
Abstract
The protein kinase C (PKC) family of serine/threonine kinases consists of ten different isoforms grouped into three subfamilies, denoted classical, novel and atypical PKCs (aPKCs). The aPKCs, PKCι/λ and PKCζ serve important roles during development and in processes subverted in cancer such as cell and tissue polarity, cell proliferation, differentiation and apoptosis. In an effort to identify novel interaction partners for aPKCs, we performed a yeast two-hybrid screen with the regulatory domain of PKCι/λ as bait and identified the Krüppel-like factors family protein TIEG1 as a putative interaction partner for PKCι/λ. We confirmed the interaction of both aPKCs with TIEG1 in vitro and in cells, and found that both aPKCs phosphorylate the DNA-binding domain of TIEG1 on two critical residues. Interestingly, the aPKC-mediated phosphorylation of TIEG1 affected its DNA-binding activity, subnuclear localization and transactivation potential.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The protein kinase C family is composed of ten related serine/threonine protein kinases, which are divided into three subfamilies including conventional, novel and atypical PKCs (aPKCs). PKCζ and ι/λ represent the aPKC isoforms, where PKCι is the human and PKCλ the murine isoform. The aPKCs differ from the other PKCs by having an N-terminal PB1 domain and their activation is independent of calcium, phorbol esters and diacylglycerol (reviewed in [1]). However, it has been demonstrated that the phorbol ester PMA activates PI3K and subsequently PKCζ [2]. The PB1 domain of aPKCs interacts with Par6 and is important for their incorporation into the apical polarity complex which contains the Par3/Par6/aPKC module (reviewed in [3]). The activity of the aPKCs is regulated by 3-phosphoinositides, phosphorylation by the phosphoinositide-dependent kinase PDK1 and through specific protein-protein interactions (reviewed in [4]). Due to their strong homology (78%) it has been difficult to distinguish the individual functions of the PKCζ and ι/λ isoforms. However, PKCι/λ is ubiquitously expressed, while PKCζ has a more restricted expression pattern [5]. The murine PKCλ differs only from the human PKCι at 10 out of 596 amino acid positions (98% sequence identity). PKCλ is essential for embryonic development in the mouse, whereas PKCζ-deficient mice develop nearly normally [6]. Conditional knock-out studies in mice and frog have shown that PKCλ plays a role in glucose transport and induction of metabolic and diabetic syndromes [6, 7], and that it controls the establishment of apicobasal polarity, adherens junctions and neuroepithelial tissue architecture in early vertebrate development [8, 9]. Both aPKC isoforms are critical for cell survival signaling, and many studies indicate a correlation between the expression or activation of aPKC and sensitivity to apoptosis (reviewed in [1, 4]). The aPKCs seem to be dynamically regulated by agents inducing cell death, and recent results indicate that they may act as a switch between cell survival and cell death (reviewed in [1, 4]).
Transforming growth factor-β-inducible early response gene 1 (TIEG1) was originally discovered as the product of a TGFβ-inducible early response gene in human osteoblasts [10]. It was later shown that overexpression of TIEG1 mimics TGFβ action and induces apoptosis and decreased proliferation in several cell lines (reviewed in [11, 12]). TIEG1 is also known as KLF10 and belongs to the Krüppel-like factors (KLF) family of proteins, constituting more than 20 family members in humans, including Sp1 as the most well-known member [13]. The C-terminal DNA-binding domain (DBD) of KLF family proteins consists of three highly conserved tandem Cys2His2 zinc finger motifs. Most KLF family proteins have similar affinity for different GC-rich promoter elements, and the amino acids that are predicted to interact with DNA are identical among several members. Importantly, competition for DNA binding has been reported for some of the factors (reviewed in [13]). In addition to DNA binding, the zinc finger motifs may also function in protein-protein interactions and are the target for post-translational modifications such as phosphorylation, acetylation and O-linked β-N-acetyl glucose amine addition (hereafter referred to as O-GlcNAc) [14]. The N-terminal regions of KLF family members are much less conserved, containing various transcriptional activation and/or repression domains. Thus, different family members vary broadly in their ability to regulate transcription. However, most members can function either as activators or repressors, depending on the specific cellular and promoter contexts [11, 13]. The KLF factor TIEG1 is reported to induce and repress the expression of multiple genes, including p21Cip1/WAF1, and to function as an inhibitor of cell proliferation and an inducer of apoptosis (reviewed in [12]). TIEG1 is suggested to function as a tumour suppressor as its expression inversely correlates with the severity and stage of breast cancer [15].
In the present study we show that TIEG1 is a substrate of the aPKCs ζ and ι/λ. Two amino acid residues in the DBD were mapped as the major targets for phosphorylation, S384 located in zinc finger 1 and T445 in zinc finger 3. Phosphorylation of S384 significantly reduced the DNA-binding activity, whereas the T445 phosphorylation enhanced the TIEG1-mediated transcriptional activation of the p21Cip1/WAF1 promoter. Importantly, both phosphorylations resulted in subnuclear redistribution of TIEG1, strengthening the observation that phosphorylation of these residues in the DBD affects protein-protein and/or protein-DNA interactions. We also show that in addition to TIEG1, the DBD of other members of the KLF family proteins may be regulated by phosphorylation mediated by the aPKCs.
Materials and methods
Antibodies and reagents
The following primary antibodies were used: rabbit polyclonal antibody to PKCζ (C-20) (SC-216), rabbit polyclonal antibody to GST (Z-5, SC-459) (Santa Cruz Biotechnology), rabbit polyclonal antibody to green fluorescent protein (GFP) (ab290) (Abcam), rabbit polyclonal to beta-actin (A2066) (Sigma-Aldrich), mouse monoclonal antibody to PKCλ (610207) and the secondary antibodies HRP-conjugated goat anti-rabbit IgG (554021), and goat anti-mouse IgG (554002) antibodies were purchased from BD Bioscience Pharmingen. PKCι, active (14-505) and PKCζ, active (14-525) are purchased from Upstate.
Yeast two-hybrid screen
The yeast two-hybrid screen was carried out with the Matchmaker system (Clontech) according to the manufacturer’s instructions. The regulatory domain of PKCλ (pGBKT7-PKCλreg) fused to the Gal4 DBD was used as bait. This bait was transformed into Saccharomyces cerevisiae strain PJ69-2A (Clontech) and then mated with the Y187 strain pretransformed with a HeLa cell cDNA library fused to the Gal4 activation domain (Clontech). Approximately 106 diploids were screened and tested for their ability to grow on yeast minimal medium lacking leucine, tryptophane, histidine, and adenine. Positive colonies were lysed by incubating them for 1–2 h in a glucuronidase-containing buffer [(50 mM Tris-HCl (pH 7.5), 10 mM EDTA, 0.3% (v/v) β-mercaptoethanol, and 1:50 glucuronidase (G7017, Sigma)] followed by vortexing with glass beads (G1145, Sigma) for 5 min. The lysates were diluted with 100 μl H2O, centrifuged briefly in a microcentrifuge at maximum speed, and 2 μl of each sample was analyzed by PCR using the REDTaq ReadyMix (R2523, Sigma). The PCR products were treated with exonuclease I (USB) and shrimp alkaline phosphatase (M820A, Promega) followed by sequencing using the BigDye sequencing kit (Applied Biosystems). The sequenced products were identified by searching the National Center for Biotechnology Information (NCBI) using the basic local alignment search tool (BLAST) algorithm. In order to verify specific interactions, clones were re-screened as described previously [16].
Plasmids
Plasmids used in this work are listed in Table 1. Point mutations were generated using the QuickChange site directed mutagenesis kit (Stratagene), and Gateway destination plasmids were made using Gateway LR recombination reactions (Invitrogen) following the manufacturer’s instructions. All plasmid constructs made in this study were verified by DNA sequencing (BigDye sequencing kit, Applied Biosystems). The oligonucleotides used for mutagenesis, PCR, and DNA sequencing were purchased from Operon.
Cell culture and transfections
HeLa cells and U2OS cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS), penicillin (100 U/ml) and streptomycin (100 μg/ml). Subconfluent cells were transfected with the different expression constructs using either Lipofectamine Plus (Invitrogen) or Metafectene Pro (Biontex) as recommended by the manufacturers.
Immunoprecipitations and immunoblots
Transfected cells were rinsed with ice-cold PBS prior to lysis in RIPA buffer [50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% NP-40 (v/v), 0.25% Triton X-100 (v/v)] supplemented with Complete Mini EDTA-free protease inhibitor cocktail tablets (1 tablet per 10 ml) (11836170001, Roche Applied Science). Lysates were cleared by centrifugation followed by 30-min incubation with protein A-agarose beads (SC-2001, Santa Cruz Biotechnology). The precleared lysates were then incubated with the indicated primary antibodies overnight at 4°C and then with Protein A-agarose beads for an additional 1 h. Precipitated immunocomplexes were washed five times with RIPA buffer, eluted by boiling for 5 min in SDS-PAGE loading buffer. Samples were subsequently resolved by SDS-PAGE and transferred to Hybond-ECL nitrocellulose membranes (Amersham). After blocking unspecific binding sites by incubating the membranes for 1 h in 5% (w/v) non-fat dry milk in TBST [10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.1% (v/v) Tween 20], blots were probed with the indicated primary antibodies overnight at 4°C and then by horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. The membranes were washed six times (5 min each) with TBST prior to detection with Western Blotting Luminal Reagent kit (SC-2048, Santa Cruz Biotechnology) and a LumiAnalyst imager (Roche Applied Sciences).
GST pulldown assays
GST protein was expressed in Escherichia coli LE392 and MBP in E. coli DB 3.1. GST and MBP-tagged proteins were expressed in E. coli BL21 STAR (DE3)pLysS cells (Invitrogen). GST and GST-fusion proteins were purified and immobilized on glutathione-coupled sepharose beads (Glutathione-sepharose 4 Fast Flow, Amersham Bioscience). MBP and MBP-tagged proteins were purified using amylose resin (New England Biolabs). GST pulldown assays were performed by incubating GST and GST-fusion proteins either with in vitro translated proteins or with mammalian cell lysate. 35S-labeled proteins were produced using the TNT T7 Quick Coupled Transcription/Translation System (Promega) in the presence of [35S] methionine (Amersham Biosciences). The synthesized proteins were diluted 20× with NET-N buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40) containing Complete Mini EDTA-free protease inhibitor cocktail. The diluted products were pre-cleared by incubating with glutathione-coupled sepharose beads for 30 min prior to incubation with purified GST or GST-fusion proteins for at least 2 h at 4°C with gentle agitation. Unbound proteins were removed by washing the resins five times with NET-N buffer. The proteins were then eluted by boiling for 5 min in SDS gel loading buffer and separated by SDS-PAGE. After vacuum drying the gel, 35S-labeled proteins were detected on a Fujifilm bioimaging analyzer BAS-5000 (Fuji). Lysates were prepared from confluent HeLa cells in 100 mm culture dishes as indicated above, except that glutathione-coupled agarose beads were used instead of protein A-agarose beads. Pulldown of PKCι/ζ from precleared HeLa cell lysates was also done as described for pulldown of 35S labeled proteins, except that proteins separated by SDS-PAGE were subjected to immunoblot analysis.
In vitro phosphorylation assays
In vitro phosphorylation assays of TIEG1 and its mutants by the aPKCs were carried out in a total volume of 30 μl, containing kinase buffer [35.5 mM Tris-HCl (pH 7.5), 10 mM MgCl, 0.5 mM EGTA (pH 8.0), 0.1 mM CaCl2, Complete Mini EDTA-free protease inhibitor cocktail and phosphatase inhibitor cocktail set II (1:100) (524625, Calbiochem)] and 50 ng recombinant active kinase. The enzyme reactions were initiated by the addition of 60 μM unlabeled ATP and 2 μCi [γ-32P] ATP (Amersham). After incubation at 30°C for 20 min, the reactions were terminated by the addition of SDS gel loading buffer and subsequent boiling for 5 min. The phosphorylated proteins were then analyzed by SDS-PAGE and autoradiography. The time courses of in vitro phosphorylations by PKCζ were performed by preparing 90 μl enzyme reactions and by withdrawing a 7-μl aliquot at the desired time points. The aliquots were transferred to microcentrifuge tubes containing SDS gel loading buffer and analyzed as described above. Where indicated, quantifications of the phosphorylated proteins and the corresponding background bands were done using the Fuji Image Gauge software (version 4.0; Fujifilm, Tokyo, Japan). The results are presented as bar graphs after background subtraction.
Gel mobility shift assay (GMSA)
The following two sets of oligonucleotides (shown 5′ to 3′) were used for GMSA: set I (for TIEG1) described in [17] and set II (for KLF4 and Sp1) described in [18].
Set I: 5′-TGCAGTGAAAAGGGGGTGTGTCAGGATGC-3′, and 5′-TGCATC CTGACACACCCCCTTTTCACTGC-3′; set II: 5′-CTTAACATTCCTTTCCCCACCCACACAGCTAGTTCCAACC-3′, and 5′-GGTTGGAACTAGCTGTGTGGGTGGGGAAAGGAATGTTAAG -3′. The DBDs of TIEG1, Sp1 and KLF4 were expressed and purified as described above and eluted in 50 mM Tris-HCl (pH 8.0) containing 10 mM reduced glutathione. The sense and antisense strands were annealed in annealing buffer [10 mM Tris-HCl (pH 8.0), 1 mM EDTA and 100 mM NaCl] and annealed by heating to 100°C for 3 min and cooling gradually to room temperature. The probes were labeled with [γ-32P] ATP using T4 polynucleotide kinase (Takara) and then purified and eluted using QIA quick nucleotide removal kit (QIAGEN) as per the recommendation of the manufacturer. The binding reaction mixtures, containing approximately 500 ng to 1 μg protein, 1 μg poly (dI-dC) and binding buffer [for TIEG1: 4% Ficoll, 10 mM HEPES (pH 7.9), 30 mM KCl, 0.1 mM EDTA, 1 mM Na2HPO4, 4 mM spermidine and 0.5 mM DTT; and for KLF4 and Sp1: 4% Ficoll, 10 mM HEPES (pH 7.9), 60 mM KCl, 1 mM EDTA, and 1 mM DTT] were incubated at room temperature for 10 min and then mixed with 3–4 × 104 cpm of each probe and incubated for an additional 20 min. The DNA-protein complexes were resolved on pre-run 5% polyacrylamide gels (39:1) in 0.5× TBE buffer (44.5 mM Tris, 44.5 mM boric acid and 2 mM EDTA) at 230 v for 3 h. Where indicated, GST-tagged proteins were incubated with 100 ng of active PKCζ for 45 min prior to GMSA under the aforementioned kinase reaction conditions except for the omission of [γ-32P] ATP from the reaction mixture.
Cell imaging
Cells were seeded at a density of 104 cells/well in eight-well coverglass slides (Nunc) and transfected with 100 ng plasmid expression vectors 24 h later using lipofectamin Plus (Invitrogen). One day after transfection cells were analyzed by live cell confocal microscopy using a Zeiss Axiovert 200 microscope with a 40×, 1.2 W C-Apochroma objective, equipped with an LSM510-META confocal module using the LSM 5 software version 3.2 (Carl Zeiss Inc.).
Fluorescence recovery after photobleaching (FRAP)
FRAP experiments were carried out with cells plated and transfected as described above and imaged using a Leica TCS SP5 confocal microscope, 63×, 1.2 W objective, equipped with an incubation chamber with CO2 and temperature control. Cells were imaged with an argon 488-nm laser at 50% laser power and a pinhole setting of one. A circular area of 5 μM in diameter of the cell nucleus was analyzed using the following conditions: 5 prebleach images per 0.113 s, 10 bleach pulses per 0.113 s, 300 postbleach images per 0.113 s, 50 postbleach images per second and finally 45 postbleach images per 5 s. For imaging, laser power was attenuated to 5% of bleach intensity. A total of ten cells were analyzed per plasmid construct for each independent experiment, and the experiments were performed at least three times. Individual FRAP curves were background subtracted and corrected for loss of fluorescence caused by photobleaching during imaging.
Reporter gene assays
U2OS cells were seeded at a density of 4 × 104 cells/well in 24-well plates and transfected 24 h later using Metafectene Pro. Transfections were performed with 100 or 250 ng of the various protein expression plasmids together with 100 ng of the reporter p21LUC. The β-galactosidase expressing pCMV-βgal vector (50 ng) (Stratagene) was included to determine transfection efficiency. Cells were harvested 24 h post transfection and luciferase activities measured using the dual light luciferase and β-galactosidase kit (TROPIX) on a Luminoskan RT dual injection luminometer (Labsystems). All reporter gene assays were carried out in independent triplicate experiments and repeated at least three times. The luciferase values varied 1–12% among the parallels.
Results
The aPKCs ι/λ and ζ interact with the zinc finger DBD of the Krüppel-like transcription factor family
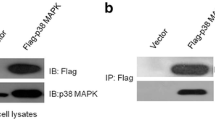
In an attempt to find novel interaction partners for the aPKCs, we performed a yeast two-hybrid screen using a HeLa cell cDNA expression library together with the regulatory domain of PKCλ as bait. Three out of 68 positive clones were identified as the C-terminal zinc finger region of the Krüppel-like transcription factor TIEG1 (Fig. 1a). The interaction was confirmed in vitro by GST pulldown assays using GST fusions of the zinc finger DBD of TIEG1 (TIEG1(DBD)) and the N-terminal proline-rich region of TIEG1 (TIEG1(1–362)), and in vitro translated PKCλ. As can be seen from Fig. 1d, no interaction was detected with the N-terminal part of TIEG1. To further evaluate the interaction between full-length TIEG1 and full-length PKCι GST pulldown assays with GST-TIEG1 and whole-cell extract isolated from HeLa cells were performed. Western blot analysis of the pulled-down proteins with an antibody recognizing both PKCι and PKCζ showed that both kinases interact with full-length TIEG1 (Fig. 1b). Finally, co-immunoprecipitation assay using U2OS cells overexpressing EGFP-TIEG1 verified that TIEG1 and PKCι interacted in HeLa cells (Fig. 1c).

PKCs ζ and ι interact with the KLF family zinc finger domain in vitro and in HeLa cells. a Schematic representation of the domain structure of TIEG1. The three zinc finger motifs constituting the DNA-binding domain are represented by filled boxes. The TIEG1 region isolated in the yeast two-hybrid screen is indicated by the solid line below the schematic. b Whole-cell extracts from HeLa cells were incubated with equal amounts of bacterially expressed GST or GST-TIEG1 immobilized on glutathione-sepharose beads. The presence of aPKCs in the pulled-down proteins were examined by immunoblotting using αPKCζ(C20) antibody that recognizes both PKCζ and PKCι. c U2OS cells were transfected with either GFP or GFP-TIEG1. The cells were lysed 24 h post transfection, and immunoprecipitations were performed using anti-GFP antibody. Western blots of the immunoprecipitates and of the cell extract were revealed using monoclonal anti-PKCλ antibody (left panel) and immunoprecipitated GFP or GFP-TIEG1 proteins were visualized using polyclonal anti-GFP antibody (right panel). d Myc-tagged PKCλ was in vitro translated in the presence of [35S] methionine and incubated with equal amounts of either glutathione-sepharose beads coupled GST or GST-tagged KLF family proteins. The pulled-down proteins together with 5% of the input were subjected to detection by autoradiography (upper panel). The levels of GST or GST-tagged proteins used in the GST pulldown assays were examined by Coomassie brilliant blue staining (lower panel). The data shown in b, c, and d correspond to a representative experiment out of two performed
TIEG1 is a member of the KLF family of three zinc finger proteins. The three zinc fingers constitute the DBD, which is highly conserved within the KLF family. In addition to DNA binding, the zinc finger motifs may also function in protein-protein interactions that modulate DNA-binding specificity [13]. Thus, to determine whether PKCλ had the ability to interact with other members of the KLF family, we conducted GST pulldown assays with PKCλ and the KLF family proteins Sp1, Sp3, BTEB1 and BTEB2. Interestingly, PKCλ was found to interact with the DBD of all KLF family proteins tested (Fig. 1d), suggesting that aPKCs may be general interaction partners of KLF family proteins.
The KLF family of transcription factors are substrates for atypical PKCι and ζ in vitro
The Krüppel-like transcription factor Sp1 has been reported to be a target for PKCζ-mediated phosphorylation in several reports (reviewed in [14]). The interaction between TIEG1 and the aPKCs ι and ζ raised the question whether TIEG1 is also a substrate for these kinases. Full-length TIEG1 expressed as a MBP fusion in E. coli was incubated with recombinant PKCι or PKCζ in an in vitro kinase assay. Both kinases had the ability to phosphorylate TIEG1 (Fig. 2a), and the phosphorylation increased linearly within a time period of 90 min (Fig. 2b). We also included the KLF proteins KLF4 and the closely related KLF11 (also called TIEG2) in a phosphorylation assay. Both proteins were PKCι/ζ substrates (Fig. 2c and data not shown), supporting the notion that the aPKCs are putative regulators of KLF family proteins. To determine whether the zinc finger DBD or the N-terminal proline-rich regions were substrates for the aPKCs, each of these regions was expressed as GST fusions and incubated with recombinant PKCζ in an in vitro kinase assay. The N-terminal proline-rich region of TIEG1 was clearly not a PKCζ substrate, whereas the zinc finger DBD was heavily phosphorylated (Fig. 2e).

The KLF family proteins are phosphorylated by the atypical PKCs ζ and ι. a, c Both PKCζ and PKCι phosphorylate TIEG1 (a) and KLF4 (c) in vitro. The phosphorylation assays were performed for 20 min at 30°C in 30-μl reaction volume containing MBP, and MBP-TIEG1 or MBP-KLF4 as substrate, 50 ng recombinant active PKCζ (upper panels) or PKCι (lower panels) 60 μM unlabeled ATP and 2 μCi [32P]-ATP. The reactions were analyzed by autoradiography. b, d The phosphorylation of TIEG1 (b) and KLF4 (d) by PKCζ is linear with time. The upper panels are bar graphs showing the quantification of the phosphorylation levels of TIEG1 and KLF4 over the indicated time points, respectively, and the lower panels are the corresponding autoradiographs. e, f The DNA-binding domains of KLF family proteins are targets for PKCζ-mediated phosphorylation. GST, GST-TIEG1(1–362) and GST-TIEG1(DBD) (e), and GST, GST-tagged DBDs of KLF family proteins (f) were subjected to in vitro phosphorylation and subsequent analysis by autoradiography. The data in a, c, e and f are representative of three independent experiments. The data in b and f are shown as the mean (± SD) of four independent experiments
The fact that the conserved DBD of TIEG1 is a PKCsι/ζ substrate encouraged us to investigate whether this would be the case for the DBD of the KLF transcription factors found to interact with PKCι and ζ in this study. All four KLFs were PKCι/ζ substrates in vitro (Fig. 2f). Sp1(DBD), which is a known PKCζ substrate, and BTEB1(DBD) were more heavily phosphorylated than Sp3(DBD) and BTEB2(DBD). Taken together, the results suggest that the aPKCs may regulate KLF family proteins by phosphorylating their DBDs.
S384 and T445 of TIEG1 are phosphorylated by PKCζ
As mentioned above, the zinc finger DBD of the KLF factors are highly conserved (Fig. 3a). Four residues within the DBD of Sp1 are reported to be phosphorylated by PKCζ: S641 in zinc finger 1, and T668, S670 and T681 in zinc finger 2 (reviewed in [14]). Phosphorylation of S641 leads to release of the inhibitor p107 from Sp1 bound to the luteinizing hormone receptor (LHR) promoter, thereby activating transcription of the LHR [19]. T668, S670 and T681 in zinc finger 2 are phosphorylated by PKCζ upon angiotensin II stimulation, leading to activation of the PDGF-D promoter [20]. In order to map the specific residues in TIEG1 phosphorylated by PKCζ, we chose to focus on S384, which is positioned in the recognition helix of zinc finger 1 similarly to S641 of Sp1 (Fig. 3a), and T445 located in the recognition helix of zinc finger 3. None of these residues are involved in direct contact with DNA bases, but they are positioned next to amino acids that are important for sequence-specific DNA binding (Fig. 3a). Substitution of S384 and T445 with alanine significantly reduced the PKCζ-mediated phosphorylation of the TIEG1 DBD in an in vitro phosphorylation assay (Fig. 3b). This was confirmed by introducing the same alanine substitutions into the full-length TIEG1 protein (supplementary material Fig. S1), where the single alanine mutants led to reduction in the phosphorylation of TIEG1. However, introducing the double mutations did not lead to a complete loss of PKCζ mediated phosphorylation (supplementary material Fig. S1). This suggests that other serine and/or threonine residues within the TIEG1 DBD may act as substrates for this kinase. T445 is located in the same position as S702 in Sp1. This residue is not a known PKCζ substrate. In order to investigate whether Sp1 S702 was a PKCζ target, an Sp1 S702A mutant was subject to an in vitro phosphorylation assay. Interestingly, PKCζ-mediated phosphorylation of the Sp1 S702A mutant was reduced to a similar degree as that of the well-known Sp1 S641A mutant (Fig. 3c), indicating Sp1 S702 as a novel target for PKCζ. The mapping of the Sp1 S702 and TIEG1 T445 residues as targets for PKCζ also explains why the Sp3- and BTEB2 DBDs were less phosphorylated than the Sp1- and TIEG1 DBDs (Fig. 2f). The Sp3- and BTEB2 DBDs have an A at the site corresponding to Sp1 S702 and TIEG1 T445 (Fig. 3a). Taken together, these results show that conserved serine and threonine residues within the recognition helices of zinc fingers 1 and 3 of TIEG1 and Sp1 are potential targets for PKCζ mediated phosphorylation.

PKCζ phosphorylates TIEG1 at Ser 384 located in zinc finger 1 and Thr 445 located in zinc finger 3. a Amino acid sequence alignment of the DBDs of the KLF family proteins. Conserved residues are shaded, and the core zinc finger domains are indicated by thick lines and labeled ZF 1–3. The locations of the phospho-acceptors Ser 384 and Thr 445 residues are boxed. Below and to the left is the structure of the zinc finger protein ZNF268 [57] with the location of the phosphorylation sites in the recognition helixes of zinc fingers 1 and 3 indicated by arrows. Below and to the right are the amino acid sequences of zinc fingers 1 and 3 in TIEG1. The amino acids within the recognition helix that are proposed to contact the DNA bases are indicated by solid lines. The prediction is based on the NMR structure of Sp1 [58]. Ser 384 and Thr 445 are shown in bold. b, c Mutations of TIEG1S384 and TIEG1T445 (b), and Sp1S641 and Sp1S702 (c) to alanine significantly reduce their PKCζ-mediated phosphorylation. Equal amounts of GST and GST-tagged TIEG1 proteins (b), GST and GST-tagged Sp1 proteins (c) were subjected to in vitro phosphorylation assays; detection and quantification as described above. Shown are the quantified phosphorylation levels as bar graphs (upper panels), autoradiographs (middle panels) and Coomassie staining indicating the levels of GST and GST-tagged proteins used in the in vitro kinase assays (lower panels). Each bar in b and c represents the mean (± SD) of four independent experiments
Phosphorylation of S384 inhibits the DNA-binding activity of TIEG1
PKCζ-mediated phosphorylation of Sp1 has been reported both to increase and decrease DNA binding (reviewed in [14]). In order to investigate whether PKCζ-mediated phosphorylation of TIEG1 would compromise or stimulate DNA binding, we performed GMSA with the S384A and T445A mutants and the double mutant S384A/T445A. Both the single and double mutants seemed to have stronger affinity for DNA than the wild-type protein (Fig. 4a). To mimic phosphorylation of these residues, the S384E, T445E and S384E/T445E mutants were generated and used in GMSA. No DNA binding could be detected for the S384E and S384E/T445E mutants. The T445E mutant, however, bound DNA with similar affinity as the wild type. These results suggest that phosphorylation of S384 impairs DNA binding, while phosphorylation of T445 has no effect on DNA binding.

PKCζ-mediated phosphorylation of TIEG1S384 impairs its DNA-binding activity. a–c Gel mobility shift assays (GMSA) demonstrating the effects of phospho-mimicking substitutions or in vitro phosphorylation on the DNA-binding ability of KLF proteins. a Substitution of Ser 384 to glutamate abolishes the DNA-binding ability of TIEG1. GMSA using GST and GST-fused TIEG1 proteins, and [32P]-ATP labeled oligonucleotides containing consensus TIEG1-binding sites described in [17]. The binding reactions were separated on non-denaturating polyacrylamide gels that were vacuum dried and analyzed by autoradiography (upper panel). The lower panel shows control protein staining indicating the amounts of proteins used in the assays. b Pre-phosphorylation of TIEG1(DBD) by PKCζ also impairs DNA binding. Here, GST and GST-fused TIEG1 proteins were pre-incubated with PKCζ in the presence or absence of ATP prior to GMSA. c PKCζ-mediated phosphorylation of the DBDs of KLF4 and Sp1 does not impair their DNA-binding activity. GMSA with GST and GST-KLF4(DBD) (left panel), and GST and GST-tagged Sp1proteins (right panel). The proteins were pre-treated with PKCζ and/or ATP prior to GMSA using [32P]-ATP labeled oligonucleotide containing consensus KLF-binding sites described in [18]. The data shown are representative of three independent experiments
In order to determine whether TIEG1 DBD phosphorylated by PKCζ would have similar DNA-binding properties as the S384E/T445E mimicking mutant, the TIEG1 DBD wild-type and double alanine mutant versions were phosphorylated by PKCζ before performing gel shift assays. Pre-phosphorylated TIEG1 DBD binds DNA with significantly less affinity than the unphosphorylated forms (Fig. 4b). Notably, the phosphorylated TIEG1 DBD S384A/T445A mutant showed less affinity for DNA than the unphosphorylated forms, indicating the presence of additional PKCζ phosphorylation sites within the TIEG1 DBD. Furthermore, the effects of PKCζ-mediated phosphorylation on DNA binding were investigated for the KLF family proteins Sp1 and KLF4. Surprisingly, phosphorylation of the KLF4 and Sp1 DBDs did not seem to affect DNA binding at all (Fig. 4c). Hence, PKCζ phosphorylation of the DBDs of KLF family proteins may increase, decrease or have no effect on DNA-binding activity depending on the specific KLF protein and the specific phosphorylation site involved. For instance, as shown here, phosphorylation of TIEG1 S384 in the recognition helix of zinc finger 1 clearly impaired its DNA-binding activity.
The phospho-mimicking mutants of TIEG1 display a redistributed subnuclear localisation
To explore whether phosphorylation of S384 and/or T445 had an impact on the nuclear localization of TIEG1, GFP fusions of wild-type TIEG1 and the six mutants S384A, S384E, T445A, T445E, S384A/T445A and S384E/T445E were expressed in HeLa and U2OS cells. The subcellular distribution of the GFP-fusion proteins was investigated by confocal fluorescence microscopy. In both cell lines, wild-type TIEG1 is localized in the nucleus but excluded from the nucleoli, with a weak staining in the nucleoplasm and enrichment in several specific irregular speckles or bodies (Fig. 5a, upper panels). Interestingly, GFP-TIEG1 S384E or GFP-TIEG1 T445E was redistributed into specific round nuclear bodies (Fig. 5a, middle panels). This redistribution into round nuclear bodies is most prominent for the double mutant GFP-TIEG1 S384E/T445E (Fig. 5a, lower panels), and the phenotype is displayed by about 50% of the transfected cell population. To address the question whether the observed subnuclear redistributions are a simple consequence of overexpression or not, we performed fluorescence recovery after photobleaching (FRAP) analysis with the wild-type and the double mutant forms of GFP-TIEG1. As shown in Fig. 5b, the round nuclear dots of GFP-TIEG1 S384E/T445E are not simple aggregates since their mobility is similar to that of the wild type and the S384A/T445A mutant. Taken together, these results suggest that phosphorylation of TIEG1 DBD may recruit the protein to specific dynamic nuclear structures.

Mutation of TIEG1 Ser 384 and Thr 445 to glutamate induces subnuclear relocalization. a The phospho-mimicking mutants of TIEG1 are enriched in round nuclear bodies. HeLa and/or U2OS cells were transfected with an expression construct for EGFP-TIEG1 or EGFP-tagged TIEG1 mutants and imaged by confocal laser fluorescence microscopy 24 h post transfection. b The TIEG1 mutants display mobility similar to the wild-type TIEG1. HeLa cells were transfected with the indicated constructs, and 24 h post transfection the cells were imaged and fluorescence signals were collected before and after photobleaching a portion of the nucleus. The recovery kinetics were calculated after double normalization of the initial fluorescence, corrected for background and loss of fluorescence caused by imaging. The graph presented here shows the average intensity of the bleach spot of 9–10 independent cells for the indicated time points. Error bars represent the SD values between various experiments
Phosphorylation of T445 enhances the transactivation potential of TIEG1
Overexpression of TIEG1 is shown to mimic TGFβ activity in several cell lines [21–23] and to enhance TGF-β1 expression [24]. TIEG1 has also been found to increase TGFβ-induced expression of the cyclin-dependent kinase inhibitor p21Cip1/WAF1 [22]. Here, we conducted reporter gene assays using the p21Cip1/WAF1 promoter cloned in front of the luciferase gene to determine whether the phosphorylation of TIEG1 DBD at S384 and/or T445 would impact its transactivation potential. As shown in Fig. 6a, overexpression of wild-type TIEG1 increased luciferase expression from the p21 Cip1/WAF1 promoter six- to nine-fold. The transactivation was strongly increased up to 25-fold when TIEG1 containing the T445E mutation was coexpressed, suggesting that phosphorylation of T445 positively regulates its transcriptional activation potential. Consistent with this notion, we observed a reduction in the luciferase activity when the T445A mutant was co-expressed. Surprisingly, the S384E and the S384E/T445E mutants, which we found had lost their DNA-binding activity, still had the capacity to stimulate expression from the p21Cip1/WAF1 promoter (Fig. 6a). A likely explanation for this observation is that the mutants with compromised DNA-binding activity are recruited to the promoter by endogenous TIEG1 and/or other KLF family proteins. The p21Cip1/WAF1 promoter contains six adjacent GC boxes close to the transcriptional start site that have the potential to recruit KLF family proteins (Fig. 6a). We found that TIEG1 (and other KLF proteins too) has a strong capability to form homo- and heterodimers with KLF family proteins via interactions involving the DBDs (Fig. 7, and supplementary material Fig. S2). This interaction is not dependent on the presence of nucleic acids as evidenced by benzonase treatment (Fig. 7c, d). The homo- and heterodimerization potential of the TIEG1 DBD is not impaired by phosphorylation of S384 or T445 (supplementary material Fig. S3). Thus, TIEG1 mutants with impaired DNA-binding activity may be recruited to specific promoter sites by interaction with endogenous KLF factors bound to the promoter.

The TIEG1T445E mutant displays enhanced transactivation potential. a U2OS cells were transiently transfected with 0.1 μg of the reporter p21-LUC, together with the expression vectors for TIEG1 and TIEG1 mutants as indicated and subjected to reporter gene assay analysis. The data represent the mean of two independent experiments performed in triplicates. Error bars represent the SD values between various experiments. b Total cell extracts from the transfected U2OS cells were prepared, and the levels of TIEG1 proteins were analyzed by Western blotting using anti-GFP antibody (upper panel) and then with anti-actin antibody as loading control (lower panel). c All KLF family proteins tested except KLF4 are potential activators of the p21Cip1/WAF1 promoter. The reporter gene assay data shown is from one experiment performed in triplicate, which is representative of three independent experiments each performed in triplicate. Error bars represent the SD values between the parallels. d Western blot analysis of extracts from transfected U2OS cells to show the expression level of the various KLF family proteins

KLF family proteins have the ability to homo- and heterodimerize via their DNA-binding domain. a–c GST pulldown assays were carried out using bacterially expressed GST, or the indicated GST-tagged proteins, and in vitro translated either full-length HA-TIEG1 (a, b) or GFP-TIEG1(DBD) (c). Autoradiographs are presented in the upper panels, whereas Coomassie brilliant blue stained SDS-polyacrylamide gels are shown in the lower panels. c, d TIEG1 homodimerization is not mediated by nucleic acids in the reaction mixture. GFP-TIEG1(DBD) was in vitro translated and divided into two. One half was used directly for GST pulldown assays (c, right panel), and the remaining half was treated with 250 units of benzonase for 20 min at room temperature prior to GST pulldown assays (c, left panel). d The benzonase used in the assay was active. The expression construct, GFP-TIEG1(DBD), was either treated with benzonase or left untreated, and the reaction analyzed on a 0.7% agarose gel followed by ethdium bromide staining. Data shown are representative of two independent experiments
Sp1 is a well-known activator of the p21Cip/WAF1 promoter. Phosphorylation of Sp1 on T453, T739 [25] or S59 [26] has been shown to induce expression of p21Cip1/WAF1 and cellular senescence. Additionally, PKC [27], PKG [28] and high-glucose induced p21Cip1/WAF1 expression is mediated by Sp1 binding to the p21Cip1/WAF1 promoter. To examine whether other KLF family proteins were potential activators of the p21Cip1/WAF1 promoter, luciferase reporter assays were performed with KLF4, KLF11 and EGR1 in addition to Sp1 and TIEG1 (Fig. 6c). Both KLF11 and EGR1 induced expression from the p21Cip1/WAF1 promoter, KLF11 (TIEG2) with a similar potency as TIEG1. KLF4, on the other hand, did not cause significant activation of the promoter. When we replaced the DBD of TIEG1 with the DBD of Sp1, the hybrid transcription factor also induced the p21Cip1/WAF1 promoter although less potently than Sp1 and TIEG1. Furthermore, we found overexpression of EGR1 to be significantly less toxic than overexpression of TIEG1, Sp1 and KLF11 (data not shown), although EGR1 also induced p21Cip1/WAF1 expression. This suggests that apoptosis induced by Sp1-like family proteins may involve additional mechanisms besides p21Cip1/WAF1 upregulation. This correlates well with reports showing that TIEG1 induces apoptosis through the mitochondrial apoptotic pathway upregulating Bax and Bim, and downregulating Bcl-2 and Bcl-XL [29], and KLF11 inducing oligodendroglial cell death by downregulation of Bcl-X expression [30].
Discussion
In this study we have shown that TIEG1 is an interaction partner and substrate for aPKCs. Two residues in the DBD, S384 and T445 in zinc fingers 1 and 3, respectively, were found to be the major sites phosphorylated by recombinant aPKCs. Substitution of S384 by the phospho-mimicking glutamate (S384E) resulted in inhibition of DNA binding, while the T445E mutant displayed an enhanced transactivation potential relative to the wild-type protein. Both mutations led to a subnuclear redistribution of the protein into round, dynamic nuclear bodies. In addition, we show that the DBDs of various KLF family proteins are targets for PKCζ/ι-mediated phosphorylation and that the KLF proteins have a strong ability to form homo- and heterodimers via their DBDs.
Sp1 is phosphorylated by PKCζ [20, 31–36] (reviewed in [14]). PKCζ-mediated phosphorylation of S641 in zinc finger 1 of Sp1 does not affect its DNA-binding activity, but leads to release of the p107 inhibitor and thereby enhanced transcriptional activation [36]. In contrast, here we report that PKCζ-mediated phosphorylation of TIEG1 at S384, which corresponds to S641 of Sp1, strongly inhibited DNA binding. However, the transcriptional activity of the TIEG1 S384E mutant on the p21Cip1/WAF1 promoter was less affected. This suggests that even if KLF family proteins are phosphorylated at corresponding amino acid residues, the biological outcome of the phosphorylation may be specific to the particular KLF factor involved.
We have also shown that corresponding residues in zinc finger 3 of TIEG1 and Sp1 are targets for PKCζ-mediated phosphorylation. The phospho-mimicking mutant TIEG1 T445E significantly enhanced TIEG1-induced activation of the p21Cip1/WAF1 promoter. Interestingly, Sp1 S702 is also reported to be modified by the monosaccharide O-linked β-N-acetylglucose amine (O-GlcNAc) [37]. O-GlcNAcylation of Sp1 has been shown to increase [37–43] or decrease [42, 44] its transcriptional activity depending on the target promoter. O-GlcNAc and phosphate modifications on substrates are shown to interact in various ways both at the global cellular protein level and at specific sites on particular proteins (reviewed in [45]). For instance, they can competitively and alternatively occupy the same serine or threonine residue, or they can regulate each other by generating large functional complexes. Hence, phosphorylation of Sp1 S702 may regulate and/or communicate with the O-GlcNAc modification of Sp1, and thereby also regulate its transcriptional activity. Furthermore, Sp1 S702 is located next to Sp1 K703, which is reported to be targeted for acetylation [46]. Acetylation of Sp1 K703 recruits HDAC1 to the Sp1 located promoter sites and represses transcription, while deacetylated K703 recruits p300 and induces transcription. Hence, phosphorylation and/or glycosylation of S702 and acetylation of K703 may interact with each other in an agonistic or antagonistic manner, and thereby integrate diverse signaling cues at the transcriptional level through Sp1 bound at promoter sites. Interestingly, we also found TIEG1 to be modified by O-GlcNAc (E. Alemu and T. Johansen, unpublished data). O-GlcNAc modifications of proteins are believed to constitute an important regulatory pathway that controls protein activity according to glucose availability. O-GlcNAc modification of Sp1 has been connected to glucose sensing, expression of glucose transporters and in various aspects of hyperglycaemic-induced glucotoxicity (reviewed in [47]), whereas TIEG1 is involved in the regulation of circadian gene expression, which is trigged by glucose [48]. Interestingly, aPKCs are heavily involved in regulation of glucose transport and insulin sensitivity [7]. Our in vitro data suggest that aPKCs at least partly may exert this regulation via post-translational modifications of Sp1 and TIEG1. Additionally, TIEG1 contains a lysine residue (K446), which lies next to the PKCζ targeted site (TIEG1 T445). Whether this lysine as well is a target for post-translational modifications, such as acetylation, has not been reported yet.
Overexpression of TIEG1 induces apoptosis by activating TGFβ-regulated signaling pathways [23, 49, 50] (reviewed in [12]) and promoting mitochondrial apoptotic pathways [29]. TIEG1 overexpression is reported to increase the expression of the cyclin-dependent kinase inhibitor p21Cip1/WAF1 and significantly decrease cellular proliferation. Here we show that TIEG1 is a potent activator of the p21Cip1/WAF1 promoter and that a phospho-mimicking mutation of TIEG1 T445 significantly enhances this activation. In general, Sp1 family transcription factors are reported as major regulators of p21Cip1/WAF1 transcription (reviewed in [51, 52]). The p21Cip1/WAF1 promoter contains six binding sites for Sp1 family proteins at the proximal promoter (bp −120 to −50), and mutations of these sites significantly affect synergistic activation by Sp1, p53 and other signals that regulate transcription of the p21Cip1/WAF1 gene [52, 53]. Importantly, the transcriptional activity of Sp1 has been reported to be regulated by phosphorylation by various kinases at different sites, and these signaling events are integrated at GC-box containing promoters (reviewed in [14]). We found that the aPKCs phosphorylate Sp1 and TIEG1 at corresponding sites in the DBD. Both proteins are reported to induce p21Cip1/WAF1 expression upon various stimuli, via the six promoter proximal GC boxes. GC box 3 is shown to mediate p21Cip1/WAF1 induction by agents such as TGFβ and histone deacetylase inhibitors, whereas GC boxes 1 and 2 are important for transcriptional activation mediated by phorbol esters and okadaic acid (reviewed in [52]). The aPKCs are involved in cell polarity processes, metabolic regulation and cell survival signaling, and PKCι is also reported to act as an oncogene when its gene is amplified in lung and ovarian cancers (reviewed in [4]). Hence, one would expect that aPKCs could have a negative effect on the suggested tumor suppressor TIEG1. This effect could be exerted via S384 phosphorylation. However, aPKC could also act positively via phosphorylation of T445. We found that the S384E/T445E double mutant strongly reduced the p21Cip1/WAF1 induction seen with the T445E mutant alone. The effect of aPKC phosphorylation of the TIEG1 DBD may therefore depend on the accessibility of these sites. The above-mentioned O-GlcNAcylation and acetylation may be involved in regulating this accessibility.
How post-translational modifications of Sp1 influence its affinity for DNA and other proteins is only beginning to become apparent. Sp1 can interact with components of the basal transcription machinery, as well as transcription factors, coactivators and corepressors. Several of these interactions are regulated by post-translational modifications of Sp1. This protein also has a strong tendency to interact with itself [54] and with other members of the KLF protein family (this work). In addition, there is direct binding competition among KLF family proteins on GC-rich promoter sequences ([55] and ref. therein). Our present results show that various activities of TIEG1 including DNA binding, transcriptional activation and subnuclear localization are also regulated by post-translational modifications, suggesting that this is most probably a general feature of KLF family proteins. Moreover, our data indicate that KLF family proteins may offer a large number of combinatorial possibilities to regulate expression from GC box containing promoters, making them important candidates for integrating various cellular signaling events at the transcriptional level.
Abbreviations
- DBD:
-
DNA-binding domain
- TIEG1:
-
Transforming growth factor-β-inducible early response gene 1
- aPKC:
-
Atypical protein kinase C
- GMSA:
-
Gel mobility shift assay
- GFP:
-
Green fluorescent protein
- FRAP:
-
Fluorescence recovery after photobleaching
References
Reyland ME (2009) Protein kinase C isoforms: multi-functional regulators of cell life and death. Front Biosci 14:2386–2399
Kim MS, Lim WK, Cha JG, An NH, Yoo SJ, Park JH, Kim HM, Lee YM (2001) The activation of PI 3-K and PKC zeta in PMA-induced differentiation of HL-60 cells. Cancer Lett 171:79–85
Assemat E, Bazellieres E, Pallesi-Pocachard E, Le Bivic A, Massey-Harroche D (2008) Polarity complex proteins. Biochim Biophys Acta 1778:614–630
Fields AP, Regala RP (2007) Protein kinase C iota: human oncogene, prognostic marker and therapeutic target. Pharmacol Res 55:487–497
Kovac J, Oster H, Leitges M (2007) Expression of the atypical protein kinase C (aPKC) isoforms iota/lambda and zeta during mouse embryogenesis. Gene Expr Patterns 7:187–196
Bandyopadhyay G, Standaert ML, Sajan MP, Kanoh Y, Miura A, Braun U, Kruse F, Leitges M, Farese RV (2004) Protein kinase C-lambda knockout in embryonic stem cells and adipocytes impairs insulin-stimulated glucose transport. Mol Endocrinol 18:373–383
Farese RV, Sajan MP, Yang H, Li P, Mastorides S, Gower WR Jr, Nimal S, Choi CS, Kim S, Shulman GI, Kahn CR, Braun U, Leitges M (2007) Muscle-specific knockout of PKC-lambda impairs glucose transport and induces metabolic and diabetic syndromes. J Clin Invest 117:2289–2301
Chalmers AD, Pambos M, Mason J, Lang S, Wylie C, Papalopulu N (2005) aPKC, Crumbs3 and Lgl2 control apicobasal polarity in early vertebrate development. Development 132:977–986
Imai F, Hirai S, Akimoto K, Koyama H, Miyata T, Ogawa M, Noguchi S, Sasaoka T, Noda T, Ohno S (2006) Inactivation of aPKClambda results in the loss of adherens junctions in neuroepithelial cells without affecting neurogenesis in mouse neocortex. Development 133:1735–1744
Subramaniam M, Harris SA, Oursler MJ, Rasmussen K, Riggs BL, Spelsberg TC (1995) Identification of a novel TGF-beta-regulated gene encoding a putative zinc finger protein in human osteoblasts. Nucleic Acids Res 23:4907–4912
Ellenrieder V (2008) TGFbeta regulated gene expression by smads and Sp1/KLF-like transcription factors in cancer. Anticancer Res 28:1531–1539
Subramaniam M, Hawse JR, Johnsen SA, Spelsberg TC (2007) Role of TIEG1 in biological processes and disease states. J Cell Biochem 102:539–548
Kaczynski J, Cook T, Urrutia R (2003) Sp1- and Kruppel-like transcription factors. Genome Biol 4:206
Tan NY, Khachigian LM (2009) Sp1 phosphorylation and its regulation of gene transcription. Mol Cell Biol 29:2483–2488
Subramaniam M, Hefferan TE, Tau K, Peus D, Pittelkow M, Jalal S, Riggs BL, Roche P, Spelsberg TC (1998) Tissue, cell type, and breast cancer stage-specific expression of a TGF-beta inducible early transcription factor gene. J Cell Biochem 68:226–236
Sjottem E, Rekdal C, Svineng G, Johnsen SS, Klenow H, Uglehus RD, Johansen T (2007) The ePHD protein SPBP interacts with TopBP1 and together they co-operate to stimulate Ets1-mediated transcription. Nucleic Acids Res 35:6648–6662
Chrisman HR, Tindall DJ (2003) Identification and characterization of a consensus DNA binding element for the zinc finger transcription factor TIEG/EGRalpha. DNA Cell Biol 22:187–199
Jiang J, Chan YS, Loh YH, Cai J, Tong GQ, Lim CA, Robson P, Zhong S, Ng HH (2008) A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat Cell Biol 10:353–360
Zhang Y, Liao M, Dufau ML (2008) Unlocking repression of the human luteinizing hormone receptor gene by trichostatin A-induced cell-specific phosphatase release. J Biol Chem 283:24039–24046
Tan NY, Midgley VC, Kavurma MM, Santiago FS, Luo X, Peden R, Fahmy RG, Berndt MC, Molloy MP, Khachigian LM (2008) Angiotensin II-inducible platelet-derived growth factor-D transcription requires specific Ser/Thr residues in the second zinc finger region of Sp1. Circ Res 102:e38–e51
Cook T, Gebelein B, Belal M, Mesa K, Urrutia R (1999) Three conserved transcriptional repressor domains are a defining feature of the TIEG subfamily of Sp1-like zinc finger proteins. J Biol Chem 274:29500–29504
Johnsen SA, Subramaniam M, Janknecht R, Spelsberg TC (2002) TGFbeta inducible early gene enhances TGFbeta/Smad-dependent transcriptional responses. Oncogene 21:5783–5790
Tachibana I, Imoto M, Adjei PN, Gores GJ, Subramaniam M, Spelsberg TC, Urrutia R (1997) Overexpression of the TGFbeta-regulated zinc finger encoding gene, TIEG, induces apoptosis in pancreatic epithelial cells. J Clin Invest 99:2365–2374
Cao Z, Wara AK, Icli B, Sun X, Packard RR, Esen F, Stapleton CJ, Subramaniam M, Kretschmer K, Apostolou I, von Boehmer H, Hansson GK, Spelsberg TC, Libby P, Feinberg MW (2009) Kruppel-like factor KLF10 targets transforming growth factor-beta1 to regulate CD4(+) CD25(−) T cells and T regulatory cells. J Biol Chem 284:24914–24924
De Siervi A, Marinissen M, Diggs J, Wang XF, Pages G, Senderowicz A (2004) Transcriptional activation of p21(waf1/cip1) by alkyl phospholipids: role of the mitogen-activated protein kinase pathway in the transactivation of the human p21(waf1/cip1) promoter by Sp1. Cancer Res 64:743–750
Kim HS, Lim IK (2009) Phosphorylated extracellular signal-regulated protein kinases 1 and 2 phosphorylate Sp1 on serine 59 and regulate cellular senescence via transcription of p21Sdi1/Cip1/Waf1. J Biol Chem 284:15475–15486
Schavinsky-Khrapunsky Y, Huleihel M, Aboud M, Torgeman A (2003) Role of protein kinase C and the Sp1–p53 complex in activation of p21(WAF-1) expression by 12-O-tetradecanoylphorbol-13-acetate in human T cells. Oncogene 22:5315–5324
Cen B, Deguchi A, Weinstein IB (2008) Activation of protein kinase G increases the expression of p21CIP1, p27KIP1, and histidine triad protein 1 through Sp1. Cancer Res 68:5355–5362
Jin W, Di G, Li J, Chen Y, Li W, Wu J, Cheng T, Yao M, Shao Z (2007) TIEG1 induces apoptosis through mitochondrial apoptotic pathway and promotes apoptosis induced by homoharringtonine and velcade. FEBS Lett 581:3826–3832
Wang Z, Spittau B, Behrendt M, Peters B, Krieglstein K (2007) Human TIEG2/KLF11 induces oligodendroglial cell death by downregulation of Bcl-XL expression. J Neural Transm 114:867–875
Dwivedi A, Slater SC, George SJ (2009) MMP-9 and -12 cause N-cadherin shedding and thereby beta-catenin signalling and vascular smooth muscle cell proliferation. Cardiovasc Res 81:178–186
Kim JI, Cordova AC, Hirayama Y, Madri JA, Sumpio BE (2008) Differential effects of shear stress and cyclic strain on Sp1 phosphorylation by protein kinase Czeta modulates membrane type 1-matrix metalloproteinase in endothelial cells. Endothelium 15:33–42
Neid M, Datta K, Stephan S, Khanna I, Pal S, Shaw L, White M, Mukhopadhyay D (2004) Role of insulin receptor substrates and protein kinase C-zeta in vascular permeability factor/vascular endothelial growth factor expression in pancreatic cancer cells. J Biol Chem 279:3941–3948
Rafty LA, Khachigian LM (2001) Sp1 phosphorylation regulates inducible expression of platelet-derived growth factor B-chain gene via atypical protein kinase C-zeta. Nucleic Acids Res 29:1027–1033
Rojo AI, Salina M, Salazar M, Takahashi S, Suske G, Calvo V, de Sagarra MR, Cuadrado A (2006) Regulation of heme oxygenase-1 gene expression through the phosphatidylinositol 3-kinase/PKC-zeta pathway and Sp1. Free Radic Biol Med 41:247–261
Zhang Y, Liao M, Dufau ML (2006) Phosphatidylinositol 3-kinase/protein kinase Czeta-induced phosphorylation of Sp1 and p107 repressor release have a critical role in histone deacetylase inhibitor-mediated derepression [corrected] of transcription of the luteinizing hormone receptor gene. Mol Cell Biol 26:6748–6761
Jackson SP, Tjian R (1988) O-glycosylation of eukaryotic transcription factors: implications for mechanisms of transcriptional regulation. Cell 55:125–133
Brasse-Lagnel C, Fairand A, Lavoinne A, Husson A (2003) Glutamine stimulates argininosuccinate synthetase gene expression through cytosolic O-glycosylation of Sp1 in Caco-2 cells. J Biol Chem 278:52504–52510
Brasse-Lagnel C, Lavoinne A, Loeber D, Fairand A, Bole-Feysot C, Deniel N, Husson A (2007) Glutamine and interleukin-1beta interact at the level of Sp1 and nuclear factor-kappaB to regulate argininosuccinate synthetase gene expression. FEBS J 274:5250–5262
Goldberg HJ, Whiteside CI, Fantus IG (2002) The hexosamine pathway regulates the plasminogen activator inhibitor-1 gene promoter and Sp1 transcriptional activation through protein kinase C-beta I and -delta. J Biol Chem 277:33833–33841
Goldberg HJ, Whiteside CI, Hart GW, Fantus IG (2006) Posttranslational, reversible O-glycosylation is stimulated by high glucose and mediates plasminogen activator inhibitor-1 gene expression and Sp1 transcriptional activity in glomerular mesangial cells. Endocrinology 147:222–231
Jochmann R, Thurau M, Jung S, Hofmann C, Naschberger E, Kremmer E, Harrer T, Miller M, Schaft N, Sturzl M (2009) O-linked N-acetylglucosaminylation of Sp1 inhibits the human immunodeficiency virus type 1 promoter. J Virol 83:3704–3718
Keembiyehetty CN, Candelaria RP, Majumdar G, Raghow R, Martinez-Hernandez A, Solomon SS (2002) Paradoxical regulation of Sp1 transcription factor by glucagon. Endocrinology 143:1512–1520
Yang X, Zhang F, Kudlow JE (2002) Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: coupling protein O-GlcNAcylation to transcriptional repression. Cell 110:69–80
Zeidan Q, Hart GW (2010) The intersections between O-GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J Cell Sci 123:13–22
Hung JJ, Wang YT, Chang WC (2006) Sp1 deacetylation induced by phorbol ester recruits p300 to activate 12(S)-lipoxygenase gene transcription. Mol Cell Biol 26:1770–1785
Issad T, Kuo M (2008) O-GlcNAc modification of transcription factors, glucose sensing and glucotoxicity. Trends Endocrinol Metab 19:380–389
Hirota T, Okano T, Kokame K, Shirotani-Ikejima H, Miyata T, Fukada Y (2002) Glucose down-regulates Per1 and Per2 mRNA levels and induces circadian gene expression in cultured Rat-1 fibroblasts. J Biol Chem 277:44244–44251
Chalaux E, Lopez-Rovira T, Rosa JL, Pons G, Boxer LM, Bartrons R, Ventura F (1999) A zinc-finger transcription factor induced by TGF-beta promotes apoptotic cell death in epithelial Mv1Lu cells. FEBS Lett 457:478–482
Ribeiro A, Bronk SF, Roberts PJ, Urrutia R, Gores GJ (1999) The transforming growth factor beta (1)-inducible transcription factor TIEG1, mediates apoptosis through oxidative stress. Hepatology 30:1490–1497
Gartel AL, Goufman E, Tevosian SG, Shih H, Yee AS, Tyner AL (1998) Activation and repression of p21(WAF1/CIP1) transcription by RB binding proteins. Oncogene 17:3463–3469
Gartel AL, Radhakrishnan SK (2005) Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res 65:3980–3985
Koutsodontis G, Moustakas A, Kardassis D (2002) The role of Sp1 family members, the proximal GC-rich motifs, and the upstream enhancer region in the regulation of the human cell cycle inhibitor p21WAF-1/Cip1 gene promoter. Biochemistry 41:12771–12784
Su W, Jackson S, Tjian R, Echols H (1991) DNA looping between sites for transcriptional activation: self-association of DNA-bound Sp1. Genes Dev 5:820–826
Choi WI, Jeon BN, Yun CO, Kim PH, Kim SE, Choi KY, Kim SH, Hur MW (2009) Proto-oncogene FBI-1 represses transcription of p21CIP1 by inhibition of transcription activation by p53 and Sp1. J Biol Chem 284:12633–12644
Datto MB, Yu Y, Wang XF (1995) Functional analysis of the transforming growth factor beta responsive elements in the WAF1/Cip1/p21 promoter. J Biol Chem 270:28623–28628
Pavletich NP, Pabo CO (1991) Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science 252:809–817
Narayan VA, Kriwacki RW, Caradonna JP (1997) Structures of zinc finger domains from transcription factor Sp1. Insights into sequence-specific protein-DNA recognition. J Biol Chem 272:7801–7809
Hamarstrøm M, Hellgren N, van Den Berg S, Berglund H, Hard T (2002) Rapid screening for improved solubility of small human proteins produced as fusion proteins in Escherichia coli. Protein Sci 11:313–321
Lamark T, Perander M, Outzen H, Kristiansen K, Overvatn A, Michaelsen E, Bjorkoy G, Johansen T (2003) Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J Biol Chem 278:34568–34581
Sjøttem E, Andersen C, Johansen T (1997) Structural and functional analyses of DNA bending induced by Sp1 family transcription factors. J Mol Biol 267:490–504
Acknowledgments
We thank the BioImaging and Proteomics FUGE core facilities at the Institute of Medical Biology, University of Tromsø, for use of instrumentation and expert assistance. This work was supported in part by grants from the FUGE program of the Norwegian Research Council, the Norwegian Cancer Society and the Blix Foundation to T.J.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Alemu, E.A., Sjøttem, E., Outzen, H. et al. Transforming growth factor-β-inducible early response gene 1 is a novel substrate for atypical protein kinase Cs. Cell. Mol. Life Sci. 68, 1953–1968 (2011). https://doi.org/10.1007/s00018-010-0541-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-010-0541-1