
Abstract
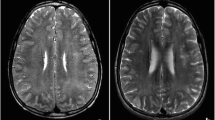
We describe on a 3-year-old child referred for evaluation and therapy of a cerebral vascular accident with residual hemiplegia and partial epilepsy. Metabolic investigations initially showed normal urinary organic acids as well as normal blood and urinary amino acids. Blood carnitine fractions had been pathological and a secondary carnitine deficiency was diagnosed and treated by oral L-carnitine supplementation. During carnitine treatment, abnormal urinary acylcarnitine profiles were noticed with excessive amounts of several carnitine esters including propionylcarnitine, butyryl-and/or isobutyryl-carnitine, isovaleryl- and/or 2-methylbutyryl-carnitine, hexanoylcarnitine and octanoyl-carnitine. Subsequently, an urinary organic acid profile suggestive of glutaric aciduria type 11 was recorded during a clinical decompensation crisis. Morphological and biochemical studies on skeletal muscle and skin fibroblasts were performed and confirmed the existence of a defect of the mitochondrial β-oxidadation pathways with lipidic myopathy, reduced palmitate and octanoate oxidation rates in cultured fibroblasts. Glutaric aciduria type 11 increases the list of metabolic disorders characterized by hemiplegia and other sequelae of brain ischaemia such as stroke-like episode, seizures, aphasia, ataxia and myoclonia, similar to those seen in MELAS.
Similar content being viewed by others


Abbreviations
- ETF :
-
electron transfer flavoprotein
- ETF:QO :
-
electron transfer flavoprotein ubiquinone oxidoreductase
- MADD :
-
multipe acyl-CoA dehydrogenase deficiency (glutaric aciduria type II)
- MELAS :
-
mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes
References
Adams RA, Lyon G (1982) Neurology of hereditary metabolic diseases of children. Hemisphere Publishing Corporation, Mc Graw-Hill Book Company, Washington
Agardh C-D, Rosén I, Ryding E (1983) Persistent vegetative state with high cerebral blood flow following profound hypoglycemia. Ann Neurol 14: 482–486
Amendt BA, Rhead WJ (1986) The multiple acyl-coenzyme A dehydrogenation disorders, glutaricacidemia type II and ethylmalonic/adipic aciduria. J Clin Invest 78: 205–213
Auer RN, Siesjo RK (1988) Biological differences between ischemia, hypoglycaemia, and epilepsy. Ann Neurol 24:699–707
Auer RN, Kalimo H, Olsson Y, Wieloch T (1985) The dentate gyrus in hypolycemia. Pathology implicating excitotoxin-mediated neuronal necrosis. Acta Neuropathol 67: 279–288
Bennett MJ, Hale DE (1992) Defects of mitochondrial -oxidation enzymes. In: Ferrari R, Dimauro S, Sherwood G (eds) L-carnitine and its role in medicine: from function to therapy. Academic Press, New York, pp 187–206
Boers GHJ, Smals AGH, Trijbels FJM, Fowler B, Bakkeren JAJM, Schoonderwaldt HC, Meijer WJ, Kloppenborg PWC (1985) Heterozygosity for homocystinuria in premature peripheral and cerebral occlusive arterial disease. NEngl J Med 313: 709–715
Cederblad G, Harper P, Lindgren K (1986) Spectrophotometry of carnitine in biological fluids and tissue with Cobas Bio centrifugal analyser. Clin Chem 32:342–346
Di Donato S, Frerman FE, Rimoldi M, Rinaldo P, Taroni F, Wiesmann UN (1986) Systemic carnitine deficiency due to lack of electron transfer flavoprotein:ubiquinone oxidoreductase. Neurology 36: 957–963
Dusheiko G, Kew MC, Joffe BI, Lewin JR, Mantagos S, Tanaka K (1979) Recurrent hypoglycemia associated with glutaric aciduria type II in an adult. NEngl J Med 301: 1405–1409
Endo H, Hasegawa K, Narisawa K, Tada K, Kagawa Y, Ohta S (1989) Defective gene in lactic acidosis: abnormal pyruvate dehydrogenase El alpha subunit caused by a frame shift. Am J Hum Genet 44: 358–364
Fontaine M, Porchet N, Largilliére C, Marrakchi S, Lhermitte M, Aubert JP, Degand P (1989) Biochemical contribution to diagnosis and study of a new case of D-glyceric acidemia/aciduria. Clin Chem 35: 2148–2151
Frerman FE, Goodman SI (1985) Deficiency of electron transfer flavoprotein or electron transfer flavoprotein: ubiquinone oxidoreductase in glutaric acidemia type II fibroblasts. Proc Natl Acad Sci USA 82: 4517–4520
Frerman FE, Goodman SI (1989) Glutaric acidemia type II and defects of the mitochondrial respiratory chain. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic basis of inherited disease, 6th edn. Mc GrawHill, New York, pp 915–931
Goodman SI, Noremberg MD, Shikes RH, Breslich DJ, Moe PG (1977) Glutaric aciduria: biochemical and morphologic considerations. J Pediatr 90: 746–750
Grauw LM de, Smit LME, Brocstedt M, Meijer Y, Klei-v. Moorsel Jvd, Jakobs C (1990) Acute hemiparesis as the presenting sign in heterozygote for ornithine transcarbamylase deficiency. Neuropediatrics 21: 133–135
Hale DE, Bennett MJ (1992) Fatty acid oxidation disorders: a new class of metabolic diseases. J Pediatr 121: 1–11
Heidenreich R, Natowicz M, Hainline BE, Berman P, Kelley RI, Hillman RE, Berry GT (1988) Acute extrapyramidal syndrome in methylmalonic acidemia: “metabolic stroke” involving the globus pallidus. J Pediatr 113: 1022–1027
Hoppel CL, Kerr DS, Dahms B, Roessmann C (1987) Deficiency of the reduced nicotinamide adenine dinucleotide dehydrogenase components of complex I of mitochondrial electron transport. Fatal infantile lactic acidosis and hypermetabolism with skeletal-cardiac myopathy and encephalopathy. JClin Invest 80: 71–77
Hurko O, Reynafarje B, Kunel R (1986) Familial mitochondrial encephalopathy, lactic acidosis, and stroke (MELAS) associated with cytochrome oxidase deficiency. Am J Genet 25: 716A
Loehr JP, Goodman SI, Frerman FE (1990) Glutaric acidemia type II: heterogeneity of clinical and biochemical phenotypes. Pediatr Res 27: 311–315
Kobayashi M, Morishita H, Sugiyama N, Yokochi K, Nakano M, Wada Y, Hotta Y, Terauchi A, Nonoaka I (1987) Two cases of NADH-Coenzyme Q reductase deficiency: relationship to MELAS syndrome. J Pediatr 110: 223–227
Koga Y, Nonaka I, Kobayashi M, Tojyo M, Nihei K (1988) Muscle pathology in complex I (NADH CoQ reductase) deficiency. Ann Neurol 24: 749–756
Kuriyama M, Umezaki H, Fukuda Y, Osame M, Koike K, Tateishi J, Igata M (1984) Mitochondrial encephalopathy with lactate-pyruvate elevation and brain infarctions. Neurology 34: 72–77
Lowes S, Rose ME (1990) Simple and unambiguous method for identifying urinary acylcarnitines using gas chromatography — mass spectrometry. Analyst 155: 511–516
MacDonald JT, Brown DR (1979) Acute hemiparesis in juvenile insulin-dependent diabetes mellitus (JIDDM). Neurology 29: 893–896
Malouf R, Brust JCM (1985) Hypoglycaemia: causes, neurological manifestations and outcome. Ann Neurol 17: 421–430
Meer A, Dubois B, Dormont D, Grimaldi A (1988) Interet dc I'im-agerie par resonnance magnétique au décours d'un coma hypoglycémique severe. Press Med 17: 1368
Pavlakis SG, Philips PC, Di Mauro S, De Vivo DC, Rowland LP (1984) Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes: a distinctive clinical syndrome. Ann Neurol 16: 481–488
Pearson DJ, Tubbs PK, Chase JFA (1974) Carnitine and acylcarnitines. In: Bergmeyer HU (ed) Methods of enzymic analysis, 2nd edn. Academic Press, New York, 4: 1762–1764
Portnoy HD (1968) Transient “ischemic” attacks produced by carotid stenosis and hypoglycemia. Neurology 18: 830–832
Schoenberger BS, Mellinger JF, Schoenberger DG (1978) Cerebrovascular disease in infants and children. A study of incidence, clinical features and survival. Neurology 28: 763–768
Scriver CR, Beaudet AL, Sly WS, Valle D (1989) The metabolic basis of inherited disease. Mc Graw-Hill, New York
Siesjö BK (1981) Cell damage in the brain: a speculative synthesis. J Cereb Blow Flow Metab 1: 155–185
Siesjö BK (1988) Hypoglycemia, brain metabolism, and brain damage. Diabetes Metab Rev 4: 113–141
Tanaka K, Mantagos S, Genel M, Seashore MR, Billings BA, Baretz BH (1977) New defect in fatty acid metabolism with hypoglycaemia and organic aciduria. Lancet II: 986–987
Vianey-Liaud C, Divry P, Gregersen N, Mathieu M (1987) The inborn errors of mitochondrial fatty acid oxidation. J Inherited Metab Dis 10: 159–198
Vianey-Saban C, Mousson B, Bertrand C, Stamm D, Dumoulin R, Zabot MT, Divry P, Floret D, Mathieu M (1993) Carnitine palmitoyltransferase I deficiency presenting as a Reye-like syndrome without hypoglycaemia. Eur J Pediatr 152: 334–338
Visser M de, Scholte HR, Schutgens RBH, Bolhuis PA, Luyt-Houwen IEM, Vaandrager-Verduin MHM, Veder HA, Oey PL (1986) Riboflavin-responsive lipid-storage myopathy and glutaric aciduria type II of early adult onset. Neurology 36: 367–372
Wallis WE, Donaldson I, Scott RS, Wilson J (1985) Hypoglycemia masquerading as cerebrovascular disease (hypoglycemic hemiplegia). Ann Neurol 18: 510–512
Wenler ID, Kerr DS, Ho L, Lusk MM, Pepin RA, Javed AA, Mole JE, Jesse BW, Tekkimkara TJ (1988) Heterogenous expression of protein and mRNA in pyruvate dehydrogenase deficiency. Proc Natl Acad Sci USA 85: 7336–7340
Yamaguchi S, Orii T, Suzuki Y, Maeda K, Oshima M, Hashimoto T (1991) Newly identified forms of electron transfer flavoprotein deficiency in two patients with glutaric aciduria type II. Pediatr Res 29: 60–63
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Vallée, L., Fontaine, M., Nuyts, JP. et al. Stroke, hemiparesis and deficient mitochondrial β-oxidation. Eur J Pediatr 153, 598–603 (1994). https://doi.org/10.1007/BF02190669
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF02190669