
Abstract
The hepatitis C virus (HCV) is a major cause of chronic liver disease globally. A chronic infection can result in liver fibrosis, liver cirrhosis, hepatocellular carcinoma and liver failure in a significant ratio of the patients. About 170 million people are currently infected with HCV. Since 80 % of the infected patients develop a chronic infection, HCV has evolved sophisticated escape strategies to evade both the innate and the adaptive immune system. Thus, chronic hepatitis C is characterized by perturbations in the number, subset composition and/or functionality of natural killer cells, natural killer T cells, dendritic cells, macrophages and T cells. The balance between HCV-induced immune evasion and the antiviral immune response results in chronic liver inflammation and consequent immune-mediated liver injury. This review summarizes our current understanding of the HCV-mediated interference with cellular immunity and of the factors resulting in HCV persistence. A profound knowledge about the intrinsic properties of HCV and its effects on intrahepatic immunity is essential to be able to design effective immunotherapies against HCV such as therapeutic HCV vaccines.
Similar content being viewed by others


Introduction
With approximately 170 million people infected with hepatitis C virus (HCV) globally, HCV represents a significant health burden. An infection with HCV is characterized by a probability of 70–80 % to develop a chronic infection accompanied by liver inflammation which causes liver fibrosis, cirrhosis and an increased risk to develop hepatocellular carcinoma in 30 % of the cases (Lauer and Walker 2001; Liang et al. 2000; Poynard et al. 2003). Since the current treatment based on pegylated interferon (IFN)-α and ribavirin can cure only about 55 % of the treated patients and is associated with various side effects, more efficient therapy options with fewer side effects are necessary. The recently developed directly acting antiviral (DAA) compounds for HCV will be able to improve the treatment success especially for the difficult-to-treat patients infected with the HCV genotype 1, but will constitute in addition to the pegylated IFN-α and ribavirin-based therapy in the first years. Thus, the development of prophylactic and therapeutic vaccines is an urgent need. A better understanding of the HCV-mediated modulation of cellular immunity is a prerequisite for the design of an effective vaccine.
HCV is an enveloped, positive-sense, single-stranded RNA virus belonging to the genus Hepacivirus in the Flaviviridae family. There are six HCV genotypes which differ in their geographic distribution and their responsiveness to antiviral therapy. The 9.6-kb RNA genome of HCV consists of a long open reading frame that is translated to a single polyprotein of approximately 3,000 amino acids. This polyprotein is co- and posttranslationally cleaved into ten structural and non-structural (NS) proteins (core, E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A and NS5B) (Bode et al. 2009). As a persistent virus, HCV has evolved mechanisms both to utilize and control cellular molecules or pathways required for the viral life cycle and to evade elimination by innate and adaptive immunity. The present review summarizes recent findings focusing on our current understanding of the mechanisms allowing HCV to interfere with host antiviral cellular immunity.
Antiviral Innate Immunity
The liver contains a large macrophage population named Kupffer cells and an unusually high frequency of natural killer (NK) and NK T (NKT) cells. In addition, several types of dendritic cells (DCs) populate the liver. When HCV infects the liver, infected hepatocytes and/or other cells react by secreting type I IFNs. The source of type I IFNs is currently unclear in vivo, but in vitro experiments suggest that it may be produced by either hepatocytes or plasmacytoid DCs (pDCs) (Shin et al. 2006; Takahashi et al. 2010). Type I IFNs not only induce cell death of the infected hepatocytes and an antiviral state in the neighboring uninfected cells, but also activate innate immune cells such as Kupffer cells, DCs, NK cells and NKT cells (Bode et al. 2008). The activated innate immune cells multiply the antiviral response by releasing pro-inflammatory cytokines and chemokines, thus inducing the activation of liver-resident immune cells and the recruitment of immune cells from the periphery. DCs have a key function in bridging innate and adaptive immunity since DCs are able to migrate from the site of infection to lymphoid tissues and to prime naïve T cells by presentation of the viral antigen. This finally results in the induction of virus-specific T and B cell responses. Several lines of evidence indicate that HCV can interfere with the activation and action of innate immune cells.
HCV-Mediated Effects on Innate Immune Cells
HCV-Mediated Modulation of the NK Cell Response
NK cells are characterized by their ability to kill virally infected and tumor cells without major histocompatibility complex (MHC) restriction or prior sensitization. Their importance for liver immunology becomes evident by the fact that their frequency in the liver (30–50 % of the lymphocytes) is much higher than in the peripheral blood (5–20 %) (Corado et al. 1997). There are two main NK cell subsets, which are distinguished by the expression level of the surface receptors CD56 and CD16: CD56dim CD16bright and CD56bright CD16dim. CD56dim CD16bright NK cells are regarded as the more mature subset and have a high cytotoxic potential by both degranulation of cytotoxic granules and activation of death receptors such as the Fas receptor and the tumor necrosis factor (TNF)-related apoptosis-inducing ligand receptor. The less mature CD56bright CD16dim NK cells have an immunomodulatory function and are able to secrete a variety of cytokines such as granulocyte–macrophage colony-stimulating factor, IFN-γ, interleukin (IL)-10, IL-13, tumor growth factor (TGF)-β and TNF-α (Andoniou et al. 2006).
NK cell function is regulated by the interplay of stimulatory and inhibitory receptors such as the killer cell immunoglobulin-like receptors (KIRs), lectin-like receptors (NKG2A-F) and natural cytotoxicity receptors (NKp30, NKp44 and NKp46). Interestingly, genetic studies could show that the combination of the gene for the inhibitory receptor KIR2DL3 and the gene for the group 1 human leukocyte antigen-C (HLA-C1) ligand are associated with both spontaneous HCV clearance and beneficial response to IFN-α/ribavirin treatment (Khakoo et al. 2004; Knapp et al. 2010; Vidal-Castineira et al. 2010). This gene combination may cause protective effects by conferring to NK cells the ability to respond faster to a viral infection (Ahlenstiel et al. 2008). Furthermore, patients homozygous for the HLA-E(R) allele were shown to be protected against chronic infection with the HCV genotypes 2 and 3 (Schulte et al. 2009).
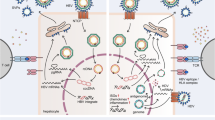
In acute HCV infection, NK cells show an activated state characterized by increased expression of the activating receptor NKG2D and enhanced cytotoxicity with no evidence of a suppressive effect of HCV on NK cell function (Amadei et al. 2010; Pelletier et al. 2010). In chronic HCV infection however, perturbations in the number, subset composition and functionality of NK cells have been found (Fig. 1). The frequency of NK cells was reduced in both the peripheral blood (Meier et al. 2005; Morishima et al. 2006; Nattermann et al. 2006) and the liver (Kawarabayashi et al. 2000) of chronic HCV patients. Furthermore, skewing of NK cell subset distribution toward increased numbers of the cytokine-producing CD56bright CD16dim population, relative to the cytotoxic CD56dim CD16bright subpopulation, was reported (Golden-Mason et al. 2008b; Lin et al. 2004; Morishima et al. 2006). This may be caused by defects in IL-15 production by DCs, since IL-15 is critical for NK cell development and maturation (Meier et al. 2005). HCV-induced interference with NK cell functionality becomes evident by changes in the cytokine profile of NK cells in chronic HCV patients. While IFN-γ release by NK cells is decreased (Ahlenstiel et al. 2010; Oliviero et al. 2009), the production of IL-10 and TGF-β is increased (De Maria et al. 2007; Jinushi et al. 2004). Thus, cytokine production by NK cells in chronic HCV infection is skewed toward secretion of Th2 type cytokines promoting an environment, which is more permissive for HCV (Fig. 1).

NK cells in acute and chronic HCV infection. a In acute HCV infection, NK cells are activated (increased NKG2D expression) and are characterized by enhanced cytotoxicity and IFN-γ production. IFN-γ and TNF-α produced by NK cells prime a Th1 response and activate DCs which provide activating signals for NK cells by secreting IL-2, IL-12, IL-15 and IL-18. b In chronic HCV patients, NK cells are reduced in their frequency and functionality (reduced cytotoxicity and IFN-γ production, enhanced NKG2A expression). Instead, they produce IL-10 and TGF-β resulting in the induction of Th2 cells and Tregs in impaired DC activation and in further production of immunosuppressive cytokines such as IL-10
The function of NK cells in chronic HCV infection may be directly impaired by the binding of the HCV E2 protein to CD81, which has an inhibitory function on NK cells (Crotta et al. 2002; Tseng and Klimpel 2002). However, reports analyzing this effect by using HCV viral particles instead of recombinant E2 proteins have been contradictory so far (Crotta et al. 2010; Yoon et al. 2009). Furthermore, the HCV core protein is able to impair NK cell activity via p53-dependent upregulation of TAP1 and consequently MHC class I surface expression. Enhanced MHC class I expression on infected hepatocytes confers them resistance to NK cell-mediated killing (Herzer et al. 2003). Additionally, a peptide derived from HCV core (HCV core 35–44) stabilizes HLA-E surface expression by binding to HLA-E and thereby impairing NK cell cytotoxicity. This may be mediated by the interaction of HLA-E with the inhibitory NK cell receptor CD94/NKG2A (Nattermann et al. 2005).
Thus, dysfunction of NK cells is critically involved in the establishment of chronic HCV infection. In particular, when one takes into consideration that NK cells not only have direct functions in the eradication of infected hepatocytes but also influence the function of DCs and T cells. Increased expression of the inhibitory receptor CD94/NKG2A in combination with enhanced release of IL-10 and TGF-β by NK cells from HCV-infected patients results in reduced capacity to activate DCs (Jinushi et al. 2004). Furthermore, the increase in NK cell-mediated IL-10 and TGF-β production is skewing the Th1/Th2 balance toward a Th2 response favoring T cell exhaustion and HCV chronicity (Fig. 1).
Role of NKT Cells in HCV Infection
NKT cells are a unique subset of T lymphocytes that co-express T cell receptors (TCRs) and NK cell markers and have both immunoregulatory and effector functions (Taniguchi et al. 2003). They recognize glycolipids presented by MHC class 1b molecules (CD1d in mice and multiple CD1 isoforms in humans) and express either a highly restricted TCR repertoire (invariant NKT cells) or a diverse TCR repertoire (variant NKT cells). When stimulated, they can secrete cytokines (IL-4, IFN-γ and TNF-α), express Fas ligand and activate other cell types such as dendritic and NK cells, which suggest that they may be involved in both clearance of virally infected cells and immune-mediated liver damage.
NKT cells are abundant in the liver comprising of about 30 % of the intrahepatic lymphocytes in mice and up to 50 % in humans (Geissmann et al. 2005; Norris et al. 1999). In chronic hepatitis C patients, decreased frequencies of intrahepatic NKT cells have been reported (Deignan et al. 2002; Kawarabayashi et al. 2000; Yamagiwa et al. 2008). Additionally, sustained response to IFN-α/ribavirin treatment is paralleled by a significant increase in the number of intrahepatic NKT cells (Yamagiwa et al. 2008). Thus, NKT cells may play a beneficial role in HCV clearance. In contrast, the number of activated NKT cells in the liver of chronic hepatitis C patients was shown to correlate with the degree of hepatocellular damage and the onset of fibrosis suggesting that NKT cells are also involved in the deleterious effects mediated by immune cells during chronic liver inflammation (de Lalla et al. 2004; Nuti et al. 1998).
Alteration of DC Functions by HCV
DCs play an essential role in the initiation of virus-specific T cell responses by taking up antigens, processing them and presenting antigen-derived peptides to T cells. After activation through antigen uptake and stimulation by inflammatory cytokines, DCs secrete chemokines, cytokines and IFNs resulting in the recruitment of inflammatory infiltrates to the sites of infection. Furthermore, they migrate to secondary lymphoid organs where they prime T cells and initiate the virus-specific T cell response. There are two major DC subsets in humans, the myeloid DCs (mDCs) and the plasmacytoid DCs (pDCs). While mDCs secrete mainly IL-10 and IL-12 and express Toll-like receptor (TLR)3 and TLR8, pDCs are characterized by IFN-α production and expression of TLR7 and TLR9.
Functional DCs seem to be of high importance to avoid HCV chronicity. Only patients who are able to increase the absolute number and percentage of circulating mDCs during acute hepatitis C are capable of eradicating the virus, while those who do not show any changes in mDCs numbers or frequencies develop viral persistence (Perrella et al. 2006a). Chronic HCV infection results in interference with both the number and the functionality of mDCs and pDCs (Fig. 2). The peripheral blood frequency of mDCs and pDCs is reduced in chronic hepatitis C patients (Kanto et al. 2004; Murakami et al. 2004; Wertheimer et al. 2004). Furthermore, mDCs and pDCs from these patients seem to be impaired in their maturation (Auffermann-Gretzinger et al. 2001; Mengshol et al. 2009).

Plasmacytoid and mDCs in chronic HCV infection. a In chronic HCV infection, plasmacytoid DCs are decreased in their frequency, their maturation and their allostimulatory capacity. Additionally, they are characterized by decreased production of IFN-α and IL-29. b In chronic HCV infection, mDCs are reduced in frequency, maturation and allostimulatory capacity. While their expression of PD-L1 and their production of IL-10 are increased, their secretion of IL-12 and IL-15 is impaired
Moreover, mDCs from HCV-infected patients are impaired in their abilities to stimulate allogeneic CD4+ T cells and to produce IL-12, while their capacity to secrete IL-10 is increased, thus creating an immunosuppressive environment (Averill et al. 2007; Kanto et al. 2004; Murakami et al. 2004). Interestingly, mDCs from HCV-infected patients are not only characterized by an increase in their own IL-10 production but also by a profound ability to prime IL-10-producing T cells (Kanto et al. 2004). Since inhibitory receptors play an important role in the development of exhausted HCV-specific T cells, the expression of receptors with inhibitory function was also investigated on DCs. And indeed, programmed death (PD)-L1 expression on mDCs from chronic hepatitis C patients was shown to be increased and inversely correlated with their allostimulatory capacity (Dolganiuc et al. 2008; Shen et al. 2010) (Fig. 3). Additionally, mDCs from these patients trigger the proliferation of regulatory T cells (Tregs), which further leads to an impairment of the HCV-specific T cell response (Dolganiuc et al. 2008). The HCV core protein may be of main importance for the described effects on mDCs, since stimulation of mDCs with core resulted in inhibition of priming of antigen-specific CD4+ and CD8+ T cells and development of IL-10-producing Tregs (Zimmermann et al. 2008). The inhibitory effect of cell culture-derived HCV on TLR ligand-mediated mDC activation of naïve CD4+ T cells was equally mediated mainly by core (Liang et al. 2009).

Factors resulting in the exhaustion of HCV-specific T cells. Factors contributing to the development of HCV-specific T cell exhaustion are: 1. inhibitory cytokines (such as IL-10 and TGF-β); 2. inhibitory T cell receptors (such as PD-1, 2B4, CD160, CTLA-4, KLRG1 and TIM-3); 3. impaired allostimulatory capacity of DCs; 4. loss of CD4+ T cell help; 5. inhibition by Treg cells
pDCs from HCV-infected patients have also been shown to have a reduced allostimulatory capacity (Kanto et al. 2004) (Fig. 3). In addition, their ability to produce IFN-α is impaired (Dolganiuc et al. 2006; Kanto et al. 2004; Murakami et al. 2004). The HCV proteins core and NS3/4A seem to play a key role in the HCV-mediated modification of pDC function and frequency. The number of intrahepatic pDCs is significantly reduced in mice with liver-specific expression of NS3/4A (Frelin et al. 2006). Furthermore, HCV core and NS3 are able to activate monocytes to produce IL-10 and TNF-α, which result in inhibition of IFN-α secretion by pDCs and pDC apoptosis (Dolganiuc et al. 2003, 2006). Recently, it was shown that the level of IL-29 (IFN-λ1), which is mainly produced by DCs, was substantially lower in patients with chronic hepatitis C as compared to both healthy controls and patients with spontaneously resolved hepatitis. Interestingly, exposure of DCs to NS3 resulted in reduced IL-29 secretion in response to stimulation with poly I:C (Langhans et al. 2011). Besides, both core and NS3 have been shown to inhibit differentiation and allostimulatory capacity of immature DCs (Dolganiuc et al. 2003).
Influence of HCV on Intrahepatic Macrophages
The liver contains a large macrophage population named Kupffer cells, which act as both phagocytes and antigen-presenting cells. Hence, they are involved in the clearance of pathogen-derived particles and toxins and in the killing of pathogens and tumor cells but also contribute to tissue damage during chronic inflammation. In chronic HCV infection, most Kupffer cells are activated and express high levels of CD80, CD40 and MHC-II, thus acquiring the phenotype of professional antigen-presenting cells. Furthermore, activated Kupffer cells display a close contact with CD4+ T cells and form Kupffer cell–T cell clusters (Burgio et al. 1998). Since they exhibit an activated phenotype, they fail to show homo- or hetero-tolerance to TLR ligands as the ones from controls or patients with non-alcoholic steatohepatitis (Dolganiuc et al. 2007). By expressing a variety of TLRs, Kupffer cells can easily sense viral pathogens and respond by secreting inflammatory cytokines such as TNF-α. In the context of HCV, the HCV proteins core and NS3 are able to induce TLR2/MyD88-dependently the secretion of IL-10 and TNF-α by monocytes/macrophages (Dolganiuc et al. 2003, 2004). In vivo, it has been shown that mice with liver-specific expression of NS3/4A are characterized by increased intrahepatic levels of CCL2 and TNF-α, which is paralleled by an enhanced intrahepatic number of macrophages. Thus, NS3/4A may induce a CCL2-mediated recruitment of macrophages to the liver resulting in increased TNF-α levels (Brenndorfer et al. 2010).
Since the expression of inhibitory receptors plays an important role in the induction of exhausted HCV-specific T cells, the expression of ligands of these receptors has also been investigated in Kupffer cells. Interestingly, galectin-9, the natural ligand for the inhibitory receptor T cell immunoglobulin domain and mucin domain protein (TIM)-3 is in the liver mainly expressed on Kupffer cells and its expression is significantly increased in patients with chronic HCV as compared to normal controls. Since galectin-9 has been shown to be involved in the expansion of Tregs, the contraction of CD4+ effector T cells and the apoptosis of HCV-specific CD8+ cells, Kupffer cell-derived galectin-9 seems to be of high importance for the suppression of the HCV-specific T cell response (Mengshol et al. 2010). Besides T cells, the inhibitory receptors PD-1 and TIM-3 are also expressed on macrophages. In chronic hepatitis C patients, PD-1 and TIM-3 were found to be overexpressed on monocytes/macrophages and to be associated with impairment in the production of IL-12 (Zhang et al. 2011a, b). Thus, Kupffer cells are involved in both the immunopathology associated with HCV by secreting high amounts of TNF-α and in the HCV-mediated interference with T cell immunity.
Antiviral Adaptive Immunity
The adaptive immune response is characterized by cellular and humoral effectors recognizing specific viral epitopes. T lymphocytes require epitope presentation by MHC molecules: CD8+ cytotoxic T lymphocytes by MHC-I and CD4+ Th lymphocytes by MHC-II molecules. CD8+ T cells are responsible for the elimination of infected cells through perforin-mediated cytolysis or activation of the death receptor pathways. While Th1 cells activate CD8+ T cells by secreting IFN-γ, IL-2 and TNF-α, Th2 cells are crucial for B cell activation and antibody secretion through the production of IL-4, IL-5, IL-6 and IL-13. When activated by antigen interaction and Th2 signals, B cells release antibodies, which are able to bind free virus and lyse infected cells. HCV infection of the liver results in production of IFN-β and IFN-α, which induce Kupffer cells to secrete MIP1α/CCL3, thereby recruiting NK cells. The recruited NK cells induce DC activation by cell–cell contact and production of IFN-γ and TNF-α. Priming of B and T cells in secondary lymphoid organs by activated mature DCs is necessary to initiate an effective antigen-specific adaptive immune response.
Humoral Immune Response During HCV Infection
Neutralizing antibodies produced by B cells are of importance for the control of many viral infections acting against both free virus and infected cells (Parren and Burton 2001). The binding of antibodies to free viral particles causes a loss of viral infectivity by inhibiting viral attachment or viral entry. Further, the Fc chain of antibodies mediates complement activation leading to opsonization of the virion. Virus uptake by professional antigen-presenting cells is also facilitated by Fc-dependent interactions resulting in the presentation of viral antigens to B and T cells and amplification of the antiviral response. The binding of antibodies to infected cells can result in both cell lysis and inhibition of viral replication, viral release or viral cell–cell transmission (Dorner and Radbruch 2007).
The role of the humoral immune response during HCV infection is still poorly understood. Acute HCV patients often do not exhibit detectable neutralizing antibody titers. Their development is significantly delayed and the existing antibody response does not correlate with viral clearance (Logvinoff et al. 2004). In contrast, most chronic HCV patients possess reactive neutralizing antibodies in their serum (Bartosch et al. 2003; Logvinoff et al. 2004). These neutralizing antibodies recognize epitopes of the viral envelope, mostly located in the hypervariable region of HCV E2 protein (Bartosch et al. 2003). The fact that the present antibodies are ineffective in terminating the ongoing HCV infection can be explained by the rapid viral evolution involving a selection of viral quasispecies, which escape the reactive HCV-specific antibodies.
Interestingly, many extrahepatic manifestations of HCV infection, e.g., cryoglobulinemia, vasculitis, non-Hodgkin lymphoma and Sjögren syndrome are related to B cells (Mayo 2003). Chronic antigen stimulation by HCV was discussed as a mechanism causing HCV-associated B cell lymphoma (de Re et al. 2007). HCV proteins involved in the promotion of B cell proliferation are HCV E2 and NS3. Clonal B cell proliferation may be initiated by HCV E2/CD81 interaction (Curry et al. 2003) and by binding of HCV NS3/IgG antigen to the B cell receptor (De Re et al. 2006). HCV possibly exerts its effects on B cells by infecting them. HCV RNA was detected in B cells from HCV-infected patients but the number of RNA copies per cell seems to be rather low (Pal et al. 2006; Radkowski et al. 2005). Furthermore, while there may be B cell-specific HCV strains, cell culture-derived HCV particles could not infect peripheral blood mononuclear cells (PBMCs) including B cells (Marukian et al. 2008).
T Cell Response in Acute HCV Infection
An acute HCV infection is usually asymptomatic. Studies in experimentally HCV-infected chimpanzees demonstrate that the viral load in the serum increases exponentially during the first weeks of infection. 4–8 weeks after the initial infection, HCV-specific T cells can be detected for the first time paralleled with an increase in alanine aminotransferase levels indicating immune-associated liver injury (Cox et al. 2005; Thimme et al. 2001; Woollard et al. 2003). The rate of spontaneous HCV clearance is estimated to be 20–25 % (Gerlach et al. 2003) and is associated with a robust, sustained and multi-specific CD4+ and CD8+ T cell response (Cooper et al. 1999; Lechner et al. 2000). That a robust CD8+ T cell response is important for viral clearance has been shown convincingly in the chimpanzee model where an early, multi-specific, intrahepatic CD8+ T cell response to HCV was detected in 2/2 chimpanzees that cleared the virus and also in 1/4 chimpanzees that became chronically infected (Cooper et al. 1999).
Depletion of CD4+ T cells and reinfection of two afore immune chimpanzees with HCV resulted in a persistent low-level viremia despite a functional intrahepatic CD8+ T cell response, indicating that CD4+ T cells are also indispensable for sufficient HCV clearance (Grakoui et al. 2003). In humans with controlled HCV infection, the diversity of virus-specific epitopes recognized by CD4+ T cells was much higher than in those who failed to control the infection (Day et al. 2002). Thus, a robust, sustained and multi-specific CD4+ and CD8+ T cell response seems to be required for viral clearance. Analysis of the cytokine profile in PBMCs and CD4+ T cells from acutely infected HCV patients further indicated that viral clearance was largely correlated to a predominant Th1 CD4+ T cell profile (production of IFN-γ and IL-2), while patients exhibiting a Th2 profile (production of IL-4 and IL-10) develop a chronic HCV infection (Tsai et al. 1997).
T Cell Response in Chronic HCV Infection
Virus-specific CD4+ and CD8+ T cells derived from patients with chronic HCV infection showed impaired effector function with reduced proliferative response, diminished peptide-specific cytotoxicity and decreased IFN-γ production (Gruener et al. 2001; Wedemeyer et al. 2002). The frequency of virus-specific CD8+ T cells from chronically infected HCV individuals was found to be about 30-fold higher in the liver as compared to the peripheral blood (He et al. 1999). However, the majority of these T cells exhibit phenotypic characteristics consistent with an incomplete differentiation (Appay et al. 2002). The question is which mechanisms are responsible for the failure of the HCV-specific T cell response to eradicate the virus and how HCV is inducing these mechanisms. We will describe the recent achievements in answering these questions in the following part of the review.
HCV-Mediated Effects on Adaptive Immunity
Viral Escape
The virally encoded HCV polymerase lacks a proofreading function so that HCV exists in each infected host as a swarm of genetically different variants called quasispecies. Pairing with a high replication rate of about 1012 virions per day (Neumann et al. 1998) allows the virus to rapidly adapt to immune pressure by selecting beneficial mutations from the pre-existing quasispecies reservoir. Selection pressure is exerted by both antibodies (Farci et al. 2000; Shimizu et al. 1994) and T cells (Chang et al. 1997; Erickson et al. 2001; Frasca et al. 1999; Seifert et al. 2004; Timm et al. 2004; Tsai et al. 1998). Regarding T cells, HCV escape has been shown to affect epitope processing (Seifert et al. 2004; Timm et al. 2004), MHC binding (Chang et al. 1997) and TCR stimulation (Chang et al. 1997; Erickson et al. 2001; Frasca et al. 1999; Tsai et al. 1998). Thus, effective T cell responses target epitopes that do not allow sequence changes because of high viral fitness costs (Uebelhoer et al. 2008).
HCV-Induced T Cell Chemotaxis
To be able to control HCV infection, the infected liver produces chemokines resulting in the recruitment of HCV-specific T cells. Chemokine receptors associated with T cell homing in the liver are CCR4, CCR5 and CXCR3. While Th1 cells express mainly CCR5 and CXCR3, Th2 cells express preferentially CCR4.
Recently, it has been shown that the expression of the CCR4 ligands, CCL17 and CCL22, is enhanced in chronic hepatitis C patients (Brenndorfer et al. 2012; Riezu-Boj et al. 2011). This results in the attraction of CCR4-expressing Th2 and Treg cells thus contributing to HCV persistence.
A variety of studies have also shown that the expression of CXCR3 ligands (CXCL9, 10 and 11) and CCR5 ligands (CCL3, 4 and 5) is increased in the liver of chronically HCV-infected patients (Zeremski et al. 2007). However, although HCV-specific effector T cells may promote HCV clearance in an early phase of the infection, they cause collateral tissue damage when the infection persists. A potential survival strategy for HCV would be to decrease the number of cytotoxic T cells in the liver to both diminish antiviral immunity and extend host survival as much as possible, thus assuring its own viability. One way to achieve this is by reducing the expression of Th1-associated chemokine receptors on CD8+ T cells (Lichterfeld et al. 2002). This mechanism is also supported by a gene association study showing the importance of CCR5 gene polymorphisms for HCV pathogenesis. Patients bearing the mutation CCR5-Δ32 that abrogates CCR5 expression are characterized by a higher HCV prevalence and when infected by a higher viral load (Woitas et al. 2002). In addition, an attenuation of intrahepatic T cell immunity is also achieved by the recruitment of CXCR3-expressing Treg cells.
Apoptosis of HCV-Specific T Cells
The liver is a unique organ due to the fact that it receives blood from both the systemic circulation and the intestine. Thus, the liver is continuously exposed to food-derived antigens and to endotoxin derived from the intestinal bacteria. To avoid constant immune activation in the liver, intrahepatic immune cells exist in a state of active tolerance, meaning that in most cases, T cell stimulation in the liver leads to Fas-mediated T cell apoptosis (Huang et al. 1994). Furthermore, it has been shown that hepatocyte-activated T cells are characterized by low expression of CD25 resulting in low production of IL-2. This causes T cell death by cytokine deprivation in a Bim-dependent manner (Holz et al. 2008).
This is of particular importance in hepatitis C, where hepatocytes are regarded as the primary site of infection. Premature death of hepatocyte-activated HCV-specific T cells diminishes the antiviral T cell repertoire and hence favors T cell tolerance and HCV persistence.
HCV-Induced T Cell Exhaustion
Chronic Hepatitis C is characterized by continuous exposure of T cells to high levels of HCV antigens resulting in chronic T cell activation and more or less dysfunctional HCV-specific T cells (Gruener et al. 2001; Lechner et al. 2000). Functions like IL-2 production and proliferative capacity are lost first, followed by TNF-α production and at the end by degranulation and IFN-γ production (Gruener et al. 2001; Lechner et al. 2000; Thimme et al. 2001; Urbani et al. 2006a; Wedemeyer et al. 2002). Interestingly, the HCV core protein can inhibit T cell proliferation through interaction with the complement receptor gC1qR (Kittlesen et al. 2000; Yao et al. 2001). Since gC1qR is expressed at higher levels on CD8+ than on CD4+ T cells, the suppressive effects of core are stronger on CD8+ T cells than on CD4+ T cells (Yao et al. 2004). The question is, which factors are contributing to which extend to the development of HCV-specific T cell exhaustion and how these exhausted T cells are characterized (Fig. 3).
Deficient CD4+ T Cell Help
In the setting of a chronic viral infection, CD4+ T cell function is critical and plays an important role in sustaining virus-specific CD8+ T cells during a chronic viral infection (Matloubian et al. 1994). Absence of functionally deficient CD4+ T cells leads to T cell exhaustion and ultimately chronic infection (Kalams and Walker 1998; Ulsenheimer et al. 2003). In chronic HCV infection, CD4+ T cell responses are often weak (Day et al. 2003) and characterized by reduced IL-2 production (Semmo et al. 2005) (Fig. 3).
Inhibitory Cytokine Milieu
Immunoregulatory cytokines are centrally involved in the functional inactivation of antiviral T cells. Especially IL-10 and TGF-β have been linked to T cell exhaustion. During chronic HCV infection, HCV-specific CD8+ T cells producing IL-10 and TGF-β with regulatory capacity occur (Abel et al. 2006; Alatrakchi et al. 2007). Furthermore, IL-10-producing (Tr1) and TGF-β-producing (Th3) CD4+ Treg cells have been described in chronic HCV patients (Cabrera et al. 2004; Ulsenheimer et al. 2003). In addition to being produced by T cells, IL-10 can also be secreted by other cell types such as macrophages, DCs, B cells and NK cells.
Persistent viral infection in mice has been shown to significantly upregulate IL-10 production by antigen-presenting cells, leading to impaired T cell responses. Blocking of the IL-10/IL-10 receptor pathway by genetic deletion or antibody treatment restored T cell function and eliminated viral infection (Brooks et al. 2006; Ejrnaes et al. 2006). IL-10 levels are also increased in patients with chronic HCV infection (Cacciarelli et al. 1996; Reiser et al. 1997). HCV-specific T cell responses in PBMCs from chronically infected patients were restored by blocking IL-10, resulting in increased IFN-γ production (Kaplan et al. 2008; Piazzolla et al. 2000). Moreover, polymorphisms of the IL-10 promotor, which are associated with increased IL-10 production, correlated with an increased susceptibility to develop chronic HCV infection (Knapp et al. 2003; Paladino et al. 2006; Persico et al. 2006).
As outlined above, IL-10 plays a crucial role for impaired DC differentiation and subsequent T cell activation in HCV-infected individuals (Auffermann-Gretzinger et al. 2001; Bain et al. 2001; Della Bella et al. 2007; Dolganiuc et al. 2003; Kanto et al. 1999). This results in suppression of CD4+ T cell response (Kanto et al. 2004, 2006), an effect which could be also mimicked by the core and NS4 protein of HCV (Brady et al. 2003; Zimmermann et al. 2008). This suppression of CD4+ T cell responses by HCV can be successfully reversed by an antibody blocking the IL-10 receptor (Rigopoulou et al. 2005).
Additionally to IL-10, HCV infection is also associated with a significant increase in TGF-β1 expression in both serum and liver (Blackard et al. 2006). Furthermore, polymorphisms of the TGF-β1 promotor were significantly associated with the HCV clearance rate (Kimura et al. 2006). HCV increases hepatocyte TGF-β1 expression through the generation of reactive oxygen species in a nuclear factor-κB-dependent manner, while HCV core and NS3–NS5 proteins seem to be involved (Bataller et al. 2004; Lin et al. 2010). Besides being a key factor in hepatic fibrosis development, TGF-β is also involved in the generation of inducible Tregs and maintenance of Treg function (Marie et al. 2005; Yamagiwa et al. 2001). Similar to IL-10, functional blockade of TGF-β enhances peripheral HCV-specific T cell responses (Alatrakchi et al. 2007).
These data suggest that both IL-10 and TGF-β play an important role for the development of HCV persistence (Fig. 3).
Inhibitory TCRs
Prolonged and/or high expression of multiple inhibitory receptors is a key feature of T cell exhaustion in both animal models and humans. Increased expression of PD-1 is a major mechanism by which virus-specific CD8+ T cells become functionally impaired and PD-1 is up-regulated by exhausted T cells during chronic HCV infections (Golden-Mason et al. 2007; Radziewicz et al. 2007). Interaction of PD-1 with PD-L1, which is expressed by liver sinusoidal cells, Kupffer cells, hepatic stellate cells and DCs (Chen et al. 2006; Iwai et al. 2003) and is IFN-α- and IFN-γ-dependently upregulated on hepatocytes (Muhlbauer et al. 2006), induces T cell apoptosis and inhibits T cell functionality (Iwai et al. 2003; Radziewicz et al. 2007). Of note, blockade of PD-L1/PD-1 interaction resulted in the functional restoration of blood-derived HCV-specific CD8+ T cell responses in chronic HCV infection (Golden-Mason et al. 2007, 2008a; Penna et al. 2007; Radziewicz et al. 2007; Urbani et al. 2006b).
Besides PD-1, many other inhibitory TCRs co-regulate T cell exhaustion. In chronic hepatitis C, HCV-specific CD8+ T cells have been reported to express inhibitory receptors such as 2B4 (CD244), CD160, CTLA-4, KLRG1 or TIM-3 (Bengsch et al. 2010; Golden-Mason et al. 2009; Nakamoto et al. 2008; Schlaphoff et al. 2011) (Fig. 3). The importance of these receptors became evident, when it was shown that PD-1 blockade alone was unable to restore the function of liver-derived HCV-specific CD8+ T cells (Nakamoto et al. 2008) but that additional blockade of CTLA-4 reinvigorated the antiviral T cell functions (Nakamoto et al. 2009). Additional studies are needed to better understand the interaction of these receptors and their role in the various stages of T cell exhaustion.
Induction of Tregs
Furthermore, HCV also interferes with Treg function, thereby contributing to dysregulation of the antiviral adaptive immune response (Fig. 3). Several reports indicate that the frequency of CD4+/CD25+ Tregs is increased in chronic HCV patients (Boettler et al. 2005; Cabrera et al. 2004; Rushbrook et al. 2005). It was shown that Tregs expand during the acute phase of HCV infection (Perrella et al. 2006b; Ulsenheimer et al. 2003), maintain their number during the chronic phase (Boettler et al. 2005; Cabrera et al. 2004; Sugimoto et al. 2003) and decrease their number to the level of healthy controls when the patients cure the HCV infection (Boettler et al. 2005; Sugimoto et al. 2003). Depletion of these cells resulted in an enhanced HCV-specific T cell response indicating that these cells are also involved in the suppression of HCV-specific T cell responses (Boettler et al. 2005; Cabrera et al. 2004; Rushbrook et al. 2005). HCV-specific Tregs use both direct and indirect mechanisms for their inhibitory functions. Cell-to-cell contact was shown to be indispensable for suppression by Tregs (Boettler et al. 2005; Cabrera et al. 2004). Furthermore, IL-10 and TGF-β secretion are important for HCV-specific Treg functions (Alatrakchi et al. 2007; Cabrera et al. 2004).
However, the mechanisms by which HCV might induce the recruitment of Tregs to the infected liver are not known. The recruitment of immune cells in the liver is mainly dependent on the release of specific chemokines. Thus, the recently reported increase of CCL17 and CCL22 expression found in the liver of HCV-infected patients may be of importance (Brenndorfer et al. 2012; Riezu-Boj et al. 2011). Contact of DCs with HCV-infected Huh7 cells strongly stimulates the expression of CCL17 and CCL22, which act as attractants for Tregs (Riezu-Boj et al. 2011). NS3/4A may be the HCV protein mediating the described effects since mice with liver-specific expression of NS3/4A are characterized by enhanced intrahepatic levels of CCL17 and CCL22 resulting in increased numbers of CCR4+ CD4+ T cells in the livers of NS3/4A-transgenic mice (Brenndorfer et al. 2012).
Immunotherapy for HCV Infection
In the last years, a substantial progress has been made in the understanding of the HCV life cycle and the mechanisms used by HCV to evade host immunity. This resulted in the discovery of various new therapy options, which may lead to the development of better immunotherapies or efficient vaccines in the future.
The current standard of care therapy for HCV-infected patients is based on pegylated IFN-α and ribavirin, which are both immunomodulatory agents. IFN-α has not only direct antiviral actions by inducing the expression of protein kinase R or 2′,5′-oligoadenylate synthetase but also effects on cellular immunity. IFN-α is known to stimulate the activation of NK cells, the maturation of DCs, the proliferation of memory T cells, the expression of MHC molecules and the promotion of Th1 cells (Maher et al. 2007). The treatment with pegylated IFN-α is thought to boost the actions of endogenous IFN-α causing a strengthening of antiviral immunity.
For ribavirin, several antiviral mechanisms have been proposed. Besides direct mechanisms resulting in the blockade of HCV replication, ribavirin is thought to increase the expression of IFN-stimulated genes (Thomas et al. 2011), modulate cytokine production by macrophages (Ning et al. 1998) and stimulate a Th1-dominated antiviral response which favors viral clearance (Hultgren et al. 1998).
HCV proteins such as NS3/4A and NS5A have not only essential functions for the viral life cycle but are also known to modulate intrahepatic immunity. NS3/4A is blocking signaling pathways in HCV-infected cells by cleaving mitochondrial antiviral signaling protein (Li et al. 2005b; Meylan et al. 2005), Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (Li et al. 2005a) and T cell protein tyrosine phosphatase (Brenndorfer et al. 2009). Furthermore, it is influencing macrophage and T cell recruitment and functionality by modulating cytokine and chemokine levels in the liver (Brenndorfer et al. 2010, 2012). NS5A was also shown to impair both the innate and adaptive hepatic immune response (Kriegs et al. 2009). Thus, future HCV therapies based on NS3/4A protease and NS5A inhibitors may exert effects beyond the viral replication by reversing HCV-mediated effects on host immunity as well.
Although the development of HCV vaccines in the last years was promising, there is still no prophylactic or therapeutic vaccine for HCV available. Vaccination experiments in chimpanzees have shown that protective immunity to both homologous and heterologous HCV strains exists (Bassett et al. 2001; Lanford et al. 2004; Weiner et al. 2001) indicating the presence of epitopes, which are conserved between genotypes. Patient studies analyzing the immune response in acute HCV infection revealed that a strong and sustained HCV-specific T cell response targeting multiple epitopes is associated with a self-limited course of infection (Rehermann 2009). Thus, a successful HCV vaccine should be able to raise these T cell responses. Since HCV NS antigens contain both highly conserved gene regions and multiple CD4+ and CD8+ T cell epitopes, most new HCV vaccine approaches have focused on inducing T cell responses to HCV NS antigens. By using an NS3/4A-transgenic mouse model that somewhat mimics the human infection with respect to a dysfunctional T cell response, it was shown that modulation of PD-1 and Tregs can restore T cell responsiveness (Chen et al. 2011). In the same model, it was shown that the recruitment of heterologous T cells to the site of T cell activation had the same effect (Chen et al. 2011). The immunogen itself and the administration routes have a central role in what type of T cells becomes activated (Gill et al. 2010; Lazdina et al. 2001; Nystrom et al. 2010).
Several HCV vaccine approaches based on peptides, DNA, vectors and recombinant proteins have recently reached phase I/II human clinical trials. A therapeutic peptide vaccine currently in clinical development is IC41 consisting of five synthetic peptides from the proteins core, NS3 and NS4 proteins which are conserved across the HCV genotypes 1 and 2 and are combined with the adjuvant poly-l-arginine (Firbas et al. 2010; Klade et al. 2008). A therapeutic DNA vaccine based on NS3/4A given in combination with in vivo electroporation is currently in a phase I/IIa clinical trial (Sallberg et al. 2009). Vector-based vaccines use the modified vaccinia Ankara (MVA) virus or adenovirus for gene delivery. A phase I clinical trial assessing the therapeutic vaccine TG4040 which is a MVA-based vaccine expressing NS3, NS4 and NS5B was recently completed (Habersetzer et al. 2011). In a further phase I clinical trial, healthy volunteers have been vaccinated with adenovirus vectors expressing NS3–NS5B (Barnes et al. 2012). To avoid preexisting immunity, rare serotype adenoviral vectors have been used. Interestingly, sustained HCV-specific immune responses responding to multiple HCV epitopes and HCV strains could be induced. Thus, this strategy may be suitable for both the use as prophylactic and therapeutic HCV vaccine. A further candidate for a prophylactic HCV vaccine based on a recombinant E1/E2 heterodimer adjuvanted with MF59C was also tested recently in a phase I trial (Frey et al. 2010).
In addition, cell-based immunotherapies using HCV antigen-presenting DCs or HCV-specific T cells have been used as vaccine approaches. In a phase I trial, DCs were harvested and then loaded and activated with HCV-specific cytotoxic T cell epitopes before they were reinjected in HCV-infected patients (Gowans et al. 2010). In a preclinical study, mice have been vaccinated against HCV with DCs transduced with an adenovirus encoding NS3 protein (Echeverria et al. 2011). Additionally, HCV TCR-transduced T cells, which have only been tested in preclinical studies till now, may be promising for the treatment of patients with chronic HCV infection (Pasetto et al. 2012; Zhang et al. 2010).
Thus, several promising vaccine trials have been completed recently and are also planned for the near future. However, since the described therapeutic HCV vaccine studies have been characterized by an induction of strong T cell responses but only a weak and transient drop of the viral load, therapeutic vaccines may be used in the next years in combination with the standard HCV therapy or future DAA regimens rather than isolated.
Conclusions
An effective cellular immune response is critical for HCV eradication during the acute phase of the infection. However, HCV has developed a big variety of mechanisms interfering with antiviral immunity so that in the majority of patients infected with HCV a chronic infection is established. In the chronic phase of the infection, persistent activation of cellular immunity without successful elimination of the virus results in chronic inflammation leading to liver injury and liver cirrhosis. HCV is preventing a higher degree of liver damage by simultaneously attenuating innate and adaptive immune responses allowing coexistence of both virus and host. Blocking the HCV-mediated impairment of cellular immunity paralleled by an effective stimulation of the HCV-specific immune response is necessary to be able to clear the virus. Since in the last years fantastic efforts have been made in the characterization of the HCV-mediated mechanisms responsible for HCV persistence, we hope that these efforts will make it possible to develop effective immunotherapies or therapeutic vaccines against HCV.
Abbreviations
- DAA:
-
Directly acting antiviral
- DC:
-
Dendritic cell
- HCV:
-
Hepatitis C virus
- HLA:
-
Human leukocyte antigen
- IFN:
-
Interferon
- IL:
-
Interleukin
- KIR:
-
Killer cell immunoglobulin-like receptor
- MHC:
-
Major histocompatibility complex
- NK cell:
-
Natural killer cell
- NKT cell:
-
Natural killer T cell
- NS:
-
Non-structural
- PBMC:
-
Peripheral blood mononuclear cell
- PD:
-
Programmed death
- TCR:
-
T cell receptor
- TGF:
-
Tumor growth factor
- Th:
-
T-helper cell
- TIM:
-
T cell immunoglobulin domain and mucin domain protein
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- Treg:
-
Regulatory T cell
References
Abel M, Sene D, Pol S et al (2006) Intrahepatic virus-specific IL-10-producing CD8 T cells prevent liver damage during chronic hepatitis C virus infection. Hepatology 44:1607–1616
Ahlenstiel G, Martin MP, Gao X et al (2008) Distinct KIR/HLA compound genotypes affect the kinetics of human antiviral natural killer cell responses. J Clin Invest 118:1017–1026
Ahlenstiel G, Titerence RH, Koh C et al (2010) Natural killer cells are polarized toward cytotoxicity in chronic hepatitis C in an interferon-alfa-dependent manner. Gastroenterology 138(325–335):e1–e2
Alatrakchi N, Graham CS, van der Vliet HJ et al (2007) Hepatitis C virus (HCV)-specific CD8+ cells produce transforming growth factor beta that can suppress HCV-specific T-cell responses. J Virol 81:5882–5892
Amadei B, Urbani S, Cazaly A et al (2010) Activation of natural killer cells during acute infection with hepatitis C virus. Gastroenterology 138:1536–1545
Andoniou CE, Andrews DM, Degli-Esposti MA (2006) Natural killer cells in viral infection: more than just killers. Immunol Rev 214:239–250
Appay V, Dunbar PR, Callan M et al (2002) Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med 8:379–385
Auffermann-Gretzinger S, Keeffe EB, Levy S (2001) Impaired dendritic cell maturation in patients with chronic, but not resolved, hepatitis C virus infection. Blood 97:3171–3176
Averill L, Lee WM, Karandikar NJ (2007) Differential dysfunction in dendritic cell subsets during chronic HCV infection. Clin Immunol 123:40–49
Bain C, Fatmi A, Zoulim F et al (2001) Impaired allostimulatory function of dendritic cells in chronic hepatitis C infection. Gastroenterology 120:512–524
Barnes E, Folgori A, Capone S et al (2012) Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci Transl Med 4(115):115ra1
Bartosch B, Bukh J, Meunier JC et al (2003) In vitro assay for neutralizing antibody to hepatitis C virus: evidence for broadly conserved neutralization epitopes. Proc Natl Acad Sci USA 100:14199–14204
Bassett SE, Guerra B, Brasky K et al (2001) Protective immune response to hepatitis C virus in chimpanzees rechallenged following clearance of primary infection. Hepatology 33:1479–1487
Bataller R, Paik YH, Lindquist JN et al (2004) Hepatitis C virus core and nonstructural proteins induce fibrogenic effects in hepatic stellate cells. Gastroenterology 126:529–540
Bengsch B, Seigel B, Ruhl M et al (2010) Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog 6:e1000947
Blackard JT, Kang M, Sherman KE et al (2006) Effects of HCV treatment on cytokine expression during HCV/HIV coinfection. J Interferon Cytokine Res 26:834–838
Bode JG, Brenndorfer ED, Haussinger D (2008) Hepatitis C virus (HCV) employs multiple strategies to subvert the host innate antiviral response. Biol Chem 389:1283–1298
Bode JG, Brenndorfer ED, Karthe J et al (2009) Interplay between host cell and hepatitis C virus in regulating viral replication. Biol Chem 390:1013–1032
Boettler T, Spangenberg HC, Neumann-Haefelin C et al (2005) T cells with a CD4+CD25+ regulatory phenotype suppress in vitro proliferation of virus-specific CD8+ T cells during chronic hepatitis C virus infection. J Virol 79:7860–7867
Brady MT, MacDonald AJ, Rowan AG et al (2003) Hepatitis C virus non-structural protein 4 suppresses Th1 responses by stimulating IL-10 production from monocytes. Eur J Immunol 33:3448–3457
Brenndorfer ED, Karthe J, Frelin L et al (2009) Nonstructural 3/4A protease of hepatitis C virus activates epithelial growth factor-induced signal transduction by cleavage of the T-cell protein tyrosine phosphatase. Hepatology 49:1810–1820
Brenndorfer ED, Weiland M, Frelin L et al (2010) Anti-tumor necrosis factor alpha treatment promotes apoptosis and prevents liver regeneration in a transgenic mouse model of chronic hepatitis C. Hepatology 52:1553–1563
Brenndorfer ED, Brass A, Soderholm J et al (2012) Hepatitis C virus non-structural 3/4A protein interferes with intrahepatic interferon-gamma production. Gut 61:589–596
Brooks DG, Trifilo MJ, Edelmann KH et al (2006) Interleukin-10 determines viral clearance or persistence in vivo. Nat Med 12:1301–1309
Burgio VL, Ballardini G, Artini M et al (1998) Expression of co-stimulatory molecules by Kupffer cells in chronic hepatitis of hepatitis C virus etiology. Hepatology 27:1600–1606
Cabrera R, Tu Z, Xu Y et al (2004) An immunomodulatory role for CD4(+)CD25(+) regulatory T lymphocytes in hepatitis C virus infection. Hepatology 40:1062–1071
Cacciarelli TV, Martinez OM, Gish RG et al (1996) Immunoregulatory cytokines in chronic hepatitis C virus infection: pre- and posttreatment with interferon alfa. Hepatology 24:6–9
Chang KM, Rehermann B, McHutchison JG et al (1997) Immunological significance of cytotoxic T lymphocyte epitope variants in patients chronically infected by the hepatitis C virus. J Clinical Invest 100:2376–2385
Chen CH, Kuo LM, Chang Y et al (2006) In vivo immune modulatory activity of hepatic stellate cells in mice. Hepatology 44:1171–1181
Chen A, Ahlen G, Brenndorfer ED et al (2011) Heterologous T cells can help restore function in dysfunctional hepatitis C virus nonstructural 3/4A-specific T cells during therapeutic vaccination. J Immunol 186:5107–5118
Cooper S, Erickson AL, Adams EJ et al (1999) Analysis of a successful immune response against hepatitis C virus. Immunity 10:439–449
Corado J, Toro F, Rivera H et al (1997) Impairment of natural killer (NK) cytotoxic activity in hepatitis C virus (HCV) infection. Clin Exp Immunol 109:451–457
Cox AL, Mosbruger T, Lauer GM et al (2005) Comprehensive analyses of CD8+ T cell responses during longitudinal study of acute human hepatitis C. Hepatology 42:104–112
Crotta S, Stilla A, Wack A et al (2002) Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J Exp Med 195:35–41
Crotta S, Brazzoli M, Piccioli D et al (2010) Hepatitis C virions subvert natural killer cell activation to generate a cytokine environment permissive for infection. J Hepatol 52:183–190
Curry MP, Golden-Mason L, Doherty DG et al (2003) Expansion of innate CD5pos B cells expressing high levels of CD81 in hepatitis C virus infected liver. J Hepatol 38:642–650
Day CL, Lauer GM, Robbins GK et al (2002) Broad specificity of virus-specific CD4+ T-helper-cell responses in resolved hepatitis C virus infection. J Virol 76:12584–12595
Day CL, Seth NP, Lucas M et al (2003) Ex vivo analysis of human memory CD4 T cells specific for hepatitis C virus using MHC class II tetramers. J Clin Invest 112:831–842
de Lalla C, Galli G, Aldrighetti L et al (2004) Production of profibrotic cytokines by invariant NKT cells characterizes cirrhosis progression in chronic viral hepatitis. J Immunol 173:1417–1425
De Maria A, Fogli M, Mazza S et al (2007) Increased natural cytotoxicity receptor expression and relevant IL-10 production in NK cells from chronically infected viremic HCV patients. Eur J Immunol 37:445–455
De Re V, Sansonno D, Simula MP et al (2006) HCV-NS3 and IgG-Fc crossreactive IgM in patients with type II mixed cryoglobulinemia and B-cell clonal proliferations. Leukemia 20:1145–1154
de Re V, Caggiari L, Simula MP et al (2007) B-cell lymphomas associated with HCV infection. Gastroenterology 132:1205–1207
Deignan T, Curry MP, Doherty DG et al (2002) Decrease in hepatic CD56(+) T cells and V alpha 24(+) natural killer T cells in chronic hepatitis C viral infection. J Hepatol 37:101–108
Della Bella S, Crosignani A, Riva A et al (2007) Decrease and dysfunction of dendritic cells correlate with impaired hepatitis C virus-specific CD4+ T-cell proliferation in patients with hepatitis C virus infection. Immunology 121:283–292
Dolganiuc A, Kodys K, Kopasz A et al (2003) Hepatitis C virus core and nonstructural protein 3 proteins induce pro- and anti-inflammatory cytokines and inhibit dendritic cell differentiation. J Immunol 170:5615–5624
Dolganiuc A, Oak S, Kodys K et al (2004) Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 127:1513–1524
Dolganiuc A, Chang S, Kodys K et al (2006) Hepatitis C virus (HCV) core protein-induced, monocyte-mediated mechanisms of reduced IFN-alpha and plasmacytoid dendritic cell loss in chronic HCV infection. J Immunol 177:6758–6768
Dolganiuc A, Norkina O, Kodys K et al (2007) Viral and host factors induce macrophage activation and loss of toll-like receptor tolerance in chronic HCV infection. Gastroenterology 133:1627–1636
Dolganiuc A, Paek E, Kodys K et al (2008) Myeloid dendritic cells of patients with chronic HCV infection induce proliferation of regulatory T lymphocytes. Gastroenterology 135:2119–2127
Dorner T, Radbruch A (2007) Antibodies and B cell memory in viral immunity. Immunity 27:384–392
Echeverria I, Pereboev A, Silva L et al (2011) Enhanced T cell responses against hepatitis C virus by ex vivo targeting of adenoviral particles to dendritic cells. Hepatology 54:28–37
Ejrnaes M, Filippi CM, Martinic MM et al (2006) Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med 203:2461–2472
Erickson AL, Kimura Y, Igarashi S et al (2001) The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity 15:883–895
Farci P, Shimoda A, Coiana A et al (2000) The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science 288:339–344
Firbas C, Boehm T, Buerger V et al (2010) Immunogenicity and safety of different injection routes and schedules of IC41, a Hepatitis C virus (HCV) peptide vaccine. Vaccine 28:2397–2407
Frasca L, Del Porto P, Tuosto L et al (1999) Hypervariable region 1 variants act as TCR antagonists for hepatitis C virus-specific CD4+ T cells. J Immunol 163:650–658
Frelin L, Brenndorfer ED, Ahlen G et al (2006) The hepatitis C virus and immune evasion: non-structural 3/4A transgenic mice are resistant to lethal tumour necrosis factor alpha mediated liver disease. Gut 55:1475–1483
Frey SE, Houghton M, Coates S et al (2010) Safety and immunogenicity of HCV E1E2 vaccine adjuvanted with MF59 administered to healthy adults. Vaccine 28:6367–6373
Geissmann F, Cameron TO, Sidobre S et al (2005) Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol 3:e113
Gerlach JT, Diepolder HM, Zachoval R et al (2003) Acute hepatitis C: high rate of both spontaneous and treatment-induced viral clearance. Gastroenterology 125:80–88
Gill HS, Soderholm J, Prausnitz MR, Sallberg M (2010) Cutaneous vaccination using microneedles coated with hepatitis C DNA vaccine. Gene Ther 17:811–814
Golden-Mason L, Palmer B, Klarquist J et al (2007) Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J Virol 81:9249–9258
Golden-Mason L, Klarquist J, Wahed AS et al (2008a) Cutting edge: programmed death-1 expression is increased on immunocytes in chronic hepatitis C virus and predicts failure of response to antiviral therapy: race-dependent differences. J Immunol 180:3637–3641
Golden-Mason L, Madrigal-Estebas L, McGrath E et al (2008b) Altered natural killer cell subset distributions in resolved and persistent hepatitis C virus infection following single source exposure. Gut 57:1121–1128
Golden-Mason L, Palmer BE, Kassam N et al (2009) Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol 83:9122–9130
Gowans EJ, Roberts S, Jones K et al (2010) A phase I clinical trial of dendritic cell immunotherapy in HCV-infected individuals. J Hepatol 53:599–607
Grakoui A, Shoukry NH, Woollard DJ et al (2003) HCV persistence and immune evasion in the absence of memory T cell help. Science 302:659–662
Gruener NH, Lechner F, Jung MC et al (2001) Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J Virol 75:5550–5558
Habersetzer F, Honnet G, Bain C et al (2011) A poxvirus vaccine is safe, induces T-cell responses, and decreases viral load in patients with chronic hepatitis C. Gastroenterology 141:890–899
He XS, Rehermann B, Lopez-Labrador FX et al (1999) Quantitative analysis of hepatitis C virus-specific CD8(+) T cells in peripheral blood and liver using peptide-MHC tetramers. Proc Natl Acad Sci USA 96:5692–5697
Herzer K, Falk CS, Encke J et al (2003) Upregulation of major histocompatibility complex class I on liver cells by hepatitis C virus core protein via p53 and TAP1 impairs natural killer cell cytotoxicity. J Virol 77:8299–8309
Holz LE, Benseler V, Bowen DG et al (2008) Intrahepatic murine CD8 T-cell activation associates with a distinct phenotype leading to Bim-dependent death. Gastroenterology 135:989–997
Huang L, Soldevila G, Leeker M et al (1994) The liver eliminates T cells undergoing antigen-triggered apoptosis in vivo. Immunity 1:741–749
Hultgren C, Milich DR, Weiland O et al (1998) The antiviral compound ribavirin modulates the T helper (Th) 1/Th2 subset balance in hepatitis B and C virus-specific immune responses. J Gen Virol 79(Pt 10):2381–2391
Iwai Y, Terawaki S, Ikegawa M et al (2003) PD-1 inhibits antiviral immunity at the effector phase in the liver. J Exp Med 198:39–50
Jinushi M, Takehara T, Tatsumi T et al (2004) Negative regulation of NK cell activities by inhibitory receptor CD94/NKG2A leads to altered NK cell-induced modulation of dendritic cell functions in chronic hepatitis C virus infection. J Immunol 173:6072–6081
Kalams SA, Walker BD (1998) The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J Exp Med 188:2199–2204
Kanto T, Hayashi N, Takehara T et al (1999) Impaired allostimulatory capacity of peripheral blood dendritic cells recovered from hepatitis C virus-infected individuals. J Immunol 162:5584–5591
Kanto T, Inoue M, Miyatake H et al (2004) Reduced numbers and impaired ability of myeloid and plasmacytoid dendritic cells to polarize T helper cells in chronic hepatitis C virus infection. J Infect Dis 190:1919–1926
Kanto T, Inoue M, Miyazaki M et al (2006) Impaired function of dendritic cells circulating in patients infected with hepatitis C virus who have persistently normal alanine aminotransferase levels. Intervirology 49:58–63
Kaplan DE, Ikeda F, Li Y et al (2008) Peripheral virus-specific T-cell interleukin-10 responses develop early in acute hepatitis C infection and become dominant in chronic hepatitis. J Hepatol 48:903–913
Kawarabayashi N, Seki S, Hatsuse K et al (2000) Decrease of CD56(+)T cells and natural killer cells in cirrhotic livers with hepatitis C may be involved in their susceptibility to hepatocellular carcinoma. Hepatology 32:962–969
Khakoo SI, Thio CL, Martin MP et al (2004) HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science 305:872–874
Kimura T, Saito T, Yoshimura M et al (2006) Association of transforming growth factor-beta 1 functional polymorphisms with natural clearance of hepatitis C virus. J Infect Dis 193:1371–1374
Kittlesen DJ, Chianese-Bullock KA, Yao ZQ et al (2000) Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation. J Clin Invest 106:1239–1249
Klade CS, Wedemeyer H, Berg T et al (2008) Therapeutic vaccination of chronic hepatitis C nonresponder patients with the peptide vaccine IC41. Gastroenterology 134:1385–1395
Knapp S, Hennig BJ, Frodsham AJ et al (2003) Interleukin-10 promoter polymorphisms and the outcome of hepatitis C virus infection. Immunogenetics 55:362–369
Knapp S, Warshow U, Hegazy D et al (2010) Consistent beneficial effects of killer cell immunoglobulin-like receptor 2DL3 and group 1 human leukocyte antigen-C following exposure to hepatitis C virus. Hepatology 51:1168–1175
Kriegs M, Burckstummer T, Himmelsbach K et al (2009) The hepatitis C virus non-structural NS5A protein impairs both the innate and adaptive hepatic immune response in vivo. J Biol Chem 284:28343–28351
Lanford RE, Guerra B, Chavez D et al (2004) Cross-genotype immunity to hepatitis C virus. J Virol 78:1575–1581
Langhans B, Kupfer B, Braunschweiger I et al (2011) Interferon-lambda serum levels in hepatitis C. J Hepatol 54:859–865
Lauer GM, Walker BD (2001) Hepatitis C virus infection. N Engl J Med 345:41–52
Lazdina U, Hultgren C, Frelin L et al (2001) Humoral and CD4(+) T helper (Th) cell responses to the hepatitis C virus non-structural 3 (NS3) protein: NS3 primes Th1-like responses more effectively as a DNA-based immunogen than as a recombinant protein. J Gen Virol 82(Pt 6):1299–1308
Lechner F, Wong DK, Dunbar PR et al (2000) Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med 191:1499–1512
Li K, Foy E, Ferreon JC et al (2005a) Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci USA 102:2992–2997
Li XD, Sun L, Seth RB, Pineda G et al (2005b) Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci USA 102:17717–17722
Liang TJ, Rehermann B, Seeff LB et al (2000) Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann Intern Med 132:296–305
Liang H, Russell RS, Yonkers NL et al (2009) Differential effects of hepatitis C virus JFH1 on human myeloid and plasmacytoid dendritic cells. J Virol 83:5693–5707
Lichterfeld M, Leifeld L, Nischalke HD et al (2002) Reduced CC chemokine receptor (CCR) 1 and CCR5 surface expression on peripheral blood T lymphocytes from patients with chronic hepatitis C infection. J Infect Dis 185:1803–1807
Lin AW, Gonzalez SA, Cunningham-Rundles S et al (2004) CD56(+dim) and CD56(+bright) cell activation and apoptosis in hepatitis C virus infection. Clin Exp Immunol 137:408–416
Lin W, Tsai WL, Shao RX et al (2010) Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology 138:2509–2518 2518.e1
Logvinoff C, Major ME, Oldach D et al (2004) Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc Natl Acad Sci USA 101:10149–10154
Maher SG, Romero-Weaver AL, Scarzello AJ et al (2007) Interferon: cellular executioner or white knight? Curr Med Chem 14:1279–1289
Marie JC, Letterio JJ, Gavin M et al (2005) TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med 201:1061–1067
Marukian S, Jones CT, Andrus L et al (2008) Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 48:1843–1850
Matloubian M, Concepcion RJ, Ahmed R (1994) CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J Virol 68:8056–8063
Mayo MJ (2003) Extrahepatic manifestations of hepatitis C infection. Am J Med Sci 325:135–148
Meier UC, Owen RE, Taylor E et al (2005) Shared alterations in NK cell frequency, phenotype, and function in chronic human immunodeficiency virus and hepatitis C virus infections. J Virol 79:12365–12374
Mengshol JA, Golden-Mason L, Castelblanco N et al (2009) Impaired plasmacytoid dendritic cell maturation and differential chemotaxis in chronic hepatitis C virus: associations with antiviral treatment outcomes. Gut 58:964–973
Mengshol JA, Golden-Mason L, Arikawa T et al (2010) A crucial role for Kupffer cell-derived galectin-9 in regulation of T cell immunity in hepatitis C infection. PLoS ONE 5:e9504
Meylan E, Curran J, Hofmann K et al (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172
Morishima C, Paschal DM, Wang CC et al (2006) Decreased NK cell frequency in chronic hepatitis C does not affect ex vivo cytolytic killing. Hepatology 43:573–580
Muhlbauer M, Fleck M, Schutz C et al (2006) PD-L1 is induced in hepatocytes by viral infection and by interferon-alpha and -gamma and mediates T cell apoptosis. J Hepatol 45:520–528
Murakami H, Akbar SM, Matsui H et al (2004) Decreased interferon-alpha production and impaired T helper 1 polarization by dendritic cells from patients with chronic hepatitis C. Clin Exp Immunol 137:559–565
Nakamoto N, Kaplan DE, Coleclough J et al (2008) Functional restoration of HCV-specific CD8 T cells by PD-1 blockade is defined by PD-1 expression and compartmentalization. Gastroenterology 134:1927–1937 1937.e1–e2
Nakamoto N, Cho H, Shaked A et al (2009) Synergistic reversal of intrahepatic HCV-specific CD8 T cell exhaustion by combined PD-1/CTLA-4 blockade. PLoS Pathog 5:e1000313
Nattermann J, Nischalke HD, Hofmeister V et al (2005) The HLA-A2 restricted T cell epitope HCV core 35–44 stabilizes HLA-E expression and inhibits cytolysis mediated by natural killer cells. Am J Pathol 166:443–453
Nattermann J, Feldmann G, Ahlenstiel G et al (2006) Surface expression and cytolytic function of natural killer cell receptors is altered in chronic hepatitis C. Gut 55:869–877
Neumann AU, Lam NP, Dahari H et al (1998) Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 282:103–107
Ning Q, Brown D, Parodo J et al (1998) Ribavirin inhibits viral-induced macrophage production of TNF, IL-1, the procoagulant fgl2 prothrombinase and preserves Th1 cytokine production but inhibits Th2 cytokine response. J Immunol 160:3487–3493
Norris S, Doherty DG, Collins C et al (1999) Natural T cells in the human liver: cytotoxic lymphocytes with dual T cell and natural killer cell phenotype and function are phenotypically heterogenous and include Valpha24-JalphaQ and gammadelta T cell receptor bearing cells. Hum Immunol 60:20–31
Nuti S, Rosa D, Valiante NM et al (1998) Dynamics of intra-hepatic lymphocytes in chronic hepatitis C: enrichment for Valpha24+ T cells and rapid elimination of effector cells by apoptosis. Eur J Immunol 28:3448–3455
Nystrom J, Chen A, Frelin L et al (2010) Improving on the ability of endogenous hepatitis B core antigen to prime cytotoxic T lymphocytes. J Infect Dis 201:1867–1879
Oliviero B, Varchetta S, Paudice E et al (2009) Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology 137:1151–1160 1160.e1–e7
Pal S, Sullivan DG, Kim S et al (2006) Productive replication of hepatitis C virus in perihepatic lymph nodes in vivo: implications of HCV lymphotropism. Gastroenterology 130:1107–1116
Paladino N, Fainboim H, Theiler G et al (2006) Gender susceptibility to chronic hepatitis C virus infection associated with interleukin 10 promoter polymorphism. J Virol 80:9144–9150
Parren PW, Burton DR (2001) The antiviral activity of antibodies in vitro and in vivo. Adv Immunol 77:195–262
Pasetto A, Frelin L, Brass A et al (2012) Generation of T-cell receptors targeting a genetically stable and immunodominant cytotoxic T-lymphocyte epitope within hepatitis C virus non-structural protein 3. J Gen Virol 93(Pt 2):247–258
Pelletier S, Drouin C, Bedard N et al (2010) Increased degranulation of natural killer cells during acute HCV correlates with the magnitude of virus-specific T cell responses. J Hepatol 53:805–816
Penna A, Pilli M, Zerbini A et al (2007) Dysfunction and functional restoration of HCV-specific CD8 responses in chronic hepatitis C virus infection. Hepatology 45:588–601
Perrella A, Atripaldi L, Bellopede P et al (2006a) Flow cytometry assay of myeloid dendritic cells (mDCs) in peripheral blood during acute hepatitis C: possible pathogenetic mechanisms. World J Gastroenterol 12:1105–1109
Perrella A, Vitiello L, Atripaldi L et al (2006b) Elevated CD4+/CD25+ T cell frequency and function during acute hepatitis C presage chronic evolution. Gut 55:1370–1371
Persico M, Capasso M, Persico E et al (2006) Interleukin-10—1082 GG polymorphism influences the occurrence and the clinical characteristics of hepatitis C virus infection. J Hepatol 45:779–785
Piazzolla G, Tortorella C, Schiraldi O, Antonaci S (2000) Relationship between interferon-gamma, interleukin-10, and interleukin-12 production in chronic hepatitis C and in vitro effects of interferon-alpha. J Clin Immunol 20:54–61
Poynard T, Yuen MF, Ratziu V et al (2003) Viral hepatitis C. Lancet 362:2095–2100
Radkowski M, Gallegos-Orozco JF, Jablonska J et al (2005) Persistence of hepatitis C virus in patients successfully treated for chronic hepatitis C. Hepatology 41:106–114
Radziewicz H, Ibegbu CC, Fernandez ML et al (2007) Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol 81:2545–2553
Rehermann B (2009) Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. J Clin Invest 119:1745–1754
Reiser M, Marousis CG, Nelson DR et al (1997) Serum interleukin 4 and interleukin 10 levels in patients with chronic hepatitis C virus infection. J Hepatol 26:471–478
Riezu-Boj JI, Larrea E, Aldabe R et al (2011) Hepatitis C virus induces the expression of CCL17 and CCL22 chemokines that attract regulatory T cells to the site of infection. J Hepatol 54:422–431
Rigopoulou EI, Abbott WG, Haigh P et al (2005) Blocking of interleukin-10 receptor—a novel approach to stimulate T-helper cell type 1 responses to hepatitis C virus. Clin Immunol 117:57–64
Rushbrook SM, Ward SM, Unitt E et al (2005) Regulatory T cells suppress in vitro proliferation of virus-specific CD8+ T cells during persistent hepatitis C virus infection. J Virol 79:7852–7859
Sallberg M, Frelin L, Diepolder H et al (2009) A first clinical trial of therapeutic vaccination using naked DNA delivered by in vivo electroporation shows antiviral effects in patients with chronic hepatitis C. J Hepatol 50:S18–S19
Schlaphoff V, Lunemann S, Suneetha PV et al (2011) Dual function of the NK cell receptor 2B4 (CD244) in the regulation of HCV-specific CD8+ T cells. PLoS Pathog 7:e1002045
Schulte D, Vogel M, Langhans B et al (2009) The HLA-E(R)/HLA-E(R) genotype affects the natural course of hepatitis C virus (HCV) infection and is associated with HLA-E-restricted recognition of an HCV-derived peptide by interferon-gamma-secreting human CD8(+) T cells. J Infect Dis 200:1397–1401
Seifert U, Liermann H, Racanelli V et al (2004) Hepatitis C virus mutation affects proteasomal epitope processing. J Clin Invest 114:250–259
Semmo N, Day CL, Ward SM et al (2005) Preferential loss of IL-2-secreting CD4+ T helper cells in chronic HCV infection. Hepatology 41:1019–1028
Shen T, Chen X, Chen Y et al (2010) Increased PD-L1 expression and PD-L1/CD86 ratio on dendritic cells were associated with impaired dendritic cells function in HCV infection. J Med Virol 82:1152–1159
Shimizu YK, Hijikata M, Iwamoto A et al (1994) Neutralizing antibodies against hepatitis C virus and the emergence of neutralization escape mutant viruses. J Virol 68:1494–1500
Shin EC, Seifert U, Kato T et al (2006) Virus-induced type I IFN stimulates generation of immunoproteasomes at the site of infection. J Clin Invest 116:3006–3014
Sugimoto K, Ikeda F, Stadanlick J et al (2003) Suppression of HCV-specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology 38:1437–1448
Takahashi K, Asabe S, Wieland S et al (2010) Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc Natl Acad Sci USA 107:7431–7436
Taniguchi M, Harada M, Kojo S et al (2003) The regulatory role of Valpha14 NKT cells in innate and acquired immune response. Annu Rev Immunol 21:483–513
Thimme R, Oldach D, Chang KM et al (2001) Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med 194:1395–1406
Thomas E, Feld JJ, Li Q et al (2011) Ribavirin potentiates interferon action by augmenting interferon-stimulated gene induction in hepatitis C virus cell culture models. Hepatology 53:32–41
Timm J, Lauer GM, Kavanagh DG et al (2004) CD8 epitope escape and reversion in acute HCV infection. J Exp Med 200:1593–1604
Tsai SL, Liaw YF, Chen MH et al (1997) Detection of type 2-like T-helper cells in hepatitis C virus infection: implications for hepatitis C virus chronicity. Hepatology 25:449–458
Tsai SL, Chen YM, Chen MH et al (1998) Hepatitis C virus variants circumventing cytotoxic T lymphocyte activity as a mechanism of chronicity. Gastroenterology 115:954–965
Tseng CT, Klimpel GR (2002) Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J Exp Med 195:43–49
Uebelhoer L, Han JH, Callendret B et al (2008) Stable cytotoxic T cell escape mutation in hepatitis C virus is linked to maintenance of viral fitness. PLoS Pathog 4:e1000143
Ulsenheimer A, Gerlach JT, Gruener NH et al (2003) Detection of functionally altered hepatitis C virus-specific CD4 T cells in acute and chronic hepatitis C. Hepatology 37:1189–1198
Urbani S, Amadei B, Fisicaro P et al (2006a) Outcome of acute hepatitis C is related to virus-specific CD4 function and maturation of antiviral memory CD8 responses. Hepatology 44:126–139
Urbani S, Amadei B, Tola D et al (2006b) PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol 80:11398–11403
Vidal-Castineira JR, Lopez-Vazquez A, Diaz-Pena R et al (2010) Effect of killer immunoglobulin-like receptors in the response to combined treatment in patients with chronic hepatitis C virus infection. J Virol 84:475–481
Wedemeyer H, He XS, Nascimbeni M et al (2002) Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J Immunol 169:3447–3458
Weiner AJ, Paliard X, Selby MJ et al (2001) Intrahepatic genetic inoculation of hepatitis C virus RNA confers cross-protective immunity. J Virol 75:7142–7148
Wertheimer AM, Bakke A, Rosen HR (2004) Direct enumeration and functional assessment of circulating dendritic cells in patients with liver disease. Hepatology 40:335–345
Woitas RP, Ahlenstiel G, Iwan A et al (2002) Frequency of the HIV-protective CC chemokine receptor 5-Delta32/Delta32 genotype is increased in hepatitis C. Gastroenterology 122:1721–1728
Woollard DJ, Grakoui A, Shoukry NH et al (2003) Characterization of HCV-specific Patr class II restricted CD4+ T cell responses in an acutely infected chimpanzee. Hepatology 38:1297–1306
Yamagiwa S, Gray JD, Hashimoto S et al (2001) A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J Immunol 166:7282–7289
Yamagiwa S, Matsuda Y, Ichida T et al (2008) Sustained response to interferon-alpha plus ribavirin therapy for chronic hepatitis C is closely associated with increased dynamism of intrahepatic natural killer and natural killer T cells. Hepatol Res 38:664–672
Yao ZQ, Nguyen DT, Hiotellis AI et al (2001) Hepatitis C virus core protein inhibits human T lymphocyte responses by a complement-dependent regulatory pathway. J Immunol 167:5264–5272
Yao ZQ, Eisen-Vandervelde A, Waggoner SN et al (2004) Direct binding of hepatitis C virus core to gC1qR on CD4+ and CD8+ T cells leads to impaired activation of Lck and Akt. J Virol 78:6409–6419
Yoon JC, Shiina M, Ahlenstiel G et al (2009) Natural killer cell function is intact after direct exposure to infectious hepatitis C virions. Hepatology 49:12–21
Zeremski M, Petrovic LM, Talal AH (2007) The role of chemokines as inflammatory mediators in chronic hepatitis C virus infection. J Viral Hepat 14:675–687
Zhang Y, Liu Y, Moxley KM et al (2010) Transduction of human T cells with a novel T-cell receptor confers anti-HCV reactivity. PLoS Pathog 6:e1001018
Zhang Y, Ma CJ, Ni L et al (2011a) Cross-talk between programmed death-1 and suppressor of cytokine signaling-1 in inhibition of IL-12 production by monocytes/macrophages in hepatitis C virus infection. J Immunol 186:3093–3103
Zhang Y, Ma CJ, Wang JM et al (2011b) Tim-3 negatively regulates IL-12 expression by monocytes in HCV infection. PLoS ONE 6:e19664
Zimmermann M, Flechsig C, La Monica N et al (2008) Hepatitis C virus core protein impairs in vitro priming of specific T cell responses by dendritic cells and hepatocytes. J Hepatol 48:51–60
Acknowledgments
E. D. Brenndőrfer was supported by grants from the Ruth and Richard Julin Foundation, the Goljes Memorial Fund, the Lars Hierta Memorial Foundation, the Swedish Royal Academy of Sciences, the Professor Nanna Svartz Fund and from Karolinska Institutet. M. Sällberg was supported by the Swedish Cancer Society, the Swedish Research Council and the Stockholm County Council.
Conflict of interest
None to declare.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Brenndörfer, E.D., Sällberg, M. Hepatitis C Virus-Mediated Modulation of Cellular Immunity. Arch. Immunol. Ther. Exp. 60, 315–329 (2012). https://doi.org/10.1007/s00005-012-0184-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-012-0184-z