
Abstract
Cell death is a fundamental ingredient of life. Thus, not surprisingly more than one form of cell death exists. Several excellent reviews on various forms of cell death have already been published but manuscripts describing interconnection and interdependence between such processes are uncommon. Here, what follows is a brief introduction on all three classical forms of cell death, followed by a more detailed insight into the role of p53, the master regulator of apoptosis, and other forms of cell death. While discussing p53 and also the role of caspases in cell death forms, we offer insight into the interplay between autophagy and apoptosis, or necrosis, where autophagy may initially serve pro-survival functions. The review moves further to present some details about less researched forms of programmed cell death, namely necroptosis, necrosis and mitoptosis. These “mixed” forms of cell death allow us to highlight the interconnected nature of cell death forms, particularly apoptosis and necrosis. The interdependence between apoptosis, autophagy and necrosis, and their significance for cancer development and treatment are also analyzed in further parts of the review. In the concluding parts, the afore-mentioned issues will be put in perspective for the development of novel anti-cancer therapies.
Similar content being viewed by others

Introduction
Cell death in its various forms shapes the essence of life. Predominantly, apoptotic cell death is of major importance during organism development as well as in regulation of the immune system and in defense response to disease stimuli (e.g. viral diseases). Cell death itself is interconnected with cell survival and cell proliferation, both at the molecular level, and in philosophical terms (Cherlonneix 2008; Maddika et al. 2007).
Several forms of cell death may typically be induced within the same tissue although apoptosis is the fastest, while other forms, like necrosis or autophagy, only become visible when apoptosis is inhibited (Los et al. 2002; Martinet et al. 2006). Apoptosis is also frequently called programmed cell death (PCD), although some researchers reserve the term PCD only to developmental process-related death. Approximately, 10 million cells per day undergo apoptosis in a healthy adult human (Curtin and Cotter 2003). Such mechanism is needed not only to preserve the homeostasis of the organism, but also to control tissue size and shape under different developmental stages, or to downsize the number of specific immune-effector cells after pathogen eradication (Los et al. 1999). Cells undergo lots of alterations during apoptosis; while chromatin condenses, cells lose their attachment to the surrounding and shrink, as the most peculiar property of apoptosis mechanism, cell membrane starts blebbing. Blebs are the progenitors of apoptotic bodies with small, nearly spherical cytoplasmic fragments encapsulated in cell membranes. Apoptotic bodies may contain functional organelles surrounded by intact plasma membranes (Elmore 2007; Ghavami et al. 2009). Phosphatidylserine, a phospholipid embedded in the plasma membrane, is exposed on the outer side of apoptotic bodies. They act as “eat me” signals, thus attracting macrophages, and are efficiently phagocytosed (Elmore 2007; Ghavami et al. 2009).
Two partly interconnected apoptotic mechanisms exist; caspase-dependent classical apoptosis and caspase-independent programmed form of cell death, sometimes called necroptosis (Kinnally et al. 2011; Smith and Yellon 2011). Both forms of cell death may be interconnected as caspases may lead to the activation of non-caspase proteases and vice versa.
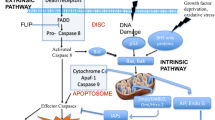
The classical, caspase-dependent apoptosis is initiated either by extrinsic or intrinsic factors (Fig. 1). The extrinsic pathway is activated by the engagement of transmembrane receptors [death receptors, i.e. CD95/APO-1/Fas, tumor necrosis factor α (TNF)-receptors, “TNF related apoptosis inducing ligand” (TRAIL)-receptors] by its ligands (APO-1/Fas, TNF, TRAIL). Upon ligand binding to death receptors their cytoplasmic death domains (usually acting as trimers) attract adaptor molecules (typically FADD) and initiate a caspase cascade. Different receptors/ligand pairs exist, i.e. Fas/FasL and TNF/TNFR1 TRAIL/DRs; they all eventually lead to the activation of caspase-8, which in turn activates downstream caspases (Elmore 2007; Los et al. 1999).

Death-receptor family signaling leading mainly to apoptosis. In general, two types of signaling complexes can form at different types of death receptors. Death-inducing signaling complexes known as DISCs are formed at CD95, TRAIL-R1 or TRAIL-R2. All these receptors recruit DISCs that have a similar basic composition (FADD, pro-caspases-8). DISC-complexes allow caspase-8 activation and transduction of the apoptotic signal. Proapoptotic Bcl2-family molecule Bid may be cleaved by caspase-8, and resulting t-Bid activates the mitochondrial (intrinsic) apoptotic pathway (omitted here to preserve figure’s transparency). The second group comprise the TNFR1/DR3/DR6 and EDAR receptors, recruit a different set of molecules (see text for further details) that transduces both apoptotic and survival signals. ASK activator of S-phase kinase, also known as DBF4A; FADD Fas-associated via death domain; FLIP flice-inhibitory protein, flice is a former name for caspase-8; Gas2 growth arrest-speciffic-2; GSK glycogen synthase kinase; IAP inhibitor of apoptosis protein; ICAD inhibitor of caspase-activated protease; IKK inhibitor of κ-light chain gene enhancer in B cells, also known as IKBKB; JNK c-Jun kinase, other names: MAPK8, SAPK1; MKK other names: MAPK mitogen-activated protein kinase, MEK; NIK nuclear factor (NF)-κB-inducing kinase; RIPK1 receptor-interacting serine/threonine kinase 1, formerly also known as RIP; t-Bid truncated BID; TRAD TNFR1-associated death domain protein; TRAF TNF-receptor-associated factor
The intrinsic pathway, also called mitochondrial pathway, is regulated by Bcl2 family of proteins. These proteins control mitochondrial membrane permeability. A heme-like structure containing protein cytochrome c located on outside of the inner mitochondrial membrane is released into cytosol, where together with (d)ATP it binds to apoptotic protease activating factor (Apaf1) and forms apoptosome complex (Barczyk et al. 2005; Elmore 2007; Los et al. 1999). Apoptosome activates caspase-9 that in turn activates down-stream elements of caspase cascade. Although mitochondrial pathway is included in caspase-dependent apoptosis mechanisms, once cytochrome c is released, it may also initiate caspase-independent apoptosis. In the caspase-independent pathway, apoptosis-inducing factor (a flavaprotein) and endonuclease-G protein are also released from mitochondria and migrate into the nucleus to condense chromatin (Elmore 2007; Saelens et al. 2004).
In the following paragraphs, we will briefly introduce two other processes, namely necrosis and autophagy that, under certain circumstances, may also constitute programmed forms of cell death.
Necrosis
Morphologically “necrotic cell death” or “necrosis” refers to a gain in cell volume, swelling of organelles, plasma membrane rupture and subsequent loss of intracellular contents. Numerous mediators, organelles and cellular processes have been implicated in necrotic cell death, but it is still unclear how they are related to each other. Biochemical behavior of apoptosis such as activation of specific proteases and oligonucleosomal DNA fragmentation may hardly happen in necrotic cells (Proskuryakov et al. 2003). Necrosis, like apoptosis, may be considered as a form of the execution phase of PCD, although the consequences of necrotic and apoptotic cell death are quite different for a whole organism. In the case of necrosis, the inflammatory response may be caused by cytosolic constituents pouring into the intercellular space through the damaged plasma membrane; in apoptosis these products are safely isolated inside macrophages. Disturbance of a fine balance between necrosis and apoptosis may be a key element in development of some diseases (Proskuryakov et al. 2003). Terms “programmed necrosis” or “necroptosis” emphasize a degree of regulation and molecular mechanism of these death processes, and are explained at further parts of the review.
Autophagy
Autophagy is a highly conserved degradation pathway for bulk cellular components; it includes macroautophagy, microautophagy and chaperone-mediated autophagy (Mizushima and Komatsu 2011). Macroautophagy is morphologically known as the formation of double membrane autophagosomes, which take the control of impaired organelles or unwanted cellular components and deliver them to lysosomes for degradation and recycling.
Autophagy is related to numerous physiological and pathological processes, including cell survival, cell death, cell metabolism, development, infection, immunity and aging (Mehrpour et al. 2010). Autophagy is closely involved in the etiology of many important human diseases, including cancer, neurodegenerative diseases and metabolic disorders (Meijer and Codogno 2009).
Autophagy predominantly serves as a cell survival mechanism, via its suppressive role on necrotic cell death, such as necroptosis and poly (ADP-ribose) polymerase-1 (PARP-1)-mediated cell death. More importantly, the anti-necrosis function of autophagy has important biological functions in various pathological processes and diseases, including cancer and ischemia/reperfusion injury (Cardinal et al. 2009; Esposti et al. 2010). “Autophagic cell death” is morphologically defined as a type of cell death that occurs in the absence of chromatin condensation but accompanied by massive autophagic vacuolization of the cytoplasm (Yu et al. 2004a, 2004b).
Interconnection Between Apoptosis and Other Cell Death Pathways
Dual Regulatory Function of p53 in Apoptosis and Autophagy
p53 protein was initially discovered as a transformation-associated protein in 1979 (Chang et al. 1979; DeLeo et al. 1979; Kress et al. 1979; Lane and Crawford 1979). Only later it became clear that the wild-type p53, in addition to its tumor suppressing activity, is also involved in regulation of wide range of cellular processes, including cell cycle control, DNA repair (Kastan et al. 1991; Levine 1997; Lu and Lane 1993), cell differentiation (Rotter et al. 1994), cell senescence (Wynford-Thomas 1999), genome stability, apoptosis (Chen et al. 1996; Ghavami et al. 2011) and autophagy (Feng et al. 2005; Ghavami et al. 2011; Tasdemir et al. 2008a).
Role of p53 in Apoptosis
p53-dependent apoptosis was first observed in irradiated mouse thymocytes (Clarke et al. 1993; Lowe et al. 1993). In addition to irradiation, oncogenes such as adenovirus E1A may activate the p53 and lead to apoptosis (Lowe and Ruley 1993). p53 contributes to apoptosis induction mostly by its transcription-dependent effects (please see next paragraphs). Nuclear p53 induces the expression of murine double minute 2 (Mdm2) gene (Barak et al. 1993; Momand et al. 1992), which negatively regulates the p53 protein activity through binding, ubiquitination, thus prompting its degradation via the proteosome-pathway. Cellular stress signals such as DNA damage interrupt Mdm2-mediated inhibition of p53, leading to accumulation of p53 both in the nucleus and in the cytoplasm.
Multiple mechanisms have been invoked to explain how p53 triggers “mitochondrial outer membrane permeabilization” (Moll et al. 2006). In apoptotic cells, p53 co-immunoprecipitates with Bcl2, Bcl-XL and Bak (Leu et al. 2004; Moll et al. 2006). According to Mihara et al. (2003), p53 binds to Bcl-XL via its DNA binding domain. However, based on a recent nuclear magnetic resonance-based binding study, Bcl-XL interacts with the N-terminal domain of p53 (Xu et al. 2006). The binding site on Bcl-XL is located in the region including α-4, the N-and C-termini of α-3, the N-terminus of α-5, and the central part of α-2 helix (Xu et al. 2006). Alternatively, cytoplasmic p53 can also induce cell death via direct activation of cytosolic Bax (Chipuk et al. 2004). Using UV-treated transformed mouse embryonic fibroblasts, Chipuk et al. (2004) showed an alternative cytosolic p53 cell death pathway in which cytosolic p53 functions analogously to the Bcl2-homology (BH) 3-only subset of proapoptotic Bcl2 proteins to activate Bax and subsequent mitochondria permeabilization and apoptosis. In later parts of the review we also discuss the role of cytoplasmic p53 at the level of mitochondria, where it acts as a repressor of autophagy (Tasdemir et al. 2008a) (Fig. 2).

p53 localization is a key regulator of p53-induced apoptosis and autophagy. Nuclear p53 induces cellular autophagy, apoptosis and repair through its transcriptional activity while cytosolic p53 inhibits autophagy and induces apoptosis
Oncogene expression, DNA damage or other forms of stress result in stabilization of p53 protein by phosphorylation or other modifications (Xu 2003). Stabilized p53 accumulates in the nucleus and binds to specific DNA sequences leading to transactivation of a number of proapoptotic genes, including those encoding members of the Bcl2 family, such as the BH3 only proteins Bax, Noxa and Puma (Chipuk et al. 2005; Miyashita and Reed 1995; Nakano and Vousden 2001; Oda et al. 2000; Xu et al. 2009) (Fig. 2). Other proapoptotic genes such as Bid, Apaf1 and PIDD (p53-induced protein with a death domain) are also defined as transcriptional targets of p53 (Moroni et al. 2001; Sax et al. 2002). Removal or inactivation of each of these genes from a particular model system produced partial resistance to p53-induced apoptosis. For instance, Jeffers et al. (2003) reported complete impairment of p53-dependent apoptosis pathway in certain cell types from PUMA (p53 upregulated modulator of apoptosis)—knock-out mice. PUMA has been also shown to disrupt the interaction of p53 with anti-apototic proteins, such as Bcl-XL, resulting in the release of p53. Displaced from Bcl-XL p53 can now induce mitochondrial permeabilization and apoptosis through interaction with other proteins such as Bax (Chipuk et al. 2005). This finding suggests that regulation of transcription by nuclear p53 may positively regulate function of cytoplasmic p53.
Role of p53 in Autophagy
p53 may either activate or repress autophagy depending on cellular energy status, and the contextual activation of other signaling pathways. The cytoplasmic p53 protein inhibits autophagy in a transcription-independent fashion, while the nuclear p53 stimulates autophagy via the transactivation of specific target genes (Tasdemir et al. 2008b) (Fig. 2). Several p53 target genes stimulate autophagy. For instance, damage-regulated autophagy modulator (DRAM) is a p53 target gene encoding a lysosomal protein that induces autophagy (Crighton et al. 2006). p53 may directly interact with DRAM and induce autophagy in response to DNA damaging agents. p53-mediated activation of autophagy is positively linked to cell death since DRAM is also required for p53-dependent induction of apoptosis (Crighton et al. 2006). Cell death is also negatively controlled by p53-dependent induction of autophagy since autophagy inhibition can enhance p53-mediated apoptosis (Crighton et al. 2006; Ghavami et al. 2011).
Sestrins are highly conserved small proteins that accumulate in the cells exposed to stress and play key roles in regulation of aging and cell metabolism (Lee et al. 2010). p53 interacts with target genes SESTRIN1 and SESTRIN2 which subsequently inhibit mammalian target of rapamycin (mTOR) through an indirect mechanisms involving stimulation of adenosine monophosphate-activated protein kinase (AMPK) (Budanov and Karin 2008; Sanli et al. 2012). Inactivation of Sestrin2 by RNA interference reduced the level of autophagy in a panel of p53-positive human cancer cell lines responding to autophagy inducers such as nutrient depletion, rapamycin, lithium and thapsigargin. Thus, Sestrin2 positively regulates autophagy in a p53-dependent fashion (Maiuri et al. 2009).
In contrast to nuclear p53, several lines of evidence indicate that cytoplasmic p53 inhibits autophagy pathways (Fig. 2). Inactivation of p53 by gene knock-out, RNA interference or chemical agents can induce autophagy in multiple model organisms including human, murine and nematode cells (Tasdemir et al. 2008a). Furthermore, p53 has a tonic inhibitory effect on autophagy pathways, which was relieved through Mdm2-dependent proteasomal degradation of p53 induced by different proautophagic stimuli including starvation and mTOR inhibition by rapamycin (Tasdemir et al. 2008a). More interestingly, the autophagy-inhibiting activity of cytoplasmic p53 seems to be distinct from its apoptosis inducing function, or even tumor suppressor activity, as long as p53 is localized in the cytoplasm, because the mutants of p53 still inhibit autophagy while multiple mutants of p53 were unable to interact with Bcl2/Bcl-XL and induce mitochondrial apoptosis (Tasdemir et al. 2008a; Tomita et al. 2006).
The autophagy-inhibitory effect of cytoplasmic p53 is cell cycle-dependent because inhibition of p53 induces autophagy mostly in the G1 and at lower extend in the S phase, but not in the G2/M phase of the cell cycle (Tasdemir et al. 2008b). This may suggest a regulatory role for cytoplasmic p53 on autophagy pathway for maintaining cellular homeostasis to increase cell survival in face of nutrient deprivation. p53 degradation in response to chronic starvation and endoplasmic reticulum stress stimulates autophagy pathways, resulting in high levels of intracellular ATP associated with better cell viability (McCormick et al. 2012; Scherz-Shouval et al. 2010). For instance, induction of autophagy in p53-deficient colorectal cancer cells exposed to prolonged nutrient deprivation maintains high ATP levels and improves the survival rate of these cells (Scherz-Shouval et al. 2010).
Interconnection Between Necrosis and Other Cell Death Forms
Necrosis has long been considered an accidental and uncontrolled, non-programmed form of cell death whereby dramatic changes in crucial cell parameters of metabolism and cell structure take place (Skulachev 2006). The accidental necrosis is in general caused by chemical or physical injury (Vandenabeele et al. 2010). Accumulating evidence shows that necrotic cell death is sometimes also controlled and programmed (Van Herreweghe et al. 2010). This is particularly frequent when a cell for one or other reason (i.e. low ATP-level) is unable to die by apoptosis (Los et al. 2002; Skulachev 2006). Necrosis is typically not associated with activation of caspases (it rather may be a result of their inhibition) (Los et al. 2002); it is characterized by swelling of the endoplasmic reticulum, mitochondria, and cytoplasm, with subsequent rupture of the plasma membrane and lysis of the cells (Schweichel and Merker 1973). Beyond this, necrosis is often associated with the disintegration of the dynamic plasma membrane of the dying cell (Leist and Jaattela 2001). Inflammatory reactions are frequently triggered in response to necrosis (Holmes 1856; Los et al. 2002). The induction of necrosis usually takes place in pathological situations of damage to cells, either in an accidental or an acute manner (Kerr et al. 1972; Wyllie et al. 1980).
Programmed necrotic cell death is the result of the interplay between several signaling cascades. Terms “programmed necrosis” or “necroptosis” collectively refer to necrosis and emphasize a degree of regulation and molecular mechanism of this death process. The main players in the propagation of necrosis are “serine/threonine kinase receptor-interacting protein” 3 (RIPK)3, Ca2+ and mitochondria. RIPK3 interacts with RIPK1 and binds to several enzymes of the carbohydrate and glutamine metabolisms (Holler et al. 2000). Ca2+ controls activation of PLA (polylactic acid), calpains and nitric oxide synthase (NOS), which induce a series of events leading to necrotic cell death. Mitochondria contribute to necrosis by excessive reactive oxygen species (ROS) formation, mitochondrial permeability transition, and ATP depletion due to mitochondrial dysfunction (Denecker et al. 2001).
Mitochondria, Necrosis and Other Cell Death Forms
Mitochondrial control of necrosis occurs mostly through their effect on cellular ATP-level. TNF plays an important role in inducing necrosis, as it also affects mitochondrial ROS production (Skulachev 2006). TNF also induces the activation of PARP-1 (presumably via mitochondrial ROS, causing DNA-damage) leading to ATP depletion and subsequent necrosis (Los et al. 2002). PARP-1 is a nuclear enzyme involved in DNA repair, DNA stability and transcriptional regulation and becomes activated by DNA damage (Leist and Jaattela 2001; Los et al. 2002). Its inhibition in cells exposed to genotoxic factors leads to decrease in the rate of DNA repair and to increase in ROS (Cieslar-Pobuda et al. 2012; Ryabokon et al. 2008). PARP-1 over-activation consumes large amounts of NAD+, resulting in a massive ATP depletion (Sims et al. 1983; Szabo and Dawson 1998). Intracellular ATP levels can affect the form of cell death: a high ATP level leads to apoptosis, whereas a low ATP level leads to necrosis, meaning that an intracellular ATP depletion switches the energy-dependent apoptotic cell death to necrosis (Eguchi et al. 1997; Los et al. 2002). But it is important that a complete ATP depletion leads to a yet another type of cell death, differing from apoptosis and necrosis (“energy-catastrophe”). This shows that both apoptosis and necrosis require some ATP (Skulachev 2006).
Mitoptosis
Apoptotic-like change inside mitochondria (mitoptosis, or death program affecting mitochondria) is a poorly understood process, described mostly by its morphologic features. Induction of mitoptosis and concomitant disruption of ATP supply by mitochondria are often followed by activation of autophagy to assure maintenance of energy supply (Fig. 3, left panels) (Jangamreddy and Los 2012; Mijaljica et al. 2010). Mitoptosis may take various forms: i.e. an inner membrane mitoptosis may occur, in which only the internal matrix and cristae are degraded while the external mitochondrial envelope remains unaltered; or an outer membrane mitoptosis may happen, where only swollen internal cristae are detected as remnants. Furthermore, the fate of the degraded mitochondria may differ under different experimental conditions. The degraded mitochondria may either end-up in autophagosomes [predominantly observed in our lab (Jangamreddy and Los 2012)], or the mitoptotic bodies may be extruded from the cell (Lyamzaev et al. 2008).

Mitoptosis: ultrastructure and some morphology features. Mitoptosis, or death program within mitochondria, is a distinctive process of mitochondria disintegration that may accompany apoptosis or (precedes) autophagy (Jangamreddy and Los 2012). Left mitoptosis as seen by electron microscopy (EM). SKBR3 cells are shown. Left normal mitochondrion, right mitoptotic mitochondria upon treatment with salinomycin. Right mitoptosis as seen by fluorescence microscopy. MCF7 cells that express cytochrome c fused to green-fluorescent protein are shown. Left cells show normal, elongated mitochondria; right cells show swollen, round mitochondria upon treatment with salinomycin (20 μM, 20 h)
During the “outer mitochondrial membrane mitoptosis”, the mitochondria undergo condensation, followed by swelling and fragmenting of cristae, and finally the outer mitochondrial membrane bursts with vesicular remnants of cristae floating in the cytoplasm. Mitochondrial swelling could be detected even at the fluorescence microscopy level, at higher resolutions when, instead of typical elongated “bean-like” shape, they appear round and swollen (Fig. 3, right panels), before they disintegrate. During the “inner mitochondrial membrane mitoptosis”, the outer mitochondrial membrane remains intact, but the cristae deteriorate. This starts with coalescence, followed by rarefaction (loss of density) of the matrix, and finally concludes with degradation of cristae. We have often observed a third, mixed form of mitoptosis, in which mitochondria undergo condensation, following by swelling and vesicular fragmenting of cristae, as in the “outer mitochondrial membrane mitoptosis”. However, instead of disruption of the outer mitochondrial membrane, the mitochondria become engulfed in autophagosomes (Jangamreddy and Los 2012). Thus, the fate of mitochondria inside stressed cells may vary, while the study of mitoptosis in different model systems and the subcellular mechanisms underlying these processes still await conclusion.
The Interconnecting Role of RIPK1 and RIPK3 Between Necrosis and Other Death Pathways
RIPK1 and RIPK3, both from serine/threonine kinases, play an important role in inducing necrosis; they are regulated by caspases and ubiquitination. RIPK have three distinct domains: an N-treminal kinase domain, an intermediary RIP homotypic interaction motif (RHIM)-domain and a C-terminal death domain (Holler et al. 2000; Vandenabeele et al. 2010). TNF or TRAIL stimulation results in induction and formation of a necrosome leading to activation of RIPK3 that interacts with enzymes controlling glycolytic flux, glutaminolysis; this would then result in formation of ROS in the mitochondria (Holler et al. 2000; Vandenabeele et al. 2010). The activity of RIPK1 is specifically associated with necrosis and not with apoptosis. The discovered necrostatin-1 (Nec-1) specifically blocks the kinase activity of RIPK1 (Degterev et al. 2008). In vitro, Nec-1 inhibits TNF-mediated necrosis in L929 cells and FasL-induced necrosis in Jurkat cells that are pretreated with zVAD-fmk caspase inhibitor or deficient in FADD (Degterev et al. 2005). RIPK is a necessary component for the activation of nuclear factor κB by TNF, but its overexpression leads to cell death. Concurrently, RIPK-deficient cell lines are resistant to caspase-independent cell death (Holler et al. 2000).
There are two hypotheses that describe the possible RIPK1-activation mechanisms. The first one is that changes in metabolism by PARP-1 activation result in ATP depletion as well as intracellular pH reduction due to lactate production under anaerobic conditions during ischemia leading to RIPK1 activation and subsequent necrotic cell death (Van Herreweghe et al. 2010). The second hypothesis describes the RIPK1 activation via the activation of a mechanism that can upregulate metabolism, e.g., by autocrine TNF production as a response to cellular stress. TNF is able to activate glycolysis (Matthews 1983). Thus, autocrine TNF may activate RIPK1. This mechanism has been shown in cellular stress induced by zVAD-fmk resulting in TNF-mediated necrosis (Hitomi et al. 2008). Nonetheless, recently it was demonstrated that RIPK3 is essential for TNF-induced necrosis (Cho et al. 2011).
RIPK1 associates with RIPK3 to form the already mentioned necrosome. Its formation needs RIPK1 activity, and is stabilized through homotypic RHIM associations between RIPK1 and RIPK3 (Van Herreweghe et al. 2010). However, under necrotic conditions, RIPK3 also binds to other metabolic enzymes, e.g., the cytosolic glycogen phosphorylase (PYGL), the cytosolic glutamate-ammonia ligase (GLUL) and the glutaminolysis initiating enzyme GLUD1, which is positively regulating RIPK3 and its enzymatic activity (Van Herreweghe et al. 2010; Zhang et al. 2009). These interactions lead to glutamine production and the regulation of glycogenolysis.
It has been proposed that RIPK1 and RIPK3 are responsible for an increased carbohydrate and glutamine metabolism of the cell, leading to a higher ROS formation and subsequent necrotic cell death (Los et al. 2009a; Van Herreweghe et al. 2010). Activity of caspase-8 blocks the necrotic cell death probably because it cleaves RIPK1 and RIPK3 (Vandenabeele et al. 2010), and downstream, through caspase-3 to -7 activation and subsequent PARP-1 cleavage (Los et al. 2002). This indicates the importance of RIPK1, -3 and PARP-1 for the induction of necrosis. Concomitantly, the inhibition of caspases leads to enhanced and accelerated ROS formation (Denecker et al. 2001; Los et al. 2001, 2002).
The release of Ca2+ to the cytosol from the endoplasmic reticulum or from the extracellular compartment may accumulate in the mitochondrial matrix, resulting in the opening of permeability transition pores in the inner mitochondrial membrane. This opening results in permeability of the membrane to low molecular mass substances, normally responsible for osmotic balance between the matrix and the intramembrane space, finally leading to swelling and disrupting of mitochondria (Skulachev 2006).
Increased intracellular Ca2+ concentrations lead to activation of calpains, which are intracellular, non-lysosomal cysteine proteases that are ubiquitously and constitutively expressed in mammalian cells (Kar et al. 2009; Stroh et al. 2002; Van Herreweghe et al. 2010). Activated calpains, for example, cleave the anti-apoptotic Bcl-XL and Bax, as well as caspase-7, -8 and -9. It is however not clear if this cleavage inhibits or stimulates caspase activity (Chua et al. 2000; Lee et al. 2006; Toyota et al. 2003; Van Herreweghe et al. 2010).
Calpains play an important role in inducing ROS-dependent, necrotic cell death because they cleave the mitochondrial Na+/Ca2+ exchanger resulting in a higher Ca2+ concentration in the mitochondria, which leads to constant ROS-production in mitochondria. Furthermore, calpains contribute to the necrotic cell death of neurons in Caenorhabditis elegans (Kar et al. 2009; Van Herreweghe et al. 2010). Ca2+ may contribute to necrosis in yet another way: it may stimulate NOS activity, and thus NO production. NO is a strong inhibitor of complex IV of the mitochondrial respiratory chain, subsequently leading to a stronger ROS production at complex III (leakage from the respiratory chain) (Van Herreweghe et al. 2010).
Other Interconnections Between Apoptotic, Necrotic and Autophagic Pathways
In previous paragraphs, we specifically focused on interconnections between apoptosis and autophagic pathways. Here, interconnection between all three-cell death mechanisms will be briefly discussed. For example, the proteins of the extrinsic death receptor pathways can also influence autophagy. The death domain of FADD in normal epithelial cells induces cell death with high levels of autophagy. The autophagy response involving FADD is much more pronounced when apoptosis is blocked, i.e., by caspase inhibition. This suggests that autophagy and apoptosis are induced simultaneously by the FADD death domain, at least in normal epithelial cells (Thorburn 2008), but since apoptotic cell death progresses faster, this is a dominantly observed form of cell death. The full onset of autophagy (or necrosis) emerges only when caspase inhibitors are applied. The usage of caspase inhibitors clearly visualizes the existence of those alternative pathways, which function as back-up if apoptosis is blocked. The caspase inhibitor zVAD-fmk may modulate all three main cell death pathways. zVAD-fmk has been mostly used for blocking apoptotic cell death. Inhibition of caspase activity by zVAD-fmk may not only shift the balance in favor of autophagy, but in some cells, such as in L929 rodent fibrosarcoma cell line it also switches apoptosis towards necrosis (Los et al. 2002). The pro-necrotic action of the caspase inhibitor under these conditions is, at least in part, the result of lack of PARP-1 cleavage/inhibition by caspase-3/caspase-7. PARP-1, that is normally otherwise cleaved by caspases, becomes hyperactivated by apoptotic DNA-damage, and consumes enormous amounts of ATP (poly-ADP-ribosylaion of DNA and proteins of DNA-repair machinery), thereby exhausting the cellular ATP-pool and leading to necrosis (Los et al. 2002).
Another molecule associated with death receptor, RIPK1 may connect all the three major death pathways: the role of RIPKs in necrosis has already been discussed in earlier paragraphs. Several studies have demonstrated that RIPK1 plays an important role in initiation of caspase-independent death. The usage of caspase inhibitors has also revealed a positive role of CypD and negative roles for catalase and caspase-8 in caspase-independent cell death pathways. Necrotic and autophagic cell death pathways are interconnected by a signaling cascade, which involves RIPK1, and is negatively regulated by caspase-8. Necrotic cell death may exhibit a rapid onset, involving ROS production, cytoplasmic ATP reduction and other cellular events. On the other hand, autophagic cell death first starts as a survival attempt by blocking necrosis and a cleanup of oxidatively damaged mitochondria (Vandenabeele et al. 2006) (Fig. 4).

Modulation of cell death mode by caspase activity, and consequences of caspase inhibition. The broad-spectrum caspase inhibitor zVAD-fmk modulates the three major types of cell death in different ways. zVAD-fmk blocks apoptotic cell death while it sensitizes cells to necrotic cell death, and autophagy, presumably by shifting the balance from apoptosis towards necrosis/autophagy. Autophagy and necrotic cell death are interconnected and may partially consist of common underlying molecular pathways involving RIPKs and negative regulation by caspase-8. Furthermore, an activatory role for cyclophilin D and an inhibitory role for catalase in caspase-independent cell death cascades have been demonstrated (see text for further details)
As discussed at the beginning, the caspase-dependent apoptosis triggered by death receptor ligation involves the assembly of death-inducing signaling complex (DISC) (Fig. 1). This process results in the induction of apoptosis through caspase-8-dependent cleavage/activation of effector caspases or through Bid cleavage and subsequent activation of the mitochondrial death pathway. Activity of RIPK1 and RIPK3, that modulate necrosis and autophagy, is limited by their cleavage by caspase-8, which results in limitation of autophagy and necrosis. Only in the absence of robust caspase-8 activity a stable complex between RIPK1 and RIPK3 is formed, promoting programmed necrosis and autophagy (instead of apoptosis) (Lu et al. 2011). Death receptor signaling via RIPK1 and RIPK3 has also been studied in a model involving the assembly of DISC in isolated membranes/pre-autophagosomal structures (PAS). Such PAS–DISC promotes caspase-8 activation and subsequent caspase-dependent apoptosis in a similar fashion as for the cell membrane-bound death receptor-DISC. Interestingly, the induction of necrosis by death receptor/DISC-complex is not affected by blockade of autophagy; however, necrosis induced by PAS/DISC can be blocked by autophagic inhibition (Walsh and Edinger 2010) (Fig. 5).

Reciprocal regulation of apoptosis and programmed necrosis/autophagy. Upon ligation of death receptor the DISC is assembled, thus leading to caspase-8 activation. This results in the caspase-8-dependent cleavage of effector caspases and activation of apoptosis with reciprocal proteolytic inhibition of RIPK1 and RIPK3. If however the DISC is formed but beside pro-caspase-8 c-FLIP is incorporated, caspase-8 activation may not occur, but the DISC will still serve as a scaffold for RIPK1 and RIPK3 complex assembly, their cross-phosphorylation and activation of necrosis and/or autophagy. Bid cleavage by caspase-8 (not shown here) will activate the mitochondrial apoptotic pathway. The threshold for apoptotic cell death is set by anti-apoptotic factors such as c-FLIP and Bcl22 (not shown here). RIPK1 and RIPK3 activity is limited by caspase-8-dependent cleavage, limiting induction of autophagy and necrosis
NAD+ and ATP levels would decrease due to metabolic stress (like for example, excessive fasting, imbalanced diet, post-exercise acidosis, or other interferences with cellular metabolism) and therapeutic stress. This could result in an increase in ROS and Ca2+ (Amaravadi and Thompson 2007; Castro et al. 2006). Cells which cannot efficiently cope with such stress undergo necrosis. The activation of AMPK improves survival of cells under low ATP conditions. mTOR is inhibited by AMPK-dependant phosphorylation; thus AMPK-activity blocks autophagy. However, AMPK may promote p53 activation, which in turn may lead to autophagy or apoptosis by activating Bax and Bak and subsequently causing cytoplasmic release of cytochrome c and activation of caspases. Stress-induced autophagy may lead either to cell death or to cell survival unlike apoptosis or necrosis that are always lethal (Amaravadi and Thompson 2007) (Fig. 6).

Cellular energy status and the interplay between necrosis, apoptosis, and autophagy. ATP depletion due to metabolic stress or noxious stimuli may cause increase of intracellular calcium (inefficient Ca-pumps), and ROS (increased mitochondrial oxidation). Cells that do not adapt to these changes undergo necrotic cell death. The activation of protective stress regulators, such as AMPK, allows cells to acutely survive these changes. AMPK-dependent phosphorylation results in the inhibition of mTOR, and thus activation of autophagy. AMPK-dependent phosphorylation also activates p53, which can lead to autophagy or apoptosis, through the activation of Bax and Bak, the mitochondrial release of cytochrome c, and caspase activation. Unlike apoptosis or necrosis, stress-induced autophagy may promote cell survival or cell death
Death Pathways and Therapy Implications in Cancer
Autophagy: Its “Janus-faced” Effect in Cancer Treatment
Autophagy is usually initiated as a pro-survival response although the net outcome of it is far from certain. Some of the cancer cells die when autophagic genes are inhibited whereas some of them die when there is an induction of autophagic process. So it mostly depends on the make-up of the tumor cell that decides the fate of autophagy. Below, some examples on both manifestations of autophagy will be presented.
Autophagy may be induced in cancer cells as an adaptation mechanism offering resistance against chemo- and radio-therapy. Inhibiting autophagy and thereby sensitizing tumor cells to apoptotic cell demise is a new therapeutic strategy targeting apoptosis-resistant tumors. Chloroquine and hydroxyl-chloroquine increase pH and thereby inhibits acidification of lysosomes thus inhibiting autophagy. Synergistic use of chloroquine and an alkylating agent showed a remarkable decrease in tumor growth in mice (Amaravadi et al. 2007). Apart from that siRNA targeted removal of “Autophagy related gene” (ATG) 5 enhanced p53-mediated cell death (Amaravadi et al. 2007). These experimental findings certainly show that inhibition of autophagy can lead to apoptosis in cancer cells. These drugs are used in combination with other drugs such as bortezomib, bevacizumab, placlitaxel, carboplatin and oxalipatin to treat a number of cancers. Chloroquine and hydroxychloroquine drugs, either alone or in combination with other chemotherapeutic drugs, are presently in clinical trails for treatment of various cancers. For details please see Table 1.
2-Deoxyglucose, a synthetic glucose analog, is a potent inducer of autophagy in human glioma cells and in prostate cancer cells (Ben Sahra et al. 2010; Wu et al. 2009). Induction of autophagy by 2-deoxyglucose is mediated by activation of elongation factor kinase-2 (eEF2-kinase). Knock-down of eEF2-kinase using siRNA leads to the inhibition of autophagy by 2-deoxyglucose (Wu et al. 2009). However, inhibition of autophagy is followed by rapid decrease in cellular ATP levels and increase in cytotoxic effects of 2-deoxyglucose by apoptosis. These results provided evidence indicating that silencing of eEF2-kinase can shift the cancer cells from the survival autophagic pathway to cell death. Targeting eEF2-kinase in cancer cells can be a good therapeutic target. In another study, reported recently, a similar strategy was adopted (Ben Sahra et al. 2010). The combination of metformin and 2-deoxyglucose has caused apoptosis cell death in prostate cancer cells. At a cellular level, combination of these two drugs resulted in p53-dependent apoptosis via energy sensor AMP pathway. 2-Deoxyglucose caused autophagy in prostate cancer cells but metformin inhibited autophagy by downregulating the expression of beclin-1 and triggering the shift from cell survival autophagy to cell death pathway (Ben Sahra et al. 2010). These results show that targeting the cell survival autophagic pathway, using small molecule inhibitors, can be a novel therapeutic strategy in treatment of cancer cells. In radiation resistant cells, the formation of autophagosomes made the cancer cells resistant to radiation therapy (Rikiishi 2012). Knock-down of autophagy related genes such as Beclin-1, ATG3, ATG4 and ATG5, using siRNAs, inhibited the formation of autophagosomes in cancer cells (Rikiishi 2012). Upon exposure to radiation, these cancer cells underwent p53-dependent apoptosis indicating that autophagy can act as a survival mechanism and deregulation of autophagic genes can lead to sensitization of cancer cells to conventional therapies. Phenylethynesulfonamide, a small molecule that targets heat shock protein 70 (Hsp-70), also inhibited autophagy and lysosomal function thereby causing the cancer cells to undergo cell death (Leu et al. 2009). The above findings show that phenylethynesulfonamide can be a potential therapeutic agent in targeting cancers where Hsp-70 is highly expressed.
Many tumor cells have reduced autophagic capacity compared to their normal counterparts. Beclin-1, one of the proteins essential for autophagy, is mono-allelically deleted in 40–75 % of sporadic human breast and ovarian cancers (Aita et al. 1999). Transfection of beclin-1 gene in MCF-7 cells containing low levels of beclin-1 showed that over expression of Beclin-1 inhibits tumor growth and tumor formation. Another study examined the role of Beclin-1 in colon cancer cells indicating that levels of Beclin-1 in colon cancer cells are variable. Beclin-1 transfection into colon cancer cells that lost beclin-1 or expressed it at low levels inhibited the growth of the cancer cells, indicating that beclin-1 expression can be used as a therapeutic strategy (Aita et al. 1999; Koneri et al. 2007). Conventional therapies such as radiation therapy, chemotherapy and targeted therapies are not suitable for tumors that have defects in apoptosis. Hence, activation of autophagy, especially in these types of tumor cells, may prove an attractive therapeutic strategy. For example, pancreatic cancer is an aggressive malignant disease often resistant to standard chemotherapeutic agents and radiation therapy. Growth of pancreatic cancer cells can be inhibited via autophagy by targeting protein kinase C-delta (PKCδ) (Akar et al. 2007). In addition, siRNA mediated knock-down of PKCδ-induced growth inhibition through autophagy without inducing apoptosis in pancreatic cancer cells.
Bcl2 proto-oncogene is expressed in ~80 % of breast cancers (Krajewski et al. 1999). Overexpression of Bcl2 makes breast cancer cells potentially resistant to radio-, chemo- and hormone-based therapy. Silencing of Bcl2 via siRNA potentiates the propensity of MCF-7 breast cancer cells to undergo autophagy (Akar et al. 2008). However, knock-down of the autophagy genes ATG5 and beclin-1 inhibits autophagy in Bcl2 knock-down cells, indicating that targeted down-regulation of Bcl2 induces autophagy. Furthermore, the results showed that doxorubicin induces autophagy at lower concentrations while at higher concentrations it induces apoptosis. Low doses of doxorubicin in combination with knock-down of Bcl2 increased autophagy in tumor cells and reduced the tumor growth (Akar et al. 2008). These results provide evidence indicating that knock-down of Bcl2 is a potential therapeutic strategy.
Renal cell carcinoma is particularly refractory to the standard therapies (Turcotte et al. 2008). The von Hippel-Lindau (VHL) tumor suppressor gene is inactivated in 75 % of the renal cell carcinomas (Turcotte et al. 2008). STF-62247 is a small compound that specifically targets the VHL deficient cancer cells, thereby inhibiting the growth of the tumor cells by inducing extensive autophagy (Turcotte et al. 2008). VHL deficient cells exhibited higher acidification of autolysosomes in response to STF 62247 and hence underwent autophagy. Knock-down of autophagy genes such as ATG5, ATG7 or ATG9 prevented the autophagy induction by STF 62247, indicating that STF 62247 induces autophagy in VHL deficient cells (Turcotte et al. 2008). These findings show that induction of autophagy using small molecules is a viable therapeutic strategy.
Potentiation of Apoptosis in Cancer Therapy
The role of apoptosis in cancer therapy has been extensively studied (Ghavami et al. 2009; Los 2009; Los et al. 2003). However, finding suitable and reliable drugs for treatment of various cancers is still a challenge for the medical community. Now it seems logical to focus on some new findings on cancer therapy and refer to drugs that target p53 and Bcl2 family of proteins that are known to affect autophagy and necrosis.
p53 is mutated and its tumor suppression function is lost in well over 50 % of cancers (Hanahan and Weinberg 2000). Thus, the restoration of p53 function is an attractive therapeutic strategy. Introduction of wild-type p53 using a replication-deficient adenoviral vector inhibited human lung cancer cell growth both in vitro and in vivo (Fujiwara et al. 1993; Vincent and Los 2011). This adenoviral-p53 under the brand name Gendicine or Advexin is in clinical trails in China and the United States. These drugs are well tolerated by patients with head and neck and lung cancer (Wang and Sun 2010); they are also successful as single agents or in combination with other chemotherapeutic drugs and radiation therapy.
A small molecule called RITA (reactivation of p53 and induction of tumor cell apoptosis) induces apoptosis in various tumor cells in a p53-dependent manner. Biochemically, RITA disrupts the interaction of p53 with its inhibitor MDM2 thereby inducing massive apoptosis both in vitro and in vivo (Ande et al. 2009). RITA-activated p53 represses the transcription of a number of anti-apoptotic proteins such as Bcl2, MAP4, Mcl-1, Survivin, and blocks Akt pathway at several levels (Grinkevich et al. 2009). Inhibition of these anti-apoptotic proteins by RITA-activated p53 induced massive apoptosis in tested cancers. These results certainly refer to potentials of RITA, as a therapeutic drug, both individually or as a combined therapy. One has to keep in mind that Akt does not always fulfill pro-survival functions. As recent reports have shown, Akt may become pro-apoptotic if it enters the nucleus (Chen et al. 2011; Los et al. 2009b; Maddika et al. 2007, 2008, 2009).
Another class of small molecules, nutlins, strongly bind to MDM2 thereby inhibiting its interaction with p53 and elevating the levels of active p53. Nutlin-3 very efficiently induces apoptosis in various tumors such as breast cancer, lymphocytic leukemia, as well as retinoblastoma and osteosarcoma (Secchiero et al. 2007; Sonnemann et al. 2011; Zhang et al. 2011). In combination with chemotherapy, nutlin-3 is very effective against lymphocytic leukemia, lung cancer, neuroblastoma and prostrate cancer (Schmitt et al. 2004). In addition, according to a more recent study (Hori et al. 2010), nutlin-3 can enhance the TRAIL-induced apoptosis through upregulation of DR5-receptor both at mRNA and protein level in human colon cancer cells and sarcoma cells (Hori et al. 2010). These results show that nutlin-3 and TRAIL synergistically elevated the levels of apoptosis (Hori et al. 2010).
The anti-apoptotic Bcl2 family of proteins, such as Bcl2, Bcl-XL, Mcl-1, Bcl-B etc., is often over-expressed in various cancers and confers resistance to anti-neoplastic drugs. The role of such proteins in regulating autophagy and necrosis was highlighted in previous paragraphs. Hence, targeting Bcl2 anti-apoptotic proteins is one of the prominent strategies to combat cancer at least in a setting where anti-apoptotic proteins are over expressed.
Gossypol is a phenolic compound found in the roots, stem and seed of the cotton plant (Kang and Reynolds 2009). Natural gossypol is a racemic mixture; however only one of its forms, the levo-gossyposl (AT101) is very effective in inhibiting anti-apoptotic proteins such as Bcl2, Bcl-XL and Mcl-1. Chronic lymphocytic leukemia is resistant to various chemotherapeutic drugs and this is due to the over expression of Bcl2 family of anti-apoptotic proteins. Administration of AT101 to the CLL cells induced apoptosis by down regulating Mcl-1 and overcame the resistance that is developed when using the other drugs (Balakrishnan et al. 2009). Another report provides evidence that AT101 markedly enhances the anti-tumor effects of chemotherapeutic agents both in vitro and in vivo (Paoluzzi et al. 2008). Synergetic treatment of AT101 with etoposide, doxorubicin and carfilzomib in mantle cell lymphoma effectively induced apoptosis and increased the efficacy of the above drugs (Paoluzzi et al. 2008). In drug-resistant severe immuno-deficient mice models of B cell lymphoma, addition of AT101 in combination with cyclophosphamide or rituximab increased efficacy of these drugs (Paoluzzi et al. 2008). These results certainly provide convincing evidence that AT101 can be used as a chemotherapeutic drug and it is presently in phase I/II stage of clinical trails.
Oblimersen is an 18-mer phosphorothiolate anti-sense oligonucleotide designed to target Bcl2 mRNA. Oblimersen effectively inhibited mRNA of Bcl2 by binding to the first six codons of Bcl2 mRNA (Moreira et al. 2006). Oblimersen induced apoptosis in tumor cells by inhibiting the Bcl2 and thereby up-regulated the expression of proapoptotic Bax (Emi et al. 2005). Apart from that, this drug also released Smac/DIABLO from mitochondria, which antagonize the inhibitors of apoptosis proteins released from mitochondria (Emi et al. 2005). Oblimersen is combined with docetaxel, adriamycin and cyclophosphamide in treatment of breast cancer and these combinations are presently in clinical trails (Rom et al. 2008). In addition, oblimersen in combination with other drugs is in clinical trails for treatment of various cancers such as multiple myeloma, melanoma, small lung cancer and non-Hodgkin’s lymphoma (Kang and Reynolds 2009).
GX15-070 (Obatoclax) is another novel pan-Bcl2 inhibitor that induces apoptosis in acute myeloid leukemia (AML) cell lines and also in primary AML cells (Konopleva et al. 2008). GX15-070 promoted the release of cytochrome c from mitochondria and released pro-apoptotic proteins such as Bak and Bim. These results show that GX15-070 can be used as a drug in treatment of AML. In addition, preclinical studies with GX15-070 on multiple myeloma cells show that GX15-070 is very effective in treatment of multiple myeloma (Trudel et al. 2007). GX15-070 inhibited binding of Bak to Mcl-1, elevating the levels of Bim. It also promoted the release of cytochrome c from mitochondria and activated caspase-3 in various human myeloma cells (Trudel et al. 2007). In some of the cancer cells Mcl-I conferred resistance to apoptotic cell death induced by either ABT-737 (A small molecule that targets Bcl2) or bortezomib (Proteosome inhibitor). Synergetic use of GX15-070 with either ABT 737 or bortezomib overcomes the apoptotic resistance in cancer cells (Nguyen et al. 2007). These experimental findings show that GX15-070 can be used in combination with other drugs and can overcome apoptosis resistance.
Closing Remarks
Cancer is a frequently occurring genetic disease (Wiechec 2011; Wiechec and Hansen 2009; Wiechec et al. 2011). Beside environmental factors, viral infections and life-style are responsible factors for its increasing frequency (Alavian et al. 2011; Gurevich-Panigrahi et al. 2009). Natural products are a frequent inspiration for the development of new anti-cancer drugs (Ghavami et al. 2010; Gokay et al. 2010; Panigrahi et al. 2012). As outlined above, much attention in recent years has been paid to experimental drugs that modulate autophagy or apoptosis. While several promising experimental anti-cancer or anti-degenerative drugs that could modulate both pathways have been developed, one has to exercise some caution with respect to autophagy modulating drugs (Alavian et al. 2011). Modulation of autophagy alone will not possibly show much clinical effect, due to its highly interconnected nature and the fact that low-level autophagy would generally promote cell survival, and that autophagy process will kill the targeted cell only when it is excessive. Therefore, as indicated in previous paragraphs, autophagy-modulating anti-cancer or anti-degenerative drugs, when clinically implemented, would most likely be used in conjunction with other therapeutics. Accordingly, such drugs would strengthen the desired effect of autophagy for pro-cell death in cancer or pro-survival in degenerative diseases when they work together. As such, despite all the limitations indicated, the authors are firmly convinced that autophagy-modulating drugs will become a clinical reality in near future.
Abbreviations
- AML:
-
Acute myeloid leukemia
- AMPK:
-
5-Prime-AMP-activated protein kinase
- Apaf1:
-
Apoptotic protease activating factor-1
- ATG:
-
Autophagy related gene
- BH:
-
Bcl2-homology
- DIABLO:
-
Direct IAP-bind protein with low pI
- DISC:
-
Death-inducing signaling complex
- DRAM:
-
Damage-regulated autophagy modulator
- eEF2-kinase:
-
Elongation factor kinase-2
- Hsp-70:
-
Heat shock protein 70
- Mcl-1:
-
Myeloid cell leukemia-1
- Mdm2:
-
Murine double minute 2
- mTOR:
-
Mammalian target of rapamycin
- Nec-1:
-
Necrostatin-1
- NOS:
-
Nitric oxide synthase
- PARP-1:
-
Poly(ADP-ribose)polymerase-1
- PCD:
-
Programmed cell death
- PKCδ:
-
Protein kinase C-δ
- PUMA:
-
p53 upregulated modulator of apoptosis
- RHIM:
-
RIP homotypic interaction motif
- RIPK:
-
Serine/threonine kinase receptor-interacting protein
- ROS:
-
Reactive oxygen species
- Smac:
-
Second mitochondria-derived activator of caspase
- TNF:
-
Tumor necrosis factor α
- TRAIL:
-
TNF related apoptosis inducing ligand
- VHL:
-
von Hippel-Lindau
References
Aita VM, Liang XH, Murty VV et al (1999) Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 59:59–65
Akar U, Ozpolat B, Mehta K et al (2007) Tissue transglutaminase inhibits autophagy in pancreatic cancer cells. Mol Cancer Res 5:241–249
Akar U, Chaves-Reyez A, Barria M et al (2008) Silencing of Bcl-2 expression by small interfering RNA induces autophagic cell death in MCF-7 breast cancer cells. Autophagy 4:669–679
Alavian SM, Ande SR, Coombs KM et al (2011) Virus-triggered autophagy in viral hepatitis—possible novel strategies for drug development. J Viral Hepat 18:821–830
Amaravadi RK, Thompson CB (2007) The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res 13:7271–7279
Amaravadi RK, Yu D, Lum JJ et al (2007) Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 117:326–336
Ande SR, Chen J, Maddika S (2009) The ubiquitin pathway: an emerging drug target in cancer therapy. Eur J Pharmacol 625:199–205
Balakrishnan K, Burger JA, Wierda WG et al (2009) AT-101 induces apoptosis in CLL B cells and overcomes stromal cell-mediated Mcl-1 induction and drug resistance. Blood 113:149–153
Barak Y, Juven T, Haffner R et al (1993) mdm2 expression is induced by wild type p53 activity. EMBO J 12:461–468
Barczyk K, Kreuter M, Pryjma J et al (2005) Serum cytochrome c indicates in vivo apoptosis and can serve as a prognostic marker during cancer therapy. J Int Cancer 116:167–173
Ben Sahra I, Laurent K, Giuliano S et al (2010) Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res 71:2465–2475
Budanov AV, Karin M (2008) p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134:451–460
Cardinal J, Pan P, Dhupar R et al (2009) Cisplatin prevents high mobility group box 1 release and is protective in a murine model of hepatic ischemia/reperfusion injury. Hepatology 50:565–574
Castro J, Ruminot I, Porras OH et al (2006) ATP steal between cation pumps: a mechanism linking Na+ influx to the onset of necrotic Ca2+ overload. Cell Death Differ 13:1675–1685
Chang C, Simmons DT, Martin MA et al (1979) Identification and partial characterization of new antigens from simian virus 40-transformed mouse cells. J Virol 31:463–471
Chen X, Ko LJ, Jayaraman L et al (1996) p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev 10:2438–2451
Chen K, Luo Z, Tang J et al (2011) A critical role of heat shock cognate protein 70 in Apoptin-induced phosphorylation of Akt. Biochem Biophys Res Commun 409:200–204
Cherlonneix L (2008) L’équivocité vive: Une nouvelle représentation du vivant [The vivid equivocity. A new representation of living]. L’Harmattan, Paris. ISBN: 978-2-296-05340-3
Chipuk JE, Kuwana T, Bouchier-Hayes L et al (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303:1010–1014
Chipuk JE, Bouchier-Hayes L, Kuwana T et al (2005) PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 309:1732–1735
Cho Y, Challa S, Chan FK (2011) A RNA interference screen identifies RIP3 as an essential inducer of TNF-induced programmed necrosis. Adv Exp Med Biol 691:589–593
Chua BT, Guo K, Li P (2000) Direct cleavage by the calcium-activated protease calpain can lead to inactivation of caspases. J Biol Chem 275:5131–5135
Cieslar-Pobuda A, Saenko Y, Rzeszowska-Wolny J (2012) PARP-1 inhibition induces a late increase in the level of reactive oxygen species in cells after ionizing radiation. Mutat Res 732:9–15
Clarke AR, Purdie CA, Harrison DJ et al (1993) Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature 362:849–852
Crighton D, Wilkinson S, O’Prey J et al (2006) DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126:121–134
Curtin JF, Cotter TG (2003) Apoptosis: historical perspectives. Essays Biochem 39:1–10
Degterev A, Huang Z, Boyce M et al (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1:112–119
Degterev A, Hitomi J, Germscheid M et al (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4:313–321
DeLeo AB, Jay G, Appella E et al (1979) Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc Natl Acad Sci USA 76:2420–2424
Denecker G, Vercammen D, Steemans M et al (2001) Death receptor-induced apoptotic and necrotic cell death: differential role of caspases and mitochondria. Cell Death Differ 8:829–840
Eguchi Y, Shimizu S, Tsujimoto Y (1997) Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res 57:1835–1840
Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35:495–516
Emi M, Kim R, Tanabe K et al (2005) Targeted therapy against Bcl-2-related proteins in breast cancer cells. Breast Cancer Res 7:R940–R952
Esposti DD, Domart MC, Sebagh M et al (2010) Autophagy is induced by ischemic preconditioning in human livers formerly treated by chemotherapy to limit necrosis. Autophagy 6:172–174
Feng Z, Zhang H, Levine AJ et al (2005) The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci USA 102:8204–8209
Fujiwara T, Grimm EA, Mukhopadhyay T et al (1993) A retroviral wild-type p53 expression vector penetrates human lung cancer spheroids and inhibits growth by inducing apoptosis. Cancer Res 53:4129–4133
Ghavami S, Hashemi M, Ande SR et al (2009) Apoptosis and cancer: mutations within caspase genes. J Med Genet 46:497–510
Ghavami S, Eshragi M, Ande SR et al (2010) S100A8/A9 induces autophagy and apoptosis via ROS-mediated cross-talk between mitochondria and lysosomes that involves BNIP3. Cell Res 20:314–331
Ghavami S, Mutawe MM, Sharma P et al (2011) Mevalonate cascade regulation of airway mesenchymal cell autophagy and apoptosis: a dual role for p53. PLoS One 6:e16523
Gokay O, Kuhner D, Los M et al (2010) An efficient approach for the isolation, identification and evaluation of antimicrobial plant components on an analytical scale, demonstrated by the example of Radix imperatoriae. Anal Bioanal Chem 398:2039–2047
Grinkevich VV, Nikulenkov F, Shi Y et al (2009) Ablation of key oncogenic pathways by RITA-reactivated p53 is required for efficient apoptosis. Cancer Cell 15:441–453
Gurevich-Panigrahi T, Panigrahi S, Wiechec E et al (2009) Obesity: pathophysiology and clinical management. Curr Med Chem 16:506–521
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Hitomi J, Christofferson DE, Ng A et al (2008) Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135:1311–1323
Holler N, Zaru R, Micheau O et al (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1:489–495
Holmes T (1856) St. George’s Hospital: case of necrosis of the ulna following diffuse inflammation after injury (and probably simple fracture): removal of a sequestrum involving the whole shaft of the bone for seven inches of its length. Assoc Med J 4:1029
Hori T, Kondo T, Kanamori M et al (2010) Nutlin-3 enhances tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis through up-regulation of death receptor 5 (DR5) in human sarcoma HOS cells and human colon cancer HCT116 cells. Cancer Lett 287:98–108
Jangamreddy JR, Los MJ (2012) Mitoptosis, a novel mitochondrial death mechanism leading predominantly to activation of autophagy. Hepat Mon 12:e6159
Jeffers JR, Parganas E, Lee Y et al (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4:321–328
Kang MH, Reynolds CP (2009) Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res 15:1126–1132
Kar P, Chakraborti T, Samanta K et al (2009) mu-Calpain mediated cleavage of the Na+/Ca2+ exchanger in isolated mitochondria under A23187 induced Ca2+ stimulation. Arch Biochem Biophys 482:66–76
Kastan MB, Onyekwere O, Sidransky D et al (1991) Participation of p53 protein in the cellular response to DNA damage. Cancer Res 51:6304–6311
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26:239–257
Kinnally KW, Peixoto PM, Ryu SY et al (2011) Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim Biophys Acta 1813:616–622
Koneri K, Goi T, Hirono Y et al (2007) Beclin 1 gene inhibits tumor growth in colon cancer cell lines. Anticancer Res 27:1453–1457
Konopleva M, Watt J, Contractor R et al (2008) Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax). Cancer Res 68:3413–3420
Krajewski S, Krajewska M, Turner BC et al (1999) Prognostic significance of apoptosis regulators in breast cancer. Endocr Relat Cancer 6:29–40
Kress M, May E, Cassingena R et al (1979) Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol 31:472–483
Lane DP, Crawford LV (1979) T antigen is bound to a host protein in SV40-transformed cells. Nature 278:261–263
Lee WK, Abouhamed M, Thevenod F (2006) Caspase-dependent and -independent pathways for cadmium-induced apoptosis in cultured kidney proximal tubule cells. Am J Physiol Renal Physiol 291:F823–F832
Lee JH, Budanov AV, Park EJ et al (2010) Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 327:1223–1228
Leist M, Jaattela M (2001) Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol 2:589–598
Leu JI, Dumont P, Hafey M et al (2004) Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol 6:443–450
Leu JI, Pimkina J, Frank A et al (2009) A small molecule inhibitor of inducible heat shock protein 70. Mol Cell 36:15–27
Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88:323–331
Los M (2009) New, exciting developments in experimental therapies in the early 21st century. Eur J Pharmacol 625:1–5
Los M, Wesselborg S, Schulze-Osthoff K (1999) The role of caspases in development, immunity, and apoptotic signal transduction: lessons from knockout mice. Immunity 10:629–639
Los M, Stroh C, Janicke RU et al (2001) Caspases: more than just killers? Trends Immunol 22:31–34
Los M, Mozoluk M, Ferrari D et al (2002) Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell 13:978–988
Los M, Burek CJ, Stroh C et al (2003) Anticancer drugs of tomorrow: apoptotic pathways as targets for drug design. Drug Discov Today 8:67–77
Los M, Maddika S, Erb B et al (2009a) Switching Akt: from survival signaling to deadly response. BioEssays 31:492–495
Los M, Panigrahi S, Rashedi I et al (2009b) Apoptin, a tumor-selective killer. Biochim Biophys Acta 1793:1335–1342
Lowe SW, Ruley HE (1993) Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev 7:535–545
Lowe SW, Schmitt EM, Smith SW et al (1993) p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 362:847–849
Lu X, Lane DP (1993) Differential induction of transcriptionally active p53 following UV or ionizing radiation: defects in chromosome instability syndromes? Cell 75:765–778
Lu JV, Weist BM, van Raam BJ et al (2011) Complementary roles of Fas-associated death domain (FADD) and receptor interacting protein kinase-3 (RIPK3) in T-cell homeostasis and antiviral immunity. Proc Natl Acad Sci USA 108:15312–15317
Lyamzaev KG, Nepryakhina OK, Saprunova VB et al (2008) Novel mechanism of elimination of malfunctioning mitochondria (mitoptosis): formation of mitoptotic bodies and extrusion of mitochondrial material from the cell. Biochim Biophys Acta 1777:817–825
Maddika S, Ande SR, Panigrahi S et al (2007) Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist Updat 10:13–29
Maddika S, Ande SR, Wiechec E et al (2008) Akt-mediated phosphorylation of CDK2 regulates its dual role in cell cycle progression and apoptosis. J Cell Sci 121:979–988
Maddika S, Panigrahi S, Wiechec E et al (2009) Unscheduled Akt-triggered activation of cyclin-dependent kinase 2 as a key effector mechanism of apoptin’s anticancer toxicity. Mol Cell Biol 29:1235–1248
Maiuri MC, Malik SA, Morselli E et al (2009) Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle 8:1571–1576
Martinet W, Schrijvers DM, Herman AG et al (2006) z-VAD-fmk-induced non-apoptotic cell death of macrophages: possibilities and limitations for atherosclerotic plaque stabilization. Autophagy 2:312–314
Matthews N (1983) Anti-tumour cytotoxin produced by human monocytes: studies on its mode of action. Br J Cancer 48:405–410
McCormick J, Knight RA, Barry SP et al (2012) Autophagy in the stress-induced myocardium. Front Biosci 4:2131–2141
Mehrpour M, Esclatine A, Beau I et al (2010) Autophagy in health and disease. 1. Regulation and significance of autophagy: an overview. Am J Physiol Cell Physiol 298:C776–C785
Meijer AJ, Codogno P (2009) Autophagy: regulation and role in disease. Crit Rev Clin Lab Sci 46:210–240
Mihara M, Erster S, Zaika A et al (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11:577–590
Mijaljica D, Prescott M, Devenish RJ (2010) Mitophagy and mitoptosis in disease processes. Methods Mol Biol 648:93–106
Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80:293–299
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147:728–741
Moll UM, Marchenko N, Zhang XK (2006) p53 and Nur77/TR3—transcription factors that directly target mitochondria for cell death induction. Oncogene 25:4725–4743
Momand J, Zambetti GP, Olson DC et al (1992) The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69:1237–1245
Moreira JN, Santos A, Simoes S (2006) Bcl-2-targeted antisense therapy (Oblimersen sodium): towards clinical reality. Rev Recent Clin Trials 1:217–235
Moroni MC, Hickman ES, Lazzerini Denchi E et al (2001) Apaf-1 is a transcriptional target for E2F and p53. Nat Cell Biol 3:552–558
Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7:683–694
Nguyen M, Marcellus RC, Roulston A et al (2007) Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci USA 104:19512–19517
Oda E, Ohki R, Murasawa H et al (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288:1053–1058
Panigrahi S, Stetefeld J, Jangamreddy JR et al (2012) Modeling of molecular interaction between apoptin, BCR-Abl and CrkL–an alternative approach to conventional rational drug design. PLoS One 7:e28395
Paoluzzi L, Gonen M, Gardner JR et al (2008) Targeting Bcl-2 family members with the BH3 mimetic AT-101 markedly enhances the therapeutic effects of chemotherapeutic agents in in vitro and in vivo models of B-cell lymphoma. Blood 111:5350–5358
Proskuryakov SY, Konoplyannikov AG, Gabai VL (2003) Necrosis: a specific form of programmed cell death? Exp Cell Res 283:1–16
Rikiishi H (2012) Novel insights into the interplay between apoptosis and autophagy. Int J Cell Biol 2012:317645
Rom J, von Minckwitz G, Eiermann W et al (2008) Oblimersen combined with docetaxel, adriamycin and cyclophosphamide as neo-adjuvant systemic treatment in primary breast cancer: final results of a multicentric phase I study. Ann Oncol 19:1698–1705
Rotter V, Aloni-Grinstein R, Schwartz D et al (1994) Does wild-type p53 play a role in normal cell differentiation? Semin Cancer Biol 5:229–236
Ryabokon NI, Goncharova RI, Duburs G et al (2008) Changes in poly(ADP-ribose) level modulate the kinetics of DNA strand break rejoining. Mutat Res 637:173–181
Saelens X, Festjens N, Vande Walle L et al (2004) Toxic proteins released from mitochondria in cell death. Oncogene 23:2861–2874
Sanli T, Linher-Melville K, Tsakiridis T et al (2012) Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PLoS One 7:e32035
Sax JK, Fei P, Murphy ME et al (2002) BID regulation by p53 contributes to chemosensitivity. Nat Cell Biol 4:842–849
Scherz-Shouval R, Weidberg H, Gonen C et al (2010) p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci USA 107:18511–18516
Schmitt E, Paquet C, Beauchemin M et al (2004) Bcl-xES, a BH4- and BH2-containing antiapoptotic protein, delays Bax oligomer formation and binds Apaf-1, blocking procaspase-9 activation. Oncogene 23:3915–3931
Schweichel JU, Merker HJ (1973) The morphology of various types of cell death in prenatal tissues. Teratology 7:253–266
Secchiero P, Zerbinati C, Melloni E et al (2007) The MDM-2 antagonist nutlin-3 promotes the maturation of acute myeloid leukemic blasts. Neoplasia 9:853–861
Sims JL, Berger SJ, Berger NA (1983) Poly(ADP-ribose) Polymerase inhibitors preserve nicotinamide adenine dinucleotide and adenosine 5′-triphosphate pools in DNA-damaged cells: mechanism of stimulation of unscheduled DNA synthesis. Biochemistry 22:5188–5194
Skulachev VP (2006) Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis 11:473–485
Smith CC, Yellon DM (2011) Necroptosis, necrostatins and tissue injury. J Cell Mol Med 15:1797–1806
Sonnemann J, Palani CD, Wittig S et al (2011) Anticancer effects of the p53 activator nutlin-3 in Ewing’s sarcoma cells. Eur J Cancer 47:1432–1441
Stroh C, Cassens U, Samraj AK et al (2002) The role of caspases in cryoinjury: caspase inhibition strongly improves the recovery of cryopreserved hematopoietic and other cells. FASEB J 16:1651–1653
Szabo C, Dawson VL (1998) Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci 19:287–298
Tasdemir E, Maiuri MC, Galluzzi L et al (2008a) Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 10:676–687
Tasdemir E, Maiuri MC, Orhon I et al (2008b) p53 represses autophagy in a cell cycle-dependent fashion. Cell Cycle 7:3006–3011
Thorburn A (2008) Apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis 13:1–9
Tomita Y, Marchenko N, Erster S et al (2006) WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J Biol Chem 281:8600–8606
Toyota H, Yanase N, Yoshimoto T et al (2003) Calpain-induced Bax-cleavage product is a more potent inducer of apoptotic cell death than wild-type Bax. Cancer Lett 189:221–230
Trudel S, Li ZH, Rauw J et al (2007) Preclinical studies of the pan-Bcl inhibitor obatoclax (GX015-070) in multiple myeloma. Blood 109:5430–5438
Turcotte S, Chan DA, Sutphin PD et al (2008) A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell 14:90–102
Van Herreweghe F, Festjens N, Declercq W et al (2010) Tumor necrosis factor-mediated cell death: to break or to burst, that’s the question. Cell Mol Life Sci 67:1567–1579
Vandenabeele P, Vanden Berghe T, Festjens N (2006) Caspase inhibitors promote alternative cell death pathways. Sci STKE 2006:pe44
Vandenabeele P, Declercq W, Van Herreweghe F et al (2010) The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal 3:re4
Vincent FC, Los MJ (2011) New potential instrument to fight hepatocellular cancer by restoring p53. Hepat Mon 11:331–332
Walsh CM, Edinger AL (2010) The complex interplay between autophagy, apoptosis, and necrotic signals promotes T-cell homeostasis. Immunol Rev 236:95–109
Wang Z, Sun Y (2010) Targeting p53 for novel anticancer therapy. Transl Oncol 3:1–12
Wiechec E (2011) Implications of genomic instability in the diagnosis and treatment of breast cancer. Expert Rev Mol Diagn 11:445–453
Wiechec E, Hansen LL (2009) The effect of genetic variability on drug response in conventional breast cancer treatment. Eur J Pharmacol 625:122–130
Wiechec E, Wiuf C, Overgaard J et al (2011) High-resolution melting analysis for mutation screening of RGSL1, RGS16, and RGS8 in breast cancer. Cancer Epidemiol Biomarkers Prev 20:397–407
Wu H, Zhu H, Liu DX et al (2009) Silencing of elongation factor-2 kinase potentiates the effect of 2-deoxy-d-glucose against human glioma cells through blunting of autophagy. Cancer Res 69:2453–2460
Wyllie AH, Kerr JF, Currie AR (1980) Cell death: the significance of apoptosis. Int Rev Cytol 68:251–306
Wynford-Thomas D (1999) Cellular senescence and cancer. J Pathol 187:100–111
Xu Y (2003) Regulation of p53 responses by post-translational modifications. Cell Death Differ 10:400–403
Xu H, Tai J, Ye H et al (2006) The N-terminal domain of tumor suppressor p53 is involved in the molecular interaction with the anti-apoptotic protein Bcl-Xl. Biochem Biophys Res Commun 341:938–944
Xu H, Ye H, Osman NE et al (2009) The MDM2-binding region in the transactivation domain of p53 also acts as a Bcl-X(L)-binding motif. Biochemistry 48:12159–12168
Yu L, Alva A, Su H et al (2004a) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304:1500–1502
Yu L, Lenardo MJ, Baehrecke EH (2004b) Autophagy and caspases: a new cell death program. Cell Cycle 3:1124–1126
Zhang DW, Shao J, Lin J et al (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325:332–336
Zhang F, Tagen M, Throm S et al (2011) Whole-body physiologically based pharmacokinetic model for nutlin-3a in mice after intravenous and oral administration. Drug Metab Dispos 39:15–21
Acknowledgments
Authors apologize to members of cell death research community for not citing several excellent papers related to cell death topic; this was simply due to space limitation. M. Los kindly acknowledges the core/startup support from Linköping University, from Integrative Regenerative Medicine Center (IGEN), and from Cancerfonden (CAN 2011/521). The authors also thank Dr. S. Ghavami for his help in writing the manuscript and Dr. P. Davoodpour for his assistance in preparing the figures.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Chaabane, W., User, S.D., El-Gazzah, M. et al. Autophagy, Apoptosis, Mitoptosis and Necrosis: Interdependence Between Those Pathways and Effects on Cancer. Arch. Immunol. Ther. Exp. 61, 43–58 (2013). https://doi.org/10.1007/s00005-012-0205-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-012-0205-y