
Abstract
Background
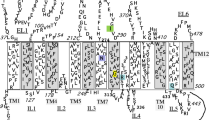
Rogers syndrome, also known as thiamine responsive megaloblastic anemia (TRMA), is an autosomal recessive disorder resulting in megaloblastic anemia, diabetes mellitus and sensorineural deafness. The gene associated with Rogers syndrome encodes for a plasma membrane thiamine transporter, THTR-1, a member of the solute carrier family that includes its homologue THTR-2 and the reduced folate carrier.
Materials and Methods
Using transient expression of wild-type and a missense mutant THTR-1 protein, derived from a TRMA family, in different cell lines and immunodetection analysis, we determined the expression, post-translational modification, and subcellular localization of the wild-type and G172D mutant THTR-1. The transport activity of the transfected THTR-1 proteins was measured using a [3H] thiamine uptake assay.
Results
The mutant THTR-1 protein was undetectable in transfected cells grown at 37°C but was readily expressed in transfected cells cultured at 28°C, thereby allowing for further biochemical and functional analysis. In contrast to its fully glycosylated wild-type mature protein, the mutant THTR-1 protein underwent only the initial stage of N-linked glycosylation. The failure to undergo a complete glycosylation resulted in the lack of plasma membrane targeting and confinement of the mutant THTR-1 to the Golgi and endoplasmic reticulum (ER) compartment. Consistently, either treatment with tunicamycin or substitution of the THTR-1 consensus N-glycosylation acceptor asparagine 63 with glutamine, abolished its glycosylation and plasma membrane targeting.
Conclusions
Taken collectively, these results suggest that the G172D mutation presumably misfolded THTR-1 protein that fails to undergo a complete glycosylation, is retained in the Golgi-ER compartment and thereby cannot be targeted to the plasma membrane. Finally, transfection studies revealed that the mutant G172D THTR-1 failed to transport thiamine. This is the first molecular and functional characterization of a missense mutant THTR-1 derived from a family with Rogers syndrome.







Similar content being viewed by others


References
Rindi G, Laforenza U. (2000) Thiamine intestinal transport and related issues: recent aspects. Proc. Soc. Exp. Biol. Med. 224: 246–255.
Labay V, Raz T, Baron D, et al. (1999) Mutations in SLC19A2 cause thiamine-responsive megaloblastic anemia associated with diabetes mellitus and deafness. Nat. Genet. 22: 300–304.
Fleming JC, Tartaglini E, Steinkamp MP, Schorderet DF, Cohen N, Neufeld EJ. (1999) The gene mutated in thiamine-responsive anemia with diabetes and deafness (TRMA) encodes a functional thiamine transporter. Nat. Genet. 22: 305–308.
Diaz GA, Banikazemi M, Oishi K, Desnick RJ, Gelb DB. (1999) Mutations in a new gene encoding a thiamine transporter cause thiamine-responsive megaloblastic anemia syndrome. Nat. Genet. 22: 309–312.
Dutta B, Huang W, Molero M, et al. (1999) Cloning of the human thiamine transporter, a member of the folate transporter family. J. Biol. Chem. 274: 31925–31929.
Rajgopal A, Edmondnson A, Goldman ID, Zhao R. (2001) SLC19A3 encodes a second thiamine transporter ThTr2. Biochim. Biophys. Acta 1537: 175–178.
Casirola D, Patrini C, Ferrari G, Rindi G. (1990) Thiamin transport by human erythrocytes and ghosts. J. Membr. Biol. 118: 11–18.
Rogers LE, Porter FS, Sidbury Jr. JB. (1969) Thiamine-responsive megaloblastic anemia. J. Pediatr. 74: 494–504.
Mandel H, Berant M, Hazani A, Naveh Y. (1984) Thiamine-dependent beriberi in the “thiamine-responsive anemia syndrome.” N. Engl. J. Med. 311: 836–838.
Rindi G, Patrini C, Laforenza U, et al. (1994) Further studies on erythrocyte thiamin transport and phosphorylation in seven patients with thiamin-responsive megaloblastic anemia. J. Inherit. Metab. Dis. 17: 667–677.
Abboud MR, Alexander D, Najjar SS. (1985) Diabetes mellitus, thiamine-dependent megaloblastic anemia, and sensorineural deafness associated with deficient alpha-ketoglutarate dehydrogenase activity. J. Pediatr. 107: 537–541.
Eudy JD, Spiegelstein O, Barber RC, Wlodarczyk BJ, Talbot J, Finnell RH. (2000) Identification and characterization of the human and mouse SLC19A3 gene: a novel member of the reduced folate family of micronutrient transporter genes. Mol. Genet. Metab. 71: 581–590.
Dixon KH, Lanpher BC, Chiu J, Kelley K, Cowan KH. (1994) A novel cDNA restores reduced folate carrier activity and methotrexate sensitivity to transport deficient cells. J. Biol. Chem. 269: 17–20.
Prasad PD, Ramamoorthy S, Leibach FH, Ganapathy V. (1995) Molecular cloning of the human placental folate transporter. Biochem. Biophys. Res. Commun. 206: 681–687.
Wong SC, Proefke SA, Bhushan A, Matherly LH. (1995) Isolation of human cDNAs that restore methotrexate sensitivity and reduced folate carrier activity in methotrexate transport-defective Chinese hamster ovary cells. J. Biol. Chem. 270: 17468–17475.
Jansen G, Pieters R. (1998) The role of impaired transport in (pre)clinical resistance to methotrexate-insights on new antifolates. Drug Resistance Updates 1: 211–218.
Lo PK, Chen JY, Tang PP, et al. (2001) Identification of a mouse thiamine transporter gene as a direct transcriptional target for p53. J. Biol. Chem. 276: 37186–37193.
Raz T, Labay V, Baron D, et al. (2000) The spectrum of mutations, including four novel ones, in the thiamine-responsive megaloblastic anemia gene SLC19A2 of eight families. Hum. Mutat. 16: 37–42.
Stagg AR, Fleming JC, Baker MA, Sakamoto M, Cohen N, Neufeld EJ. (1999) Defective high-affinity thiamine transporter leads to cell death in thiamine-responsive megaloblastic anemia syndrome fibroblasts. J. Clin. Invest. 103: 723–729.
Naviaux RK, Costanzi E, Haas M, Verma IM. (1996) The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 70: 5701–5705.
Hibi M, Lin A, Smeal T, Minden A, Karin M. (1993) Identification of an oncoprotein and UV responsive kinase that binds and potentiates the activity of the c-jun activation domain. Genes Dev. 7: 2135–2148.
Wong SC, Zhang L, Proefke SA, Matherly LH. (1998) Effects of the loss of capacity for N-glycosylation on the transport activity and cellular localization of the human reduced folate carrier. Biochim. Biophys. Acta 1375: 6–12.
Sharif KA, Goldman ID. (2000) Rapid determination of membrane transport parameters in adherent cells. Biotechniques 28: 926–928, 930, 932.
Gahmberg CG, Tolvanen M. (1996) Why mammalian cell surface proteins are glycoproteins. Trends Biochem. Sci. 21: 308–311.
Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. (1992) Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 358: 761–764.
Featherstone C. (1998) Coming to grips with the Golgi. Science 282: 2172–2174.
Fleming JC, Steinkamp MP, Kawatsuji R, et al. (2001) Characterization of a murine high-affinity thiamine transporter, Slc19a2. Mol. Genet. Metab. 74: 273–280.
Valerio G, Franzese A, Poggi V, Tenore A. (1998) Long-term follow-up of diabetes in two patients with thiamine-responsive megaloblastic anemia syndrome. Diabetes Care 21: 38–41.
Klausner RD, Sitia R. (1990) Protein degradation in the endoplasmic reticulum. Cell 62: 611–614.
Verma R, Deshaies RJ. (2000) A proteasome howdunit: the case of the missing signal. Cell 101: 341–344.
Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. (1995) Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83: 129–135.
Loo TW, Clarke DM. (1994) Prolonged association of temperature-sensitive mutants of human P-glycoprotein with calnexin during biogenesis. J. Biol. Chem. 269: 28683–28689.
Pind S, Riordan JR, Williams DB. (1994) Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 269: 12784–12788.
Drori S, Jansen G, Mauritz R, Peters GJ, Assaraf YG. (2000) Clustering of mutations in the first transmembrane domain of the human reduced folate carrier in GW1843U89-resistant leukemia cells with impaired antifolate transport and augmented folate uptake. J. Biol. Chem. 275: 30855–30863.
Zhao R, Sharina IG, Goldman ID. (1999) Pattern of mutations that results in loss of reduced folate carrier function under antifolate selective pressure augmented by chemical mutagenesis. Mol. Pharmacol. 56: 68–76.
Acknowledgments
We wish to thank Drs Y. C. Broder and O. Shenkar for technical assistance with cell staining and confocal microscopy, respectively; Ms. A. Cohen for technical assistance; Dr G. Spira for the use of laboratory equipment; Drs V. Labay and D. Kornizer for fruitful suggestions; and Dr L. H. Matherly for the human RFC-HA expression plasmid.
This work was partially supported by the Israeli Academy of Sciences and Juvenile Diabetes International Foundation to N. C. and by Technion internal grants to A. A.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Baron, D., Assaraf, Y.G., Cohen, N. et al. Lack of Plasma Membrane Targeting of a G172D Mutant Thiamine Transporter Derived from Rogers Syndrome Family. Mol Med 8, 462–474 (2002). https://doi.org/10.1007/BF03402026
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/BF03402026