting neurons in vitro. Nature 404:992–995
Liu YW, Li J, Ye JH (2010) Histamine regulates activities of neurons in the ventrolateral preoptic nucleus. J Physiol 588:4103–4116
Takahashi K, Kayama Y, Lin JS, Sakai K (2010) Locus coeruleus neuronal activity during the sleep-waking cycle in mice. Neuroscience 169:1115–1126
Sakai K, Takahashi K, Anaclet C, Lin JS (2010) Sleep-waking discharge of ventral tuberomamillary neurons in wild-type and histidine decarboxylase knock-out mice. Front Behav Neurosci 4:1–10
Borbely AA (1982) A two process model of sleep regulation. Hum Neurobiol 1:195–204
Borbely AA, Achermann P (1999) Sleep homeostasis and models of sleep regulation. J Biol Rhythms 14:557–568
Borbely AA, Tobler I (1989) Endogenous sleep-promoting substances and sleep regulation. Physiol Rev 69:605–670
Radulovacki M, Virus RM, Djuricic-Nedelson M, Green RD (1984) Adenosine analogs and sleep in rats. J Pharmacol Exp Ther 228:268–274
Huston JP, Haas HL, Boix F, Pfister M, Decking U, Schrader J, Schwarting RK (1996) Extracellular adenosine levels in neostriatum and hippocampus during rest and activity periods of rats. Neuroscience 73:99–107
Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW (1997) Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science 276:1265–1268
Bjorness TE, Greene RW (2009) Adenosine and sleep. Curr Neuropharmacol 7:238–245
Sergeeva OA, Klyuch BP, Fleischer W, Eriksson KS, Korotkova TM, Siebler M, Haas HL (2006) P2Y receptor-mediated excitation in the posterior hypothalamus. Eur J Neurosci 24:1413–1426
Haas HL, Selbach O (2000) Functions of neuronal adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol 362:375–381
Benington JH, Kodali SK, Heller HC (1995) Stimulation of A1 adenosine receptors mimics the electroencephalographic effects of sleep deprivation. Brain Res 692:79–85
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lin, JS., Anaclet, C., Sergeeva, O.A. et al. The waking brain: an update. Cell. Mol. Life Sci. 68, 2499–2512 (2011). https://doi.org/10.1007/s00018-011-0631-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-011-0631-8