
Abstract
Pompe disease or glycogen storage disease type II (OMIM 232300) is a metabolic myopathy with a broad clinical spectrum. Generalised muscle weakness combined with cardiomegaly presents within the first 3 months after birth, if the lysosomal α-glucosidase (AGLU) deficiency is complete. Residual enzyme activity prevents cardiac involvement and delays onset of muscle weakness. Enzyme therapy, by intravenous administration of acid AGLU, aims to supplement the missing enzyme activity. At the SHS symposium on Glycogen Storage Diseases Type I and II, in Fulda, two interim accounts were given of studies on the efficacy of enzyme therapy for Pompe disease; one with recombinant human acid AGLU produced in Chinese hamster ovary cells and the other with the same enzyme produced in the milk of transgenic rabbits.Conclusion: this review focuses on the latter study, discusses the scientific, technological and commercial aspects of the enterprise, and addresses the prospects and challenges of enzyme therapy for Pompe disease.
Similar content being viewed by others
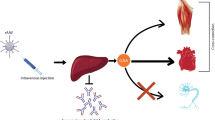

Abbreviations
- AGLU :
-
α-glucosidase
- CHO :
-
Chinese hamster ovary
- CRIM :
-
cross-reactive immunological material
- M6P :
-
mannose-6-phosphate
References
Amalfitano A, Bengur AR, Morse RP, Majure JM, Case LE, Veerling DL, Mackey J, Kishnani P, Smith W, McVie-Wylie A, Sullivan JA, Hoganson GE, Phillips JA, 3rd, Schaefer GB, Charrow J, Ware RE, Bossen EH, Chen YT (2001) Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med 3: 132–138
Ashwell G, Morell AG (1974) The role of surface carbohydrates in the hepatic recognition and transport of circulating glycoproteins. Adv Enzymol Relat Areas Mol Biol 41: 99–128
Barton NW, Furbish FS, Murray GJ, Garfield M, Brady RO (1990) Therapeutic response to intravenous infusions of glucocerebrosidase in a patient with Gaucher disease. Proc Natl Acad Sci U S A 87: 1913–1916
Barton NW, Brady RO, Dambrosia JM, Di Bisceglie AM, Doppelt SH, Hill SC, Mankin HJ, Murray GJ, Parker RI, Argoff CE et al (1991) Replacement therapy for inherited enzyme deficiency—macrophage-targeted glucocerebrosidase for Gaucher's disease, N Engl J Med 324: 1464–1470
Baudhuin P, Hers HG, Loeb H (1964) An electron microscopic and biochemical study of type II glycogenosis. Lab Invest 13: 1139–1152.
Bijvoet AG, Kroos MA, Pieper FR, De Boer HA, Reuser AJ, Van der Ploeg AT, Verbeet MP (1996) Expression of cDNA-encoded human acid alpha-glucosidase in milk of transgenic mice. Biochim Biophys Acta 1308: 93–96
Bijvoet AG, Kroos MA, Pieper FR, Van der Vliet M, De Boer HA, Van der Ploeg AT, Verbeet MP, Reuser AJ (1998) Recombinant human acid alpha-glucosidase: high level production in mouse milk, biochemical characteristics, correction of enzyme deficiency in GSDII KO mice. Hum Mol Genet 7: 1815–1824
Bijvoet AG, Van Hirtum H, Kroos MA, Van de Kamp EH, Schoneveld O, Visser P, Brakenhoff JP, Weggeman, M, van Corven EJ, Van der Ploeg AT, Reuser AJ (1999) Human acid alpha-glucosidase from rabbit milk has therapeutic effect in mice with glycogen storage disease type II. Hum Mol Genet 8: 2145–2153
De Duve C, Wattiaux R (1966) Functions of lysosomes. Annu Rev Physiol 28: 435–492
Eng CM, Banikazemi M, Gordon RE, Goldman M, Phelps R, Kim L, Gass A, Winston J, Dikman S, Fallon JT, Brodie S, Stacy CB, Mehta D, Parsons R Norton K, O'Callaghan M, Desnick RJ (2001) A phase 1/2 clinical trial of enzyme replacement in Fabry disease: pharmacokinetic, substrate clearance, and safety studies. Am J Hum Genet 68: 711–722
Fuller M, Van der Ploeg A, Reuser AJ, Anson DS, Hopwood JJ (1995) Isolation and characterisation of a recombinant, precursor form of lysosomal acid alpha-glucosidase. Eur J Biochem 234: 903–909
Grabowski GA, Leslie N, Wenstrup R (1998) Enzyme therapy for Gaucher disease: the first 5 years. Blood Rev 12: 115–133
Hers GH (1963) Alpha-glucosidase deficiency in generalized storage disease (Pompe's disease), Biochem J 86: 11–16
Hickman S, Shapiro LJ, Neufeld EF (1974) A recognition marker required for uptake of a lysosomal enzyme by cultured fibroblasts. Biochem Biophys Res Commun 57: 55–61
Hobbs JR (1992) Bone marrow transplants in genetic diseases. Eur J Pediatr 151[Suppl 1]: S44-S49
Hoefsloot LH, Hoogeveen-Westerveld M, Kroos MA, van Beeumen J, Reuser AJ, Oostra BA (1988) Primary structure and processing of lysosomal alpha-glucosidase; homology with the intestinal sucrase-isomaltase complex. EMBO J 7: 1697–1704
Hoefsloot LH, Hoogeveen-Westerveld M, Reuser AJ, Oostra BA (1990) Characterization of the human lysosomal, alpha-glucosidase gene. Biochem J 272: 493–497
Houdebine LM (1994) Production of pharmaceutical proteins from transgenic animals. J Biotechnol 34: 269–287
Hug G, Schubert WK (1967) Lysosomes in type II, glycogenosis. Changes during administration of extract fromAspergillus niger. J Cell Biol 35: C1-C6
Kakkis ED, Muenzer J, Waber L, Belmont J, Passage M, Izykowski B, Phillips J, Doroshow R, Walot I, Hoft R, Neufeld EF (2001) Enzyme-replacement therapy in mucopolysacchari-dosis I. N Engl J Med 344: 182–188
Kaplan A, Achord DT, Sly WS (1977) Phosphohexosyl components of a lysosomal enzyme are recognized by pinocytosis receptors on human fibroblasts. Proc Natl Acad Sci U S A 74: 2026–2030
Kikuchi T, Yang HW, Pennybacker M, Ichihara N, Mizutani M, Van Hove JLK, Chen YT (1998) Clinical and metabolic correction of Pompe disease by enzyme therapy in acid maltase-deficient quail. J Clin Invest 101: 827–833
Mancini GMS, Verheijen FW (1995) Lysosomal storage diseases In: Bittar EE, Bittar N (eds) Principles, of Medical Biology, vol. 3. JAI Press, Stamford, pp 133–155
Martiniuk F, Mehler M, Tzall S, Meredith G, Hirschhorn R (1990) Sequence of the cDNA and 5′-flanking region for human acid alpha-glucosidase, detection of an intron in the 5′ untranslated leader sequence, definition of 18-bp polymorphisms, and differences with previous cDNA and amino acid sequences. DNA Cell Biol 9: 85–94
Martiniuk F, Bodkin M, Tzall S, Hirschhorn R (1991) Isolation and partial characterization of the structural gene for human acid alpha glucosidase. DNA Cell Biol 10: 283–292
Reuser AJ, Kroos MA, Ponne NJ, Wolterman RA, Loonen MC, Busch HF, Visser WJ, Bolhuis PA (1984) Uptake and stability of human and bovine acid alpha-glucosidase in cultured fibroblasts and skeletal muscle cells from glycogenosis type II patients. Exp Cell Res 155: 178–189
Reuser AJ, Kroos MA, Hermans MM, Bijvoet AG, Verbeet MP, Van Diggelen OP, Kleijer WJ, Van der Ploeg AT (1995) Glycogenosis type II (acid maltase deficiency). Muscle and Nerve 3: S61-S69
Schiffmann R, Murray GJ, Treco D, Daniel P, Sellos-Moura M, Myers M, Quirk JM, Zirzow GC, Borowski M, Loveday K, Anderson T, Gillespie F, Oliver KL, Jeffries NO, Doo E, Liang TJ, Kreps C, Gunter K, Frei K, Crutchfield K, Selden RF, Brady RO (2000) Infusion of alpha-galactosidase A reduces tissue, globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci U S A 97: 365–370
Shepherd VL, Stahl PD (1984) Macrophage receptors for lysosomal enzymes. In: Dingle JT, Dean RT, Sly WS (eds) Lysosomes in biology and pathology. North Holland Press, Amsterdam, pp 83–98
Tager JM, Hamers MN, Schram AW, Van den Bergh FA, Rietra PJ, Loonen C, Koster, JF, Slee R (1980) An appraisal of human trials in enzyme replacement therapy of genetic diseases. Birth Defects: Orig Art Ser 16: 343–359
Van den Hout H, Reuser AJ, Vulto AG, Loonen MC, Cromme-Dijkhuis A, Van der Ploeg AT (2000) Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet 356: 397–398
Van der Ploeg AT, Kroos M, van Dongen JM, Visser WJ, Bolhuis PA, Loonen MC, Reuser AJ (1987) Breakdown of lysosomal glycogen in cultured fibroblasts from glycogenosis type II patients after uptake of acid alpha-glucosidase. J Neurol Sci 79: 327–336
Van der Ploeg AT, Bolhuis PA, Wolterman RA, Visser JW, Loonen MC, Busch HF, Reuser AJ (1988) Prospect for enzyme therapy in glycogenosis II variants: a study on cultured muscle cells. J Neurol 235: 392–396
Van der Ploeg AT, Loonen MC, Bolhuis PA, Busch HM, Reuser AJ, Galjaard H (1988) Receptor-mediated uptake of acid alpha-glucosidase corrects lysosomal glycogen storage in cultured skeletal muscle. Pediatr Res 24: 90–94
Van der Ploeg AT, Kroos MA, Willemsen, R, Brons NH, Reuser AJ (1991) Intravenous administration of phosphorylated acid alpha-glucosidase leads to uptake of enzyme in heart and skeletal muscle of mice. J Clin Invest 87: 513–518
Van Hove JL, Yang HW, Wu JY, Brady RO, Chen YT (1996) High-level production of recombinant human lysosomal acid alpha-glucosidase in Chinese hamster ovary cells which targets to heart muscle and corrects glycogen accumulation in fibroblasts from patients with Pompe disease. Proc Natl Acad Sci U S A 93: 65–70
Author information
Authors and Affiliations
Corresponding author
Additional information
Published online: 13 August 2002
Rights and permissions
About this article

Cite this article
Reuser, A.J.J., Van den Hout, H., Bijvoet, A.G.A. et al. Enzyme therapy for Pompe disease: from science to industrial enterprise. Eur J Pediatr 161, S106–S111 (2002). https://doi.org/10.1007/BF02680006
Published:
Issue Date:
DOI: https://doi.org/10.1007/BF02680006