
Abstract
The separation of fine mineral particles, especially using environmentally friendly approaches, is one of the main problems in the processing of low-grade ores and the re-processing of mining tailings. This work assesses the potential of biosurfactants as collectors in the flotation of ultrafine (smaller than 20 μm) particles of hematite and malachite. As biosurfactants, we test acetylated acidic (ac-ASL) and lactonic sophorolipids (ac-LSL). In addition, n-dodecyl-β-D-maltoside (DDM) is used as a model non-ionic alkyl disaccharide surfactant, and sodium oleate (NaOl) is used as a reference. The biosurfactants are characterized using surface tension and foam analysis. The interaction of the minerals with the surfactants is characterized using zeta potential, solubility, and single-mineral flotation. The collecting properties of the surfactants are compared for the ultrafine (− 20 μm) and coarser (38–90 µm) particle size in the two-mineral flotation of hematite and malachite against quartz. The ultrafine particle size improves the grade in the oleate flotation of hematite, as well as the grades in the DDM flotation of hematite and malachite, which is explained by the weak interactions of the metal oxides with fatty acids and DDM. At the same time, the flotation with ac-LSL and ac-ASL is highly tolerant to the ultrafine particle size. These results indicate that biosurfactants are an interesting alternative to conventional petroleum-based surfactants in the flotation of Fe and Cu oxides. Moreover, a proper selection of surfactants can help combat the problem of fines.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The supply risks and the increasing demand for minerals critical for the energy transition have stimulated their domestic production from both primary (ores) and secondary (waste) resources. This economic shift has in turn spurred more efforts toward the development of novel technological solutions to boost resource efficiency and drastically reduce the carbon footprint of flotation.
Flotation is a physicochemical method that has widely been employed for separating minerals from ores for more than a century. Its selectivity is achieved using surfactants (collectors) that make the target mineral particles hydrophobic. During the long history of flotation, tremendous progress has been achieved in developing collectors for different types of minerals, along with a general framework of the collector-mineral interactions [1,2,3,4]. However, most conventional collectors originate from non-renewable resources (petrochemicals). In addition, some of them or products of their chemical degradation are hazardous and pose risks to human health and the environment. The quintessential examples are xanthates [5, 6] and amines [7]. Therefore, there is a growing trend to extend the share of green reagents in flotation [8,9,10,11]. The adjective “green,” or “eco-friendly,” implies that the reagents comply across their life cycle with a set of principles of green chemistry, including production from renewable feedstocks, minimization of hazards, and benign degradation after the use [12, 13]. It has been suggested that the true “greenness” of collectors should be measured over four major life cycle stages, (i) manufacturing, (ii) storage and transportation, (iii) usage and processing, and (iv) post-usage and processing, for which most reagents are still unavailable [14].
Flotation already employs two important classes of green reagents—fatty acids and polysaccharides—which are mainly used as collectors and depressants of iron oxides, respectively [3, 15, 16].
The fatty acid flotation of iron oxides depends on pH, with a maximum being at pH 6–8 [3, 16, 17]. This maximum can be explained by the physisorption-chemisorption model [18, 19]. In this model, fatty acids are chemisorbed via one O atom of their carboxylate groups. The electrostatic repulsion between the co-adsorbed carboxylate groups is screened by co-adsorbed neutral molecules, which improves the packing density and hydrophobicity of the surfactant layer. Chemisorbed fatty acids become more important at pH above the iso-electric point (IEP) of iron oxides, but their concentration rapidly declines due the relatively weak adsorption strength. Fatty acids have also commercially been used in the flotation of oxide copper minerals from siliceous ores, where “copper oxide” is a generic term for non-sulfide copper minerals [20]. In contrast to iron oxides, the adsorption of fatty acids on Cu oxides is typically explained in terms of chemisorption [21, 22], though, to the best of our knowledge, the adsorption mechanism has not been studied thoroughly. However, the application of fatty acids as collectors is limited by their relatively low selectivity [4, 23]. As a result, there is ongoing research to find more selective alternatives, mainly among chelating petroleum-based surfactants [21].
Starch (a natural polysaccharide) is a common depressant of iron oxides. Spectroscopic results suggest that starch interacts with hematite (α-Fe2O3) via complexation with the surface Fe-OH groups [24], which can explain the preferable chemisorption of starch on hematite vs quartz [25]. In the hematite-starch-oleate system, starch covers the hematite surface almost fully, thereby preventing the adsorption of oleate [26], which suggests that starch interacts with hematite more strongly than oleate does. The specific interaction of starch with hematite agrees with the dependence of the affinity of hematite to starch on the hematite crystallographic plane, which decreases in the order of (1 1 \(\overline{2 }\) 0) > (1 \(\overline{1 }\) 0 0) > (0 0 1 1) [27]. This dependence has been explained by multisite complexation between starch molecules and oxygen surface sites. In contrast, a non-ionic sugar-based surfactant n-dodecyl-β-d-maltoside (DDM) is physisorbed on hematite through hydrogen bonding with surface hydroxyl groups [28,29,30], which can be associated with the strong hydration of the maltose headgroup [31]. In contrast, DDM is likely to chemisorb on hematite at a highly alkaline pH [30]. Interestingly, DDM adsorbs on hematite, but very little on silica [28].
At the same time, there is very limited knowledge about the potential of biosurfactants as green collectors, not to mention a general framework that classifies biosurfactants in terms of the minerals that they can float [8, 9, 11, 13, 32]. Biosurfactants are surfactants that are either extracted from plants or produced by biocatalysis (using enzymes) or non-pathogenic microbial processes from renewable biological sources (biomass) [33, 34]. These surfactants are increasingly introduced into personal care, pharma, detergency, and cosmetics due to their non-hazardous nature as well as higher biocompatibility and biodegradability compared to conventional surfactants.
The most studied biosurfactants are glycolipids, which are built of different sugar groups linked to ß-hydroxy fatty acids [34]. Their sugar headgroups and auxiliary chains have multiple O-donor atoms that can coordinate to a metal center. The large hydrophilic sugar groups can improve the solubility of biosurfactants and their complexes with metal ions and hence make biosurfactants more tolerant to higher levels of total dissolved solids compared to conventional surfactants. An example is sophorolipids which can be produced with various molecular structures and easily functionalized [33]. Another important benefit of sophorolipids is that they hold the largest share of the biosurfactant market while their production from food waste improves their economic and environmental sustainability [35, 36].
So far, sophorolipids have been studied in detail only in solutions and at the air–water interfaces [33], while their interaction with minerals has been addressed only in a handful of works [32, 37,38,39,40]. In particular, it has been concluded that an asymmetrical bipolar (“bola”) acidic sophorolipid (ASL) renders copper sulfides hydrophobic through the dissolution–precipitation mechanism that forms hydrophobic Cu-ASL precipitates on the sulfide surface [32]. The hydrophobicity of the precipitates has been explained by a loop-like chelating coordination of both the sophorose and carboxylate headgroups of ASL to Cu cations. This mechanism allows ASL to float copper sulfides from a pyrite-poor copper ore similarly to or even better than conventional thiol collectors [13]. The chelating structure of the Cu-ASL complexes can explain the strong leaching activity of ASL toward Cu sulfides and metallic Cu [32, 38]. In contrast, only the carboxylate headgroup of ASL coordinates to iron oxide nanoparticles during the precipitation of iron oxide nanoparticles in ASL solutions, while the sophorose headgroup is left free which renders the nanoparticles hydrophilic [39]. The different coordination of ASL to copper sulfides and iron oxides can underpin its selectivity to Cu and Fe oxides. Thus, given that O-donors (“oxyhydrils”) are conventional collectors of iron and copper oxides [3, 4, 41, 42], it is of interest to test the performance of sophorolipids in the flotation of these minerals, starting from model synthetic systems.
Apart from selectivity, a common problem in the flotation of iron and copper oxides is the so-called problem of fines, that is, difficulty to float fine (smaller than 38 m) particles using standard schemes. Fine particles are naturally produced in significant amounts during the grinding of non-sulfide ores due to their mechanical properties. In addition, the gradual depletion of easy-to-extract high-grade ores has steered the mineral processing industry toward low-grade ores and secondary resources such as flotation tailings [43,44,45,46]. Due to the complex intergrowth and ultrafine-grained dissemination of mineral phases, these resources need to be ground down to ultrafine sizes to liberate valuable minerals.
The negative impact of fines on flotation is caused by several chemical and physical mechanisms [15, 47, 48]. In particular, fine particles have lower collision rates with bubbles, which results in slower flotation kinetics [15]. Due to their higher specific surface areas, fine particles can consume a higher amount of the collector per unit weight and be more reactive (change its redox state and dissolve faster) compared to the particles with the conventional flotation size (38–150 m) [15, 48]. The entrainment of fine particles in the froth and their heterocoagulation with other minerals can drop selectivity and increase mineral loss [47, 48]. Entrainment is the mechanical (non-selective) mass transfer of suspended particles to the water between bubbles which brings them to the collected flotation froth. This process can involve hydrophilic particles of other minerals and is linked to the amount of water carried in the froth and the foam stability [49]. Fine particles can also be attached to and monopolize the surface of the air bubbles, preventing them from attaching to other minerals [15]. Even though there are technical solutions to the problem of fines [4, 15, 47, 50], flotation chemistry can be a part of these solutions. At the same time, there is limited information on how different collectors respond to the fine particle size.
Herein, we study the effect of particle size on the efficiency of two sophorolipids as collectors of hematite α-Fe2O3 and malachite CuCO3·Cu(OH)2 in synthetic two-mineral systems with quartz with an outlook for their application to real mineral processing systems which are yet to be defined. Hematite is the main resource of iron on Earth [16]. Malachite is the most common copper oxide mineral. It is usually associated with the weathering of copper ores [20, 21]. Quartz is used as a model gangue mineral given its abundance in iron oxide and malachite-containing copper oxide ores [16, 20, 23]. To delineate the effect of particle size, we compare the flotation of ultrafine (− 20 m) and larger (+ 38 − 90 µm) particles. Even though the larger particles are in the conventional size range of flotation, we call them “coarse” hereafter to distinguish them from the ultrafine particles.
The sophorolipids studied are acetylated acidic sophorolipid (ac-ASL) and acetylated lactonic sophorolipid (ac-LSL). Ac-ASL is an anionic surfactant with an acetylated sophorose headgroup attached to the C17 carbon of oleic acid (Fig. 1a). In contrast, ac-LSL is non-ionic as it has a closed C17 carbon chain with the carboxylate group of oleic acid linked to the sophorose headgroup through the ester bond (Fig. 1b). As a result, ac-LSL has a stiffer geometry and is less soluble in water than ac-ASL. Lactonic sophorolipids have lower surface tension and critical micelle concentration (CMC) and stronger bactericide properties, while acidic sophorolipids are better foamers [51].

Surfactants used in this study: a acetylated acidic sophorolipid (ac-ASL); b acetylated lactonic sophorolipid (ac-LSL); c n-dodecyl-β-D-maltoside (DDM); d sodium oleate (NaOl)
For comparison purposes, we included in our study sodium oleate (NaOl) (Fig. 1d) and DDM (Fig. 1c). NaOl is used as both a reference and a model fatty acid given that fatty acids are among the main collectors of iron and copper oxides [3, 16, 17]. Moreover, oleate is a structural unit of both ac-ASL and ac-LSL. Hence, their comparison with NaOl could inform us about the involvement of the carboxylate and sophorose groups into the phenomena studied. DDM (Fig. 1c) is used for comparison with ac-LSL as both are non-ionic disaccharides. However, the longer hydrocarbon chain, its fixed loop-like shape, and acetylation of the sophorose headgroup make ac-LSL more hydrophobic and rigid, which is expected to affect solubility of ac-LSL and its self-assembly at interfaces [31, 52]. The three ester groups can also make ac-LSL chemically reactive. It has been reported that the acetyl groups and ester bonds are irreversibly hydrolyzed during long-term storage at pH values higher than 7.0–7.5 [51]. Finally, the interaction of DDM with hematite has already been studied [28,29,30, 53], which can help interpret our results. As research methods, we use static surface tension measurements, dynamic foam analysis, ζ-potential, and single- and two-mineral flotation.
2 Materials and Methods
2.1 Materials
Diacetylated ac-LSL and diacetylated ac-ASL were provided by Bio Base Europe Pilot Plant, Ghent, Belgium. The chemical structures of ac-ASL and ac-LSL are confirmed by FTIR as described in the “Supplementary Information” and shown in Fig. S1. Sodium oleate (NaOl) was produced by J. T. Baker and DDM was produced by Avanti Polar Lipids, Alabaster, Alabama. All the surfactants were used without further purification. Milli-Q water was used for all experiments. NaOH and HNO3 were used for pH adjusting, and NaNO3 and KCl were used as background electrolyte solutions. These reagents were purchased from VWR™.
2.2 Minerals
Hematite was provided by Rana Gruber, Norway. Malachite was purchased from Richard Tayler Minerals, UK. The origin of quartz was Kyshtym, South Ural, Russia.
The minerals were crushed and milled in a planetary mill with a stainless-steel jar and stainless steel 30-mm balls, followed by wet sieving to obtain a − 20-µm fraction. To remove contaminations, − 20-µm hematite and quartz particles were cleaned in a 0.01 M HCl solution followed by washing in Milli-Q water until neutral pH is reached. The cleaning procedure for malachite was eliminated due to the mineral dissolution at acidic pH. The coarse fraction (+ 38 − 90 m) was prepared by dry sieving, followed by the cleaning procedure used for the fine fraction. The samples were split by Retsch mini splitter into three batches—two for flotation (for duplication) and one for analysis.
2.3 Surfactant Solutions
A 1-g/L stock solution of ac-ASL was prepared by dissolving the surfactant in water. The stock solution of ac-ASL had pH 5.6. Ac-LSL was dissolved at a concentration of 0.6 g/L in a 0.002 M NaOH solution to increase its solubility. We limited pH of the stock solution by 9.5 which can cause de-acetylation of ac-LSL.
2.4 Flotation
Flotation tests were conducted using a XFG II flotation machine at surfactant concentrations of 50 µM. These tests were conducted without adding a frother. DowFrother™ 200 at a concentration of 50 µM was used only in single-mineral flotation to create a flotation baseline without surfactants. As NaOl does not generate a measurable froth at 50 μM, we collected the hydrophobic layer of particles on the surface of the flotation cell. In the other cases, we collected the froth.
In the single-mineral flotation of ultrafine particles, 1 g of particles was placed into the 100-mL flotation cell. Then, 90 mL Milli-Q water was added, and the pulp was stirred for 2 min. After 2 min, pH was adjusted to the required value and the surfactant solution at the same pH was added. pH was readjusted after 4 min followed by the flotation in 1 min. The reported pH values were measured under flotation conditions. All these steps were implemented under continuous stirring at 1500 rpm. Flotation time was 2 min at an airflow rate of 100 L/h (approximately 1650 mL/min). Floated and non-floated fractions were collected and filtered using filter paper of a 589/3 grade followed by drying in a heating chamber. The weight of the dried particles was used to determine recoveries. The grade was measured using portable X-ray fluorescence (XRF). Since this method has limited accuracy, each sample was measured three times and the results were averaged. All the flotation tests were repeated twice, and the average values are reported. The same procedure was repeated for mix-mineral flotation. The ratio of minerals was 1:1, where 1 g of each mineral was used for the ultrafine fraction, and 1.5 g of each for the coarse fraction.
2.5 Surface Tension
Static surface tension was measured by Du Nouy’s ring method using a Biolin Scientific Sigma 702 instrument. The ring was repeatedly flamed until it glowed red-hot in an ethanol flame and washed with deionized water to ensure the complete removal of impurities. The instrument was first calibrated with water (72 ± 1 mN/m). The stock solutions of ac-LSL or ac-ASL were adjusted to specific pH, and then added to water with the same pH. The pH of the final solution did not drift by more than 0.2 pH. Each reported surface tension data point is an average of 10 measurements in the same solution. Each set of experiments was repeated in two solutions. Differences between the duplicates were insignificant.
2.6 Foaming Properties
Foaming properties were studied by a Kruss Dynamic Foam Analyzer DFA 100. The surfactant solutions were prepared in 0.001 M KCl at pH 5 and pH 10 at a surfactant concentration of 50 µM. KCl was added to provide the solution with the conductivity needed for the analyzer sensors. The volume of each solution sample was 50 mL, and each experiment was repeated twice.
2.7 Solubility
To study the effect of the surfactants on the solubility of ultrafine hematite and malachite, we measured the Fe and Cu concentrations in the supernatant after conditioning 0.5 g of the mineral particles in 50 mL of the 50 M surfactant solutions at pH 5.2 and pH 9.7 for 3 h.
2.8 FTIR Spectroscopy
ATR FTIR spectra of bulk dry surfactants were recorded using a Bruker Vertex 80v FTIR spectrometer. A typical spectrum was an average of 200 scans measured at a 4 cm−1 resolution.
2.9 ICP-MS
Metal ions in the surfactant solutions before and after interaction with minerals were analyzed using ICP-HR-MS Element 2 (Thermo) equipped with an auto-sampler SC2 DX dust-covered with a ULPA filter. For these experiments, 0.5 g of ultrafine mineral particles was added to 50 mL of a 50 μM surfactant solution prepared at different pH. pH was readjusted after 30 min, and the dispersions were conditioned for 3 h on a shaking table. The resulting supernatant was filtered through a syringe membrane (0.2 µm), digested by HNO3, and analyzed by ICP-MS. The error was no more than 10% as estimated by duplicating random points.
2.10 Zeta Potential
The zeta (ζ) potential of minerals in water and surfactant solutions was measured using a Malvern ZetaSizer Nano Z (laser Doppler micro-electrophoresis) instrument. Mineral particles were first dispersed in a 0.001 M NaNO3 background solution at 0.1 wt.% by sonication for 15 min. This dispersion was split into five parts to prepare dispersions with the four surfactants and one blank (mineral particles in 0.001 M NaNO3). Then, the prepared samples were split into seven 30-mL samples followed by pH adjustment of each sample. The pH-adjusted samples were equilibrated on a shaking table overnight. Afterward, pH was readjusted with 0.01 M NaOH and HNO3 solutions. Final pH was measured for each sample before measuring its ζ-potential. As the material settings in ZetaSizer, we used refractive indices of 3.0, 3.6, and 1.5 and absorption of 0.8, 0.7, and 0.2 for hematite, malachite, and quartz respectively. ζ-potential of NaOl and ac-LSL was measured in water using a refractive index of 1.0 and an absorption index of 0.1. Each ζ-potential data point is an average of 3 replicate points where each point was an average of 10 scans each. The ζ-potential uncertainty is ± 2 mV and the pH uncertainty is within 0.1 pH.
2.11 The BET (Brunauer, Emmett, and Teller) Surface Area and Particle Size Distribution
The BET surface areas were measured using a Micromeritics Tristar 3000 Analyzer. All samples were degassed at 250 °C under helium flow for 3 h. The particle size distribution was measured by the dynamic light scattering method using a Mastersizer 3000E.
2.12 XRD
X-ray diffractometry (XRD) was used to determine the phase composition of the minerals. For the quantitative phase analysis, ultrafine particles were additionally ground in ethanol by agate milling rods. Bruker D8 Advance Series 2 XRD equipment was used. The samples were scanned in the θ range of 5–80° under the Co Kα radiation.
2.13 SEM
The morphology analysis of mineral particles was performed using a Hitachi SU6600 field emission SEM. A copper double-sided tape was used as a support. Materials were placed on individual tapes and coated with carbon to eliminate charging.
3 Results and Discussion
3.1 Characterization of Minerals
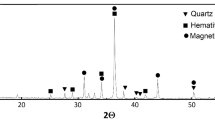
The XRD analysis shows that malachite and quartz used in our study are mineralogically pure (> 99%) (Fig. 2). However, the hematite diffractogram has a small peak at 31° corresponding to a quartz impurity of about 1%. The particle size distribution of the ultrafine (− 20 µm) fraction is from 0.8 to 24 μm (Table 1). The specific surface area of ultrafine hematite and ultrafine quartz is 1.5 m2/g, while that of ultrafine malachite is three times larger (Table 1). The specific surface areas of coarse (+ 38 − 90 µm) hematite and malachite particles are 0.31 m2/g and 0.38 m2/g, respectively, closer one to another than in the case of ultrafine particles.

XRD of (black line) ultrafine hematite, (blue line) ultrafine malachite, and (red line) ultrafine quartz. Malachite and quartz have a purity > 99%. Hematite ultrafine particles have ca. 1% quartz impurity
Ultrafine hematite particles have irregular morphologies and cleavage surfaces with sharp edges (Fig. 3a), while coarse particles have rounder shapes (Fig. 3b). Both fractions contain submicron particles (“slimes”), the percentage of which is higher for the coarse particles (Table 1). The ultrafine fraction of malachite includes 10–20-µm particles with a fibrous structure and multistep rectangular edges, as well as a large portion of micron and submicron particles with shapes of broken rectangular rods (Fig. 3c). The coarse fraction of malachite contains large particles with interlayered ruptured-plate morphology typical of malachite, which comes along with an admixture of micron and submicron particles (Fig. 3d). Ultrafine quartz particles (Fig. 3e, 3f) have shapes similar to those of the ultrafine hematite particles (Fig. 3a).

SEM images of a ultrafine hematite, b coarse hematite, c ultrafine malachite, d coarse malachite, e, f ultrafine quartz particles. The ultrafine particles are smaller than 20 µm. The coarse particles are in the 38–90-µm size range
3.2 Surface Tension and Foaming Properties
We measured the surface tension of ac-ASL and ac-LSL to characterize their propensity to self-assemble in solution and at the air–water interface. The interfacial properties of DDM and NaOl have been studied earlier [31, 54, 55]. At low ionic strengths, CMC and the minimum surface tension of DDM in water is 0.15–0.18 mM and 35.5 mN/m, respectively [31, 54]. CMC of NaOl (defined as the inflection point of the surface tension) increases from 17 to 90 M in the pH range from 7 to 12, while the minimum surface tension varies non-monotonically between 26.4 and 30.1 mN/m [55].
The surface tension of ac-ASL depends on pH, while there is no significant effect of pH on the surface tension of ac-LSL (Fig. 4). These results are consistent with the presence of the carboxylate headgroup in the former and the non-ionic character of the latter (Fig. 1a, b). The CMC of ac-ASL of 0.18 mM at pH 6 and pH 10 is higher than that of non-acetylated ASL of 40 μM and 0.10 mM at pH 7 and pH 11, respectively (Table 2) [32]. It follows that acetyl groups hamper the self-assembly of ASL in solution and at the air–water interface. In contrast to CMC of non-acetylated ASL [32], CMC of ac-ASL does not depend on pH (Fig. 4a). This difference suggests that acetyl groups impose steric constraints on the carboxyl group to contact water when ac-ASL is most densely packed at the air–water interface.

Surface tension of a ac-ASL, and b ac-LSL at pH 6 ± 0.3 and 10 ± 0.2. Dashed lines show surface tension at a concentration of 50 µM used in our study
Since ac-LSL is more hydrophobic than ac-ASL, it is expected to have lower values of CMC and the minimum surface tension than ac-ASL. In fact, CMC of diacetylated LSL at pH 7.4 and 25 °C has been reported to be 20 mg/L (29 M) [56]. A similar value of 15 mg/L has been obtained at pH 7 and 25 °C for LSL with an unspecified degree of acetylation [52]. In contrast, CMC of another LSL, also with the unknown acetylation degree, is 73 mg/L (0.12 mM) at pH 8.94 and 20 °C [57], which is close to CMC of crude sophorolipids [52]. However, the surface tension curve of the ac-LSL sample used in our study does not reach the minimum plateau at concentrations up to 0.4–0.6 mM where the solution becomes visually turbid (Fig. 4b). Hence, instead of the classical adsorption/desorption mechanism at the air–water interface, ac-LSL used in our study is likely to follow the colloidal interfacial adsorption similar to high-molecular-weight biosurfactants [33]. In fact, the formation of a colloidal solution by ac-LSL at 50 μM is detected by ζ-potential measurements (Section 3.3.2). Another reason can be the de-acetylation of ac-LSL stock solution at pH 9.5. At the same time, the lowest surface tension achieved by ac-LSL in our study, which is 39 and 38 mN/m at pH 6 and pH 10, respectively (Fig. 4b), is within the range of 34–41 mN/m reported for LSL [52, 56, 57].
The foaming properties of surfactants are also important for flotation as this process is more effective with less stable wet foams. Foaming properties can affect flotation as they control the attachment of hydrophobic particles to bubbles, the entrainment of the foreign particles with the aqueous solution, and their entrapment between particles attached to air bubbles. Hence, we studied the foaming properties (foamability and foam stability) of ac-ASL, ac-LSL, DDM, and NaOl. The study was performed at the surfactant concentration of 50 μM, which is well below CMC of ac-ASL and DDM but within the CMC range of NaOl (Section 3.2). We also assessed the liquid content and structure of their foams.
NaOl does not produce a stable and measurable foam at a concentration of 50 μM and pH 5 and 10. In contrast, DDM has the strongest foamability and foam stability at pH 10.9 (Fig. 5a and Table 3). Next in the ranking is ac-LSL at pH 5.4. The foaming properties of ac-ASL and ac-LSL at pH 10 are much weaker than at pH 5.3 and cannot be assessed by the foam analyzer under the same foam generation conditions. Therefore, we measured their foaming properties at pH 10 at a twice as high flow rate and a half interval as compared to the rest surfactants. The purpose of these measurements was to characterize the foam structures, given that a change in the airflow rate did not affect the bubble size distribution of foams generated by rhamnolipids [58].

Foam properties of 50 μM ac-ASL, ac-LSL, and DDM: a the foam heights as a function of time (dash lines mark the airflow stop); b the structure of the foams when airflow was stopped (30 s for ac-ASL and ac-LSL at pH 10, and 60 s for the rest). The scale bar is 1 mm
In terms of the liquid content, foams generated by ac-LSL are wet at both pH, while foams generated by DDM are dry (Table 3). As shown in Fig. 5b, ac-LSL at pH 10.1 generates bubbled water rather than an actual foam, consistent with the poor foam properties of this surfactant. Similarly, the foam produced by ac-ASL at pH 9.9 has a higher liquid content than at pH 5.3 (Fig. 5b). More foam images are reported in Figs. S3 and S4.
3.3 Mineral-Surfactant Interaction
In this section, we study the interaction of ac-ASL, ac-LSL, DDM, and NaOl with ultrafine hematite and malachite particles using their effect on the ζ-potential, solubility, and single-mineral flotation properties of the metal oxides and quartz. This study was performed at a surfactant concentration of 50 M at which the single-mineral flotation of hematite and malachite with NaOl has a maximum [17, 22, 59, 60].
3.3.1 ζ-Potential
Ζ-potential is used to determine how the four surfactants affect the surface charge of ultrafine hematite, malachite, and quartz. It is expected that their adsorption will cause a change in surface charge. In addition, ionic surfactants will shift IEP of the minerals. Surface charge controls important physical-chemistry phenomena underpinning flotation, such as the surfactant adsorption on the mineral surface, the heterocoagulation of different types of mineral particles in the pulp, the dispersion-coagulation of ultrafine mineral particles, and the attachment of particles to bubbles [61]. Hence, ζ-potential can help interpret flotation results.
Figure 6a shows the ζ-potential of ultrafine hematite particles in the absence and presence of 50-µM surfactants. In water, IEP of the particles is pH ca. 6, which is below the IEP range of 6.7–9.2 reported for pure hematite [19, 62]. The lower IEP value can be explained by the quartz impurities in the hematite used in our study (Fig. 2) [62]. All the surfactants shift IEP of hematite to more acidic pH, which indicates that they adsorb on the positively charged hematite surface. The most pronounced impact is made by NaOl. It renders the hematite surface most negatively charged, shifting IEP to the lowest pH value of 2.7 (Fig. 6a). This IEP is typical of hematite in oleate solutions [63]. Surprisingly, next in the ranking is non-ionic ac-LSL. IEP of hematite in its presence is pH 3.4, while ζ-potential is less negative than in the presence of NaOl (Fig. 6a). Despite their different electrostatic properties, anionic ac-ASL and non-ionic DDM have similarly weak impact on ζ-potential. They shift IEP of hematite to pH ca. 5, while the DDM-adsorbed hematite is somewhat less negatively charged than the ac-ASL counterpart. The effect of all the surfactants on ζ-potential diminishes at pH above 10, which suggests that their surface densities become low when the hematite surface is highly negatively charged.

ζ-potential of ultrafine (− 20 µm) a hematite, b malachite, and c quartz particles in the absence and presence of 50 µM (▼) ac-LSL, (▪) ac-ASL, (
 ) DDM, and (◼) NaOl in 0.001 M NaNO3; d ζ-potential of surfactant solutions (▼) ac-LSL and (■) NaOl at a concentration of 50 µM. The vertical error bars do not exceed 4 mV and 6 mV for the minerals and the surfactants, respectively
) DDM, and (◼) NaOl in 0.001 M NaNO3; d ζ-potential of surfactant solutions (▼) ac-LSL and (■) NaOl at a concentration of 50 µM. The vertical error bars do not exceed 4 mV and 6 mV for the minerals and the surfactants, respectively
ζ-potential of malachite was measured at pH above 6 because this mineral becomes increasingly soluble at lower pH [64, 65]. Figure 6b shows that IEP of malachite in water is around 9.5, which is at the upper limit of the reported range of 8.5–9.5 [59, 64, 66, 67]. A shift of IEP to higher pH can be explained by the dissolution of malachite followed by re-adsorption of Cu(II) cations or insufficient equilibration with atmospheric CO2 [66, 68]. As in the case of hematite, NaOl renders the malachite surface most negatively charged, causing an IEP shift from pH 9.5 to ca. 6.0 (Fig. 6b), which qualitatively agrees with a previous report [59]. Even though ac-LSL causes a similar IEP shift as NaOl, the ζ-potential of malachite at pH above 6.0 is less negative (Fig. 6a). Ac-ASL reduces the positive surface charge of malachite at acidic pH less strongly compared to ac-LSL while the ζ-potential curves of malachite in the solutions of ac-ASL and ac-LSL converge at pH above 7.3. The weakest deviation of the ζ-potential of malachite is caused by DDM. It shifts IEP from 9.5 in water to pH 8.8. Thus, the impact of the four surfactants on the surface charge and IEP of hematite and malachite decreases in the order of NaOl > ac-LSL > /≈ ac-ASL > DDM. We interpret this order below.
The ζ-potential of mineral particles in oleate solutions at pH below 5 is commonly explained following Laskowski et al. by the heterocoagulation of the particles with colloids (droplets) of non-dissolved oleic acid [69]. Only 10−7.8 M of oleic acid dissolves at pH below 5 (pKa of oleic acid) reaching 50 M only at pH 9 [69]. At 50 M, IEP of oleic acid colloids is around pH 2.0 (Fig. 6d), which is in good agreement with the reported value of pH 2.5 [69]. Hence, adhered colloids of non-dissolved oleic acid dominate the surface charge of hematite and malachite at pH below 9. The negative surface charge at pH above 9 is consistent with the physisorption-chemisorption model of the fatty acid adsorption on iron (hydr)oxides [18, 19]. The relatively weak binding strength of a chemisorbed fatty acid results in the steep decline of the absorption densities at pH above IEP [17,18,19], which explains the convergence of the ζ-potential curves of hematite in the absence and presence of oleate (Fig. 6a).
The adsorption of fatty acids on malachite is currently poorly understood. The effect of NaOl on the ζ-potential of malachite has been explained by chemisorption [59]. The rationale is that malachite is sparingly soluble, while chemisorption is among the mechanisms of the oleate adsorption on salt-type (sparingly soluble) minerals. However, chemisorption does not exclude additional contributions of physisorption and precipitation of the cation-oleate complexes, which also have been proposed for the adsorption of fatty acids on salt-type minerals [70, 71].
The adsorption of non-ionic surfactants is typically interpreted in terms of physisorption [72]. The neutral surfactant layer screens surface charge and shifts the shear plane farther from the Stern plane, which reduces the absolute values of ζ-potential of carbon without significantly affecting IEP of the adsorbent [73]. However, this picture is observed neither for ac-LSL nor DDM. Given the low solubility of ac-LSL, an alternative mechanism can be surface precipitation of the surfactant colloids as in the case of NaOl. To verify this hypothesis, we measured the ζ-potential of colloids in the colloidal solution of 50 M ac-LSL. As shown in Fig. 6d, IEP of these colloids is in the pH 2.5–3.5 range. Hence, these colloids can be deposited at higher pH on the positively charged hematite particles. On this basis, we conclude that the effect of ac-LSL on the ζ-potential of the minerals at acidic pH is dominated by adhered ac-LSL colloids.
In contrast to NaOl and ac-LSL, both ac-ASL and DDM are well soluble in water. Given that DDM is physisorbed on hematite at acidic pH [28, 29], the IEP shift of hematite to pH 5 by adsorbed DDM implies that its maltose group is strongly polarized upon adsorption. At the same time, a similar effect of ac-ASL on the ζ-potential of hematite is likely caused by chemisorption and/or precipitation of ac-ASL complexes as ac-ASL can form complexes with Fe(III) ions (Section 3.3.2). Chemisorption and precipitation are also possible for the adsorption of ac-ASL and DDM on malachite at acidic pH (Section 3.3.2).
Finally, IEP of ultrafine quartz in water is pH 2–3 (Fig. 6c), which is in good agreement with reported values of pH 1.5–4 [74]. The impact of the four surfactants on ζ-potential of quartz is insignificant. Hence, in contrast to hematite and malachite, within the detection limit of ζ-potential, quartz does not adsorb these surfactants. This result agrees with the general notion that negatively charged silicates have a negligible affinity to carboxylate and sugar groups [28, 75].
The ζ-potential dependences show the pH ranges where the metal oxides and quartz have surface charges with the same sign, which would hamper their heterocoagulation (Fig. 6a–c). This feature offers an opportunity to selectively float the metal oxides from quartz, which we confirm in Section 3.4.
3.3.2 Leaching of Ultrafine Hematite and Malachite by Biosurfactants
We studied the effect of ac-ASL, ac-LSL, DDM, and NaOl on the dissolution of ultrafine hematite and malachite to delineate possible contributions of chemisorption and surface precipitation to the surfactant adsorption. Surfactants can enhance dissolution of a mineral by acting as ligands in the ligand-controlled/promoted dissolution mechanism. It starts with the chemisorption of a ligand followed by the detachment of the ligand–metal complexes from the surface [76]. In addition, surfactant-metal complexes can be formed at the mineral-solution interface followed their adsorption or precipitation on the mineral surface. Surface precipitation of surfactant-metal complexes has been observed during the interaction of octyl hydroxamate with hematite, ASL with djurleite, and NaOl with salt-type (sparingly soluble) minerals such as apatite, scheelite, calcite, and fluorite [32, 70, 71, 77]. Hence, if the adsorption of a surfactant is accompanied by an increase in the solubility of the mineral, this surfactant can potentially be chemisorbed and/or its metal complexes can precipitate on the mineral surface.
To study the effect of the surfactants on the solubility of hematite and malachite, we measured the Fe and Cu concentrations in the supernatant after conditioning 0.5 g ultrafine mineral particles in 50 mL of 50 M surfactant solutions at pH 5.2 and pH 9.7 for 3 h. As the baseline, we used the mineral particles stirred in water under the same conditions. The results are reported in Table 4.
Hematite
Anhydrous hematite is practically insoluble under oxic conditions in the pH range from 4 to 14 [77, 78]. Accordingly, ultrafine hematite releases in water at pH 5.2 only 11 g/L Fe (Table 4). The Fe concentration is not affected by non-ionic DDM and ac-LSL. In contrast, anionic ac-ASL and NaOl increase it to 17 g/L and 15 g/L, respectively. The inertness of DDM can be explained by its physisorption on hematite at acidic pH [28,29,30]. However, the converse is not necessarily true for ac-LSL. The mechanism of its adsorption calls for a separate study. The leaching activity of ac-ASL and NaOl can be attributed to the capacity of their carboxylate groups to form inner-sphere complexes with iron (hydr)oxides [18, 19, 39].
The natural solubility of hematite is almost doubled at pH 9.7 vs. pH 5.2, as follows from the Fe concentration in water of 20 g/L (Table 4). At highly alkaline pH, the FeOOH-like surface layer created by hydration is dissolved by hydroxyl ions [78, 79]:
NaOl does not have impact on the Fe concentration due to the low absorption density of oleate on hematite at this pH (Section 3.3.1). In contrast, all three sugar-based surfactants increase the Fe concentration in the order of DDM = ac-LSL < ac-ASL, the Fe concentration in the ac-ASL solution reaching 86 g/L (Table 4). Hence, these surfactants can be chemisorbed and/or precipitated at pH 9.7. This conclusion is in line with the findings that (i) sugars form strong chemical bonds with hydroxylated multivalent metals at basic pH [80, 81], (ii) DDM is chemisorbed on hematite at pH 12 [30], and (iii) starch is chemisorbed on hematite [24, 27]. The stronger leaching properties of ac-ASL compared to as-LSL at pH 9.7 are likely to be caused by two effects. Ac-ASL can form stronger complexes with Fe ions by coordinating both its headgroups as postulated earlier for the ASL complexes with Cu [32]. In addition, due to the carboxylate group, ac-ASL-Fe complexes can be more soluble compared to ac-LSL-Fe complexes.
Malachite
Ultrafine malachite is sparingly soluble in water at pH 5.2, releasing 800 g/L Cu (Table 4). The corresponding dissolution reaction reads as [65]
The natural solubility of malachite is explained by the ionic character of its Cu–O bonds, which is also behind the high hydrophilicity of this mineral [21]. The Cu concentration is increased by NaOl, DDM, ac-ASL, and ac-LSL to 860, 900, 1070, and 1740 mg/L, respectively. In contrast, malachite is practically insoluble in water at pH 9.7 (the Cu concentration is only 6.4 g/L). This result agrees with the stability diagram [65] and experimental data [64]. It is explained by increasing natural concentration of (bi)carbonate with increasing pH of air-saturated solutions. The solubility of malachite at pH 9.7 is slightly inhibited by DDM, the Cu concentration being dropped to 5.3 g/L. The other three surfactants increase it in the order of ac-ASL < NaOl < ac-LSL, the Cu concentration with ac-LSL being 15 g/L.
Thus, the strongest lixiviant of hematite and malachite is ac-ASL and ac-LSL, respectively. The leaching by the sugar-based surfactants at pH 9.7 is consistent with their chemisorption. However, the strongest leaching activity of ac-LSL toward malachite at pH 5.2 is a curious phenomenon given that this non-ionic surfactant is less soluble than ac-ASL and lacks the additional complexing carboxylate group. This result suggests that ac-LSL chemically reacts with malachite also at pH 5.2.
3.3.3 Single-Mineral Flotation of Ultrafine Mineral Particles
Single-mineral flotation (floatability) was used to characterize the hydrophobicity of ultrafine minerals. Mineral particles that adhere to bubbles in flotation (float) are commonly referred to as hydrophobic (water-repelling). Other conventional methods to characterize hydrophobicity include contact angle, water permeation, and partitioning of mineral particles to a non-polar solvent. The advantage of floatability is that it characterizes hydrophobicity in situ under dynamic flotation conditions without affecting the critical properties that affect the amenability of mineral particles to flotation such as the particle size and the aqueous environment [42]. At the same time, it should be kept in mind that the floatability of ultrafine particles can be affected by entrainment [13].
The flotation tests were conducted on ultrafine hematite, malachite, and quartz in 50 µM solutions of ac-ASL, ac-LSL, DDM, and NaOl. The in the pH range for hematite and quartz was from 2 to 10. The low pH limit for malachite was 6 due to the increased solubility of this mineral at more acidic pH.
Hematite
Figure 7a shows that a maximum recovery of 70% can be achieved for hematite with all four surfactants, though at different pH. Specifically, the oleate flotation has a maximum at pH 6, being depressed at pH 2 and 10. This pH dependence is typical of the single-mineral flotation of hematite at low concentrations oleate [16, 17]. It can be explained by the interplay of the chemisorption and physisorption of the fatty acid and acid–base properties of hematite, which impart maximum hydrophobicity to hematite at pH close to its IEP [18, 19]. Non-ionic ac-LSL and DDM achieve the 70% flotation maximum at pH 4–6 and 6–8, respectively. In contrast, the ac-ASL flotation monotonically increases with pH, reaching 70% at pH 10. This increase can be explained by the transition of ac-ASL from the hydrophilic open configuration with the sophorose group dangling in the solution to the hydrophobic closed configuration in which both the headgroups are coordinated to the metal cation (Section 3.3.2). It should be noted that the surface coverages of the surfactants at pH 10 are low as follows from the converging of the ζ-potential curves in the surfactant solutions and water (Fig. 6a). However, these small surface coverages are sufficient for DDM and ac-ASL to float hematite (Fig. 7a).

Effect of pH on single-mineral flotation of − 20 µm fraction of a hematite and b malachite with 50 µM of (▼) ac-LSL, (■) ac-ASL, (
 ) DDM, and (●) NaOl. The line “no collector” corresponds to flotation using 50 µM DawFroth 200 (a frothing agent)
) DDM, and (●) NaOl. The line “no collector” corresponds to flotation using 50 µM DawFroth 200 (a frothing agent)
Malachite
The floatability of malachite with NaOl, ac-ASL, and ac-LSL in the pH range from 6 to 10 is higher by 20–30% and much less responsive to pH compared to that of hematite in the same pH range (Fig. 7b). The highest recovery of 98% is achieved by ac-LSL at pH 8. The floatability with NaOl and ac-ASL monotonically increases to 90% at pH 10 from 85 and 75% at pH 6, respectively. A similarly weak pH dependence was reported earlier for the malachite flotation with 50 µM NaOl [60]. The higher hydrophobicity in a wider pH range generally suggests that the mineral interacts with a surfactant more strongly, which results in its higher adsorption density and better packing. We should note this effect is observed even though malachite has the threefold higher BET surface area than hematite (Table 1). In contrast, there is no difference in the floatability of malachite and hematite with DDM.
The trends in the flotation of hematite and malachite do not correlate with the activity of the surfactants at the air–water interface. For example, ac-ASL and ac-LSL float at pH 10 70% and 40% of hematite, respectively (Fig. 7a), while their surface tension at 50 µM and pH 10 is similar (Fig. 4). The collecting power of ac-LSL at pH 6 increases to 70%, while there is no difference in surface tension at pH 6 and 10.
At the same time, there is a correlation between the floatability/hydrophobicity and solubility of the minerals in some systems. In particular, the floatability of hematite at pH 10 increases in the order of NaOl < ac-LSL < DDM < ac-ASL (Fig. 7a), which is similar to NaOl < ac-LSL = DDM < ac-ASL observed for the solubility of hematite at pH 9.7 (Table 4). Also, ac-LSL is both the strongest collector and the strongest lixiviant of malachite at pH 6 and 10, while DDM is both the weakest collector and the weakest lixiviant at pH 10. Hence, the main hydrophobization mechanism these systems is likely to be the chemisorption of the surfactants and/or precipitation of their complexes with metals. The absence of such a correlation at acidic pH indicates the critical role of physisorption. In addition, the flotation at acidic pH can be influenced by entrainment since the foaming properties of the sugar-based surfactants are significantly better at pH 5.2–5.4 than at pH 10–11 (Fig. 5).
Quartz
As shown in Fig. 8, all the surfactants do not float ultrafine quartz at pH 5 and 10, which can be explained by their negligible adsorption (Section 3.3.1).

Recovery of ultrafine (− 20 µm) quartz particles by 50 µM ac-LSL, ac-ASL, DDM, and NaOl at pH 5 (blue bar) and pH 10 (green bar). The uncertainty of pH is within 0.2 units
3.4 The Effect of Particle Size on the Flotation of Metal Oxides in Two-Mineral Systems with Quartz
In this section, we study the collecting properties of ac-LSL, ac-ASL, DDM, and NaOl in the flotation of ultrafine (− 20 µm) and coarse (+ 38 − 90 µm) hematite and malachite in their binary mixtures with ultrafine (− 20 µm) quartz. The tests were conducted at pH 5 (hematite) and pH 6 (malachite), as well as pH 10 (both) at a surfactant concentration of 50 µM. This concentration is chosen as in the above studies to keep the same surfactant-metal oxide ratio given that the surfactant adsorption on quartz is negligible within the detection level of ζ-potential (Fig. 6c).
3.4.1 Hematite
As seen in Fig. 9, the best collector of ultrafine hematite from quartz at pH 5 and 10 is NaOl. At pH 5, it recovers 91% of hematite at a grade of 87%. NaOl is followed by ac-LSL with a 98% recovery and a 68% grade, and then by ac-ASL with a 93% recovery and a 61% grade (Fig. 9a). The weakest collector at both pH is DDM. It recovers 63% of hematite at a grade of 56%. The decrease in the grade correlates well with ζ-potential of hematite at pH 5, which becomes less negative in the same order of NaOl > ac-LSL > ac-ASL > DDM (Fig. 6a). This correlation suggests that the main reason for the grade loss is the heterocoagulation of hematite with quartz.

Recovery and grade in the flotation of a 1:1 (wt) mixture of hematite and ultrafine quartz with 50 µM (▼▽) ac-ASL, (▲△) ac-LSL, (●○) DDM, and (■□) NaOl at (a) pH 5 ± 0.3 and (b) pH 10 ± 0.3. The filled and empty symbols correspond to ultrafine and coarse hematite particles, respectively. The error of the grade and recovery data points without the error bars is less than 5%. A different representation of the dependencies is shown in Fig. S5
The recovery of ultrafine hematite with all four surfactants decreases at pH 10 as compared to pH 5 while there is no significant change in the grade (Fig. 9b and Fig. S5). The same trend is observed in the single-mineral flotation with NaOl, ac-LSL, and DDM (Fig. 7a). However, there is a discrepancy between the trends in the single- and mixed mineral flotation with ac-ASL.
The ranking of the collectors is slightly different for coarse hematite (Fig. 9). Here, the best at both pH is ac-LSL. It recovers 98% of hematite at a grade of 65–68%. The next is NaOl with a 90% recovery at similar grades. DDM is the worst at both pH. At pH 5, it recovers only 24% of coarse hematite at a grade as low as 24%. Such a poor performance of DDM suggests a low surface density of the physisorbed surfactant on coarse hematite. However, the performance of DDM is improved at pH 10, where it recovers 55% of coarse hematite at a grade of 42%. Taking into accounts the leaching results (Section 3.3.2), this improvement can be explained by the activation of the chemical interaction between the maltose headgroup and the hematite at basic pH.
The comparison of the results for ultrafine and coarse hematite shows that ac-LSL flotation is tolerant to the particle size at pH 5 (Fig. 9a). In the case of ac-ASL, the ultrafine particle size drops the grade by 10% while increasing the recovery by 25%. In contrast, the ultrafine particle size significantly improves the collecting properties of NaOl and DDM. At the same recovery of 90%, the hematite grade in the oleate flotation at pH 5 is by 22% higher for ultrafine compared to coarse particles. DDM practically does not float coarse hematite at pH 5 but recovers ultrafine hematite at a recovery of 62% and a grade of 56%. In the NaOl and DDM flotation at pH 10, the ultrafine particle size improves the grade by 16% though the recovery is compromised, especially in the NaOl flotation.
The promoting effect of the ultrafine particle size on the oleate flotation of hematite can be explained by the relatively weak (monodentate) coordination of fatty acids to the crystallographically smooth surfaces of iron (hydr)oxides [18, 19]. A decrease in the particle size would increase the adsorption density of fatty acids due to the associated increase in the concentration of surface defects (surface energy). A higher adsorption density of an anionic surfactant would make the hematite particles more negatively charged, thereby enhancing their repulsion from the negatively charged quartz particles and hence improving the grade. Surface defects are also expected to improve the physisorption of DDM on hematite at acidic pH as it is increased with the surface concentration of OH groups [28,29,30]. As a result, the positive charge of the hematite particles would be screened better.
3.4.2 Malachite
The ranking of the collectors for ultrafine malachite is the same as for ultrafine hematite (Fig. 10 and Fig. S6). At pH 6, NaOl recovers 75% of malachite at a grade of 80%. Even though ac-LSL achieves a better recovery of 86% than NaOl, the corresponding grade is 71%. DDM floats malachite at the same grade of 61% as ac-ASL but at a lower recovery of 56% vs. 72%. The principal difference with ultrafine hematite is that pH 10 improves the recovery of ultrafine malachite vs. that at pH 6 with all the four surfactants, while the grades in the NaOl and ac-LSL flotation decrease by 7–10%. This difference is likely to originate from the difference in the adsorption mechanisms of these surfactants on hematite and malachite. These mechanisms remain largely unknown for all the surfactants on malachite, as well as for ac-LSL on hematite.

Recovery and grade in the flotation of a 1:1 (wt) mixture of malachite and ultrafine quartz with 50 µM (▼▽) ac-ASL, (▲△) ac-LSL, (●○) DDM, and(■□) at (a) pH 6 ± 0.3 and (b) pH 10 ± 0.3. The filled and empty symbols correspond to ultrafine and coarse malachite particles, respectively. The error of the grade and recovery data points without the error bars is less than 5%. A different representation of the dependencies is shown in Fig. S6
There is a good correlation between the grade and the impact of the surfactants on the ζ-potential of malachite: The grade at pH 6 decreases in the same order of NaOl > ac-LSL > ac-ASL > DDM as the difference between IEP of malachite in water and in the surfactant solutions (Fig. 6b). Hence, as in the case of hematite, the main reason for the loss of selectivity is the heterocoagulation with quartz, while the contribution of the quartz activation by adsorbed metal–surfactant complexes is insignificant. In addition, the recovery of ultrafine malachite in the two-mineral flotation decreases in the same order of ac-LSL > NaOl ≈ ac-ASL > DDM (Fig. 10 and Fig. S6) as in the single-mineral flotation (Fig. 7b). This fact suggests that and the non-selective entrainment of the ultrafine particles to the two-mineral flotation is insignificant too.
The coarse particle size improves the recovery of malachite in the oleate flotation at pH 6 and 10 by 15–30% but does not affect the grade (Fig. 10 and Fig. S6). This result is different from that observed for hematite, where the coarse size decreases the grade while hardly affecting the recovery (Fig. 9b and Fig. S5). This difference suggests that an increase in the surface density of oleate on the more defective ultrafine malachite particles is not critical for preventing their heterocoagulation with quarts.
As in the case of hematite, ac-LSL and ac-ASL demonstrate good tolerance to the particle size. The difference in the grade and recovery for the two particle sizes is less than a 10% in the ac-LSL flotation at pH 6 and 10, as well as in the ac-ASL flotation at pH 10. Also, the DDM flotation of coarse malachite is inferior to ultrafine malachite, which suggests the promoting role of surface defects (see above).
Finally, it is instructive to compare the performance of the biosurfactants and hydroxamates in the flotation of ultrafine malachite. Hydroxamates are chelating O-donors widely used in oxide flotation as more selective alternatives to fatty acids [41, 66]. Among different hydroxamates tested, benzohydroxamate has demonstrated the best figures of merit (a 95% recovery at a 95% grade) in the flotation of coarse malachite (− 150 + 38 µm) against quartz at pH 8 [66]. However, the fine (− 38 µm) particle size drops the recovery and the grade to 75% and 35–40%, respectively. To compare, ac-LSL floats the (+ 38 − 90 µm) fraction of malachite at pH 6 at a 90% recovery and a 75% grade, which remain the same for the ultrafine (− 20 µm) fraction (Fig. 10 and Fig. S6). The recovery and grade in the ac-ASL flotation of ultrafine malachite at pH 10 are 82% and 62%, respectively, practically the same as for coarse particles. Hence, hydroxamates lack the tolerance to fines inherent to ac-LSL and ac-ASL. A mechanistic insight into this interesting result could help design collectors for ultrafine particles.
4 Conclusions
This study aimed at extending the knowledge base on biosurfactants as eco-friendly collectors.
Without a frother in synthetic mineral systems, NaOl, ac-LSL, ac-ASL provide similarly high grades in the flotation of coarse hematite against quartz. For coarse malachite, NaOl achieves somewhat higher grades compared to ac-LSL and ac-ASL. DDM is the worst collector of both the metal oxides under the conditions studied.
The ultrafine particle size significantly improves the grade of hematite in the two-mineral flotation against quartz with NaOl and DDM at pH 5 and 10, as well as in the two-mineral flotation of malachite against quartz with DDM at pH 5. In contrast, the ultrafine particle size practically does not affect the high grade of malachite in oleate flotation, as well as the high grades of both the oxides in ac-LSL and ac-ASL flotation. The beneficial effect of the ultrafine particle size on the NaOl and DDM flotation can be explained by the promoting role of surface defects on the adsorption of NaOl and DDM.
There is no direct correlation between the flotation results, the activity of the surfactants at the air–water interface, and their foaming properties. At the same time, the grades of hematite and malachite in the two-mineral flotation against quartz can be explained by the heterocoagulation of the minerals with quartz. The surfactants hydrophobize the minerals at pH 10 through chemisorption and/or precipitation as complexes with metal ions, while there is contribution of physisorption at acidic pH. Malachite adsorbs NaOl, ac-LSL, and ac-ASL more strongly than hematite, while the affinity of these two oxides to DDM is similar. In contrast, quartz does not adsorb all the four surfactants.
Given the low selectivity of fatty acids, the good collecting properties of ac-LSL and ac-ASL along with their tolerance to the ultrafine particle size make these biosurfactants attractive for further studies on natural metal oxide ores in comparison with conventional surfactants. The results of this study can also be used as a benchmark for the development of green collectors for ultrafine mineral particles.
Data Availability
The essential data is available in the main text and in the supplementary file. The additional data is available from the first or corresponding author by reasonable request.
References
Fuerstenau DW (2019) Pradip. A century of research leading to understanding the scientific basis of selective mineral flotation and design of flotation collectors. Min Metall Explor 36(1):3–20. https://doi.org/10.1007/s42461-018-0042-6
Nagaraj DR, Farinato RS (2016) Evolution of flotation chemistry and chemicals: a century of innovations and the lingering challenges. Miner Eng 96–97:2–14. https://doi.org/10.1016/j.mineng.2016.06.019
Ramachandra Rao S, Leja J (2004) Surface chemistry of froth flotation. Vol. 2 Reagents and mechanisms. 2nd ed. New York: Kluwer
Nagaraj DR, Farinato RS, Arinaitwe E (2019) Flotation chemicals and chemistry. In: Dunne RC, Kawatra KS, Young CA (eds) SME Mineral processing and extractive metallurgy handbook. Society for Mining, Metallurgy & Exploration, Englewood, Colorado, pp 967–1010
Webb M, Ruber H, Leduc G (1976) The toxicity of various mining flotation reagents to rainbow trout (Salmo gairdneri). Water Res 10(4):303–306. https://doi.org/10.1016/0043-1354(76)90171-8
Rocchi L (2018) Xanthates in mining (update). https://www.rshq.qld.gov.au/safety-notices/mines/xanthates-in-mining-update. Accessed
Greim H, Bury D, Klimisch HJ, Oeben-Negele M, Ziegler-Skylakakis K (1998) Toxicity of aliphatic amines: Structure-activity relationship. Chemosphere 36(2):271–295. https://doi.org/10.1016/S0045-6535(97)00365-2
Jain G, Havskjold H, Dhar P, Ertesvåg H, Chernyshova I, Kota HR (2020) Green Foam-based methods of mineral and ion separation. In: Chernyshova I, Ponnurangam S, Liu Q (eds) Multidisciplinary Advances in Efficient Separation Processes. American Chemical Society, pp 265–301
Oulkhir A, Lyamlouli K, Danouche M, Ouazzani J, Benhida R (2022) A critical review on natural surfactants and their potential for sustainable mineral flotation. Rev Environ Sci Bio Technol. https://doi.org/10.1007/s11157-022-09639-8
Hartmann R, Beaumont M, Pasquie E, Rosenau T, Serna-Guerrero R (2022) N-Alkylated chitin nanocrystals as a collector in malachite flotation. ACS Sustain Chem Eng 10(32):10570–10578. https://doi.org/10.1021/acssuschemeng.2c01978
Asgari K, Huang Q, Khoshdast H, Hassanzadeh A (2022) A review on bioflotation of coal and minerals: classification, mechanisms, challenges, and future perspectives. Mineral Process Ext Metall Rev 1–31. https://doi.org/10.1080/08827508.2022.2121919
Höfer R, Bigorra J (2007) Green chemistry—a sustainable solution for industrial specialties applications. Green Chem 9(3):203–212. https://doi.org/10.1039/b606377b
Dhar P, Thornhill M, Roelants S, Soetaert W, Chernyshova IV, Rao Kota H (2021) Linking molecular structures of yeast-derived biosurfactants with their foaming, interfacial, and flotation properties. Miner Eng 174:107270. https://doi.org/10.1016/j.mineng.2021.107270
Shen Y, Lo C, Nagaraj DR, Farinato R, Essenfeld A, Somasundaran P (2016) Development of Greenness Index as an evaluation tool to assess reagents: Evaluation based on SDS (Safety Data Sheet) information. Miner Eng 94:1–9. https://doi.org/10.1016/j.mineng.2016.04.015
Zhang X, Gu X, Han Y, Parra-Álvarez N, Claremboux V, Kawatra SK (2021) Flotation of Iron Ores: A Review. Miner Process Extr Metall Rev 42(3):184–212. https://doi.org/10.1080/08827508.2019.1689494
Nakhaei F, Irannajad M (2018) Reagents types in flotation of iron oxide minerals: a review. Miner Process Extr Metall Rev 39(2):89–124. https://doi.org/10.1080/08827508.2017.1391245
Kulkarni RD, Somasundaran P (1980) Flotation chemistry of hematite/oleate system. Colloids Surf 1(3):387–405. https://doi.org/10.1016/0166-6622(80)80025-4
Chernyshova IV, Ponnurangam S, Somasundaran P (2011) Adsorption of Fatty acids on iron (hydr)oxides from aqueous solutions. Langmuir 27(16):10007–10018. https://doi.org/10.1021/la2017374
Ponnurangam S, Chernyshova IV, Somasundaran P (2012) Rational design of interfacial properties of ferric (hydr)oxide nanoparticles by adsorption of fatty acids from aqueous solutions. Langmuir 28(29):10661–10671. https://doi.org/10.1021/la300995g
Bulatovic SM (2010) 19 - Flotation of Oxide Copper and Copper Cobalt Ores. In: Bulatovic SM (ed) Handbook of flotation reagents: chemistry, theory and practice. Elsevier, Amsterdam, pp 47–65
Feng Q, Yang W, Wen S, Wang H, Zhao W, Han G (2022) Flotation of copper oxide minerals: a review. Int J Min Sci Technol. https://doi.org/10.1016/j.ijmst.2022.09.011
Li Z, Rao F, Escudero García R, Li H, Song S (2018) Partial replacement of sodium oleate using alcohols with different chain structures in malachite flotation. Miner Eng 127:185–190. https://doi.org/10.1016/j.mineng.2018.08.022
Araujo AC, Viana PRM, Peres AEC (2005) Reagents in iron ores flotation. Miner Eng 18(2):219–224. https://doi.org/10.1016/j.mineng.2004.08.023
Moreira GF, Peçanha ER, Monte MBM, Leal Filho LS, Stavale F (2017) XPS study on the mechanism of starch-hematite surface chemical complexation. Miner Eng 110:96–103. https://doi.org/10.1016/j.mineng.2017.04.014
RohemPeçanha E, da Fonseca de Albuquerque MD, AntounSimão R, de Salles Leal Filho L, de Mello Monte MB (2019) Interaction forces between colloidal starch and quartz and hematite particles in mineral flotation. Colloids Surf A Physicochem Eng Asp 562:79–85. https://doi.org/10.1016/j.colsurfa.2018.11.026
Li L, Zhang C, Yuan Z, Xu X, Song Z (2019) AFM and DFT study of depression of hematite in oleate-starch-hematite flotation system. Appl Surf Sci 480:749–758. https://doi.org/10.1016/j.apsusc.2019.02.224
Félix LL, Moreira GF, Filho LSL, Stavale F (2022) Starch adsorption on hematite surfaces: Evidence of the adsorption mechanism dependence on the surface orientation. Miner Eng 178. https://doi.org/10.1016/j.mineng.2022.107429
Zhang L, Somasundaran P, Maltesh C (1997) Adsorption ofn-Dodecyl-β-d-maltoside on Solids. J Colloid Interface Sci 191(1):202–208. https://doi.org/10.1006/jcis.1997.4923
Zhang L, Somasundaran P, Mielczarski J, Mielczarski E (2002) Adsorption Mechanism of n-dodecyl-β-D-maltoside on Alumina. J Colloid Interface Sci 256(1):16–22. https://doi.org/10.1006/jcis.2001.7858
Mielczarski E, Mielczarski JA, Zhang L, Somasundaran P (2004) Structure of adsorbed n-dodecyl-beta-D-maltoside layers on hematite. J Colloid Interface Sci 275(2):403–409. https://doi.org/10.1016/j.jcis.2004.02.083
Stubenrauch C, Claesson PM, Rutland M, Manev E, Johansson I, Pedersen JS et al (2010) Mixtures of n-dodecyl-beta-D-maltoside and hexaoxyethylene dodecyl ether - Surface properties, bulk properties, foam films, and foams. Adv Colloid Interface Sci 155(1–2):5–18. https://doi.org/10.1016/j.cis.2009.12.002
Dhar P, Havskjold H, Thornhill M, Roelants S, Soetaert W, Kota HR et al (2021) Toward green flotation: Interaction of a sophorolipid biosurfactant with a copper sulfide. J Colloid Interface Sci 585:386–399. https://doi.org/10.1016/j.jcis.2020.11.079
Baccile N, Seyrig C, Poirier A, Alonso-de Castro S, Roelants SLKW, Abel S (2021) Self-assembly, interfacial properties, interactions with macromolecules and molecular modelling and simulation of microbial bio-based amphiphiles (biosurfactants). A tutorial review. Green Chemistry 23(11):3842–3944. https://doi.org/10.1039/D1GC00097G
Nikolova C, Gutierrez T (2021) Biosurfactants and their applications in the oil and gas industry: current state of knowledge and future perspectives. Front Bioeng Biotechnol 9. https://doi.org/10.3389/fbioe.2021.626639
Hu X, Subramanian K, Wang H, Roelants SLKW, To MH, Soetaert W, et al (2021) Guiding environmental sustainability of emerging bioconversion technology for waste-derived sophorolipid production by adopting a dynamic life cycle assessment (dLCA) approach. Environ Pollut 269. https://doi.org/10.1016/j.envpol.2020.116101
Hu X, Subramanian K, Wang H, Roelants SLKW, Soetaert W, Kaur G et al (2021) Bioconversion of food waste to produce industrial-scale sophorolipid syrup and crystals: dynamic life cycle assessment (dLCA) of emerging biotechnologies. Bioresour Technol 337. https://doi.org/10.1016/j.biortech.2021.125474
Baccile N, Cuvier A-S, Valotteau C, Van Bogaert INA (2013) Practical methods to reduce impurities for gram-scale amounts of acidic sophorolipid biosurfactants. Eur J Lipid Sci Technol 115(12):1404–1412. https://doi.org/10.1002/ejlt.201300131
Castelein M, Verbruggen F, Van Renterghem L, Spooren J, Yurramendi L, Du Laing G et al (2021) Bioleaching of metals from secondary materials using glycolipid biosurfactants. Miner Eng 163. https://doi.org/10.1016/j.mineng.2020.106665
Baccile N, Noiville R, Stievano L, Bogaert IV (2013) Sophorolipids-functionalized iron oxide nanoparticles. Phys Chem Chem Phys 15(5):1606–1620. https://doi.org/10.1039/c2cp41977g
Peyre J, Hamraoui A, Faustini M, Humblot V, Baccile N (2017) Surface-induced assembly of sophorolipids. Phys Chem Chem Phys 19(23):15227–15238. https://doi.org/10.1039/C7CP01339F
Lee JS, Nagaraj DR, Coe JE (1998) Practical aspects of oxide copper recovery with alkyl hydroxamates. Miner Eng 11(10):929–939. https://doi.org/10.1016/S0892-6875(98)00080-6
Wills BA, Finch JA (2016) Chapter 12 - Froth Flotation. In: Wills BA, Finch JA (eds) Wills’ Mineral Processing Technology, 8th edn. Butterworth-Heinemann, Boston, pp 265–380
Abaka-Wood GB, Addai-Mensah J, Skinner W (2021) The use of mining tailings as analog of rare earth elements resources: part 1 – characterization and preliminary separation. Miner Process Extr Metall Rev 1–15. https://doi.org/10.1080/08827508.2021.1920410
Ceniceros-Gómez AE, Macías-Macías KY, de la Cruz-Moreno JE, Gutiérrez-Ruiz ME, Martínez-Jardines LG (2018) Characterization of mining tailings in México for the possible recovery of strategic elements. J S Am Earth Sci 88:72–79. https://doi.org/10.1016/j.jsames.2018.08.013
Alcalde J, Kelm U, Vergara D (2018) Historical assessment of metal recovery potential from old mine tailings: A study case for porphyry copper tailings. Chile Miner Eng 127:334–338. https://doi.org/10.1016/j.mineng.2018.04.022
Tang C, Li K, Ni W, Fan D (2019) Recovering iron from iron ore tailings and preparing concrete composite admixtures. Minerals 9(4). https://doi.org/10.3390/min9040232
Farrokhpay S, Filippov L, Fornasiero D (2020) Flotation of fine particles: a review. Miner Process Extr Metall Rev 1–11. https://doi.org/10.1080/08827508.2020.1793140
Sivamohan R (1990) The problem of recovering very fine particles in mineral processing — a review. Int J Miner Process 28(3–4):247–288. https://doi.org/10.1016/0301-7516(90)90046-2
Melo F, Laskowski JS (2006) Fundamental properties of flotation frothers and their effect on flotation. Miner Eng 19(6):766–773. https://doi.org/10.1016/j.mineng.2005.09.031
Hassanzadeh A, Safari M, Hoang DH, Khoshdast H, Albijanic B, Kowalczuk PB (2022) Technological assessments on recent developments in fine and coarse particle flotation systems. Miner Eng 180. https://doi.org/10.1016/j.mineng.2022.107509
Van Bogaert INA, Zhang J, Soetaert W (2011) Microbial synthesis of sophorolipids. Process Biochem 46(4):821–833. https://doi.org/10.1016/j.procbio.2011.01.010
Li G, Yi X, Jiang J, Zhang Y, Li Y (2020) Dynamic surface properties and dilational rheology of acidic and lactonic sophorolipids at the air-water interface. Colloids Surf B Biointerfaces 195. https://doi.org/10.1016/j.colsurfb.2020.111248
Lu S, Bian Y, Zhang L, Somasundaran P (2007) pH dependence of adsorption of n-dodecyl-β-d-maltoside on solids. J Colloid Interface Sci 316(2):310–316. https://doi.org/10.1016/j.jcis.2007.08.063
Zhang L, Somasundaran P, Maltesh C (1996) Electrolyte Effects on the Surface Tension and Micellization of n-Dodecyl β-d-Maltoside Solutions. Langmuir 12(10):2371–2373. https://doi.org/10.1021/la950670w
Theander K, Pugh RJ (2001) The influence of ph and temperature on the equilibrium and dynamic surface tension of aqueous solutions of sodium oleate. J Colloid Interface Sci 239(1):209–216. https://doi.org/10.1006/jcis.2000.7543
Otto RT, Daniel HJ, Pekin G, Müller-Decker K, Fürstenberger G, Reuss M et al (1999) Production of sophorolipids from whey. Appl Microbiol Biotechnol 52(4):495–501. https://doi.org/10.1007/s002530051551
Hirata Y, Ryu M, Oda Y, Igarashi K, Nagatsuka A, Furuta T et al (2009) Novel characteristics of sophorolipids, yeast glycolipid biosurfactants, as biodegradable low-foaming surfactants. J Biosci Bioeng 108(2):142–146. https://doi.org/10.1016/j.jbiosc.2009.03.012
Díaz De Rienzo MA, Kamalanathan ID, Martin PJ (2016) Comparative study of the production of rhamnolipid biosurfactants by B. thailandensis E264 and P. aeruginosa ATCC 9027 using foam fractionation. Process Biochem 51(7):820–7. https://doi.org/10.1016/j.procbio.2016.04.007
Choi J, Choi SQ, Park K, Han Y, Kim H (2016) Flotation behaviour of malachite in mono- and di-valent salt solutions using sodium oleate as a collector. Int J Miner Process 146:38–45. https://doi.org/10.1016/j.minpro.2015.11.011
Li H, Liu M, Liu Q (2018) The effect of non-polar oil on fine hematite flocculation and flotation using sodium oleate or hydroxamic acids as a collector. Miner Eng 119:105–115. https://doi.org/10.1016/j.mineng.2018.01.004
Fuerstenau DW (2005) Pradip. Zeta potentials in the flotation of oxide and silicate minerals. Adv Colloid Interface Sci 114–115:9–26. https://doi.org/10.1016/j.cis.2004.08.006
Quast K (2006) Flotation of hematite using C6–C18 saturated fatty acids. Miner Eng 19(6–8):582–597. https://doi.org/10.1016/j.mineng.2005.09.010
Quast K (2016) The use of zeta potential to investigate the interaction of oleate on hematite. Miner Eng 85:130–137. https://doi.org/10.1016/j.mineng.2015.11.007
Li Z, Rao F, Song S, Uribe-Salas A, López-Valdivieso A (2019) Effects of common ions on adsorption and flotation of malachite with salicylaldoxime. Colloids Surf A 577:421–428. https://doi.org/10.1016/j.colsurfa.2019.06.004
Preis W, Gamsjäger H (2002) Solid–solute phase equilibria in aqueous solution. XVI. Thermodynamic properties of malachite and azurite—predominance diagrams for the system Cu 2+– H2 O– CO 2. J Chem Thermodyn 34(5):631–50. https://doi.org/10.1006/jcht.2002.0928
Marion C, Jordens A, Li R, Rudolph M, Waters KE (2017) An evaluation of hydroxamate collectors for malachite flotation. Sep Purif Technol 183:258–269. https://doi.org/10.1016/j.seppur.2017.02.056
Li F, Zhong H, Xu H, Jia H, Liu G (2015) Flotation behavior and adsorption mechanism of α-hydroxyoctyl phosphinic acid to malachite. Miner Eng 71:188–193. https://doi.org/10.1016/j.mineng.2014.11.013
Wang H, Wen S, Han G, He Y, Feng Q (2022) Adsorption behavior and mechanism of copper ions in the sulfidization flotation of malachite. Int J Min Sci Technol 32(4):897–906. https://doi.org/10.1016/j.ijmst.2022.06.006
Laskowski JS (1993) Electrokinetic measurements in aqueous solutions of weak electrolyte type surfactants. J Colloid Interface Sci 159(2):349–353. https://doi.org/10.1006/jcis.1993.1333
Pawlik M (2022) Fundamentals of froth flotation. ChemTexts 8(4):19. https://doi.org/10.1007/s40828-022-00170-5
Rao KH, Cases JM, Forssberg KSE (1991) Mechanism of oleate interaction on salt-type minerals: V Adsorption and precipitation reactions in relation to the solid/liquid ratio in the synthetic fluorite—sodium oleate system. J Colloid Interface Sci 145(2):330–48. https://doi.org/10.1016/0021-9797(91)90365-F
Chang Z, Chen X, Peng Y (2018) The adsorption behavior of surfactants on mineral surfaces in the presence of electrolytes – a critical review. Miner Eng 121:66–76. https://doi.org/10.1016/j.mineng.2018.03.002
Sis H, Birinci M (2009) Effect of nonionic and ionic surfactants on zeta potential and dispersion properties of carbon black powders. Colloids Surf A 341(1):60–67. https://doi.org/10.1016/j.colsurfa.2009.03.039
Kosmulski M (2006) pH-dependent surface charging and points of zero charge. J Colloid Interface Sci 298(2):730–741. https://doi.org/10.1016/j.jcis.2006.01.003
Feng Q, Wen S, Zhao W, Chen Y (2018) Effect of calcium ions on adsorption of sodium oleate onto cassiterite and quartz surfaces and implications for their flotation separation. Sep Purif Technol 200:300–306. https://doi.org/10.1016/j.seppur.2018.02.048
Stumm W, Morgan JJ (1996) Aquatic chemistry, 3rd edn. Wiley, New York
Fuerstenau MC, Harper RW, Miller JD (1970) Hydroxamate vs. fatty acid flotation of iron oxide. Transactions SME/AIME 247:69–73
Diakonov II, Schott J, Martin F, Harrichourry J-C, Escalier J (1999) Iron(III) solubility and speciation in aqueous solutions. experimental study and modelling: part 1. hematite solubility from 60 to 300°C in NaOH–NaCl solutions and thermodynamic properties of Fe(OH)4−(aq). Geochim Cosmochim Acta 63(15):2247–61. https://doi.org/10.1016/S0016-7037(99)00070-8
Jang JH, Dempsey BA, Burgos WD (2007) Solubilitv of hematite revisited: Effects of hydration. Environ Sci Technol 41(21):7303–7308. https://doi.org/10.1021/es070535t
Liu Q, Zhang Y, Laskowski JS (2000) The adsorption of polysaccharides onto mineral surfaces: an acid/base interaction. Int J Miner Process 60(3–4):229–245. https://doi.org/10.1016/S0301-7516(00)00018-1
Liu Q, Laskowski JS (1989) The interactions between dextrin and metal hydroxides in aqueous solutions. J Colloid Interface Sci 130(1):101–111. https://doi.org/10.1016/0021-9797(89)90081-7
Acknowledgements
We acknowledge the financial support of the Research Council of Norway (NFR), FRINATEK Project No.: 274691, and the Department of Geoscience and Petroleum, NTNU. We thank Bio Base Europe Pilot Plant for providing biosurfactants and Laurentius Tijhuis for the XRD and ICP-MS analyses. We also thank the reviewer for many suggestions on how to improve this work.
Funding
Open access funding provided by NTNU Norwegian University of Science and Technology (incl St. Olavs Hospital - Trondheim University Hospital)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Slabov, V., Jain, G., Larsen, E. et al. Eco-Friendly Collectors for Flotation of Fine Hematite and Malachite Particles. Mining, Metallurgy & Exploration 40, 475–492 (2023). https://doi.org/10.1007/s42461-023-00743-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42461-023-00743-z