
Abstract
3-Hydroxyacyl-CoA dehydrogenase deficiency is a newly recognised fatty acid oxidation disorder with a usually fatal outcome. We present a further patient who presented with hypoketotic hypoglycaemia, hepatopathy, secondary carnitine deficiency and increased plasma long-chain acylcarnitines. 3-Hydroxydicarboxylic aciduria was present and the diagnosis confirmed in cultured skin fibroblasts. Our patient is compared with those reported in the literature with respect to clinical symptoms, differential diagnosis and possible therapeutic regimens.
Similar content being viewed by others
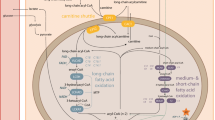

Abbreviations
- CoA :
-
co-enzyme A
- LCAD :
-
long-chain acyl-CoA dehydrogenase
- LCHAD :
-
long-chain hydroxyacyl-CoA dehydrogenase
- MCAD :
-
medium-chain acyl-CoA dehydrogenase
- MCT :
-
medium-chain triglycerides
- SCAD :
-
short-chain acyl-CoA dehydrogenase
- VLCAD :
-
very long-chain acyl-CoA dehydrogenase
References
Bertini E, Dionisi-Vici C, Garavaglia B, Burlina AB, Sabatelli M, Rimoldi M, Bartuli A, Sabetta G, DiDonato S (1992) Peripheral sensory-motor polyneuropathy, pigmentary retinopathy, and fatal cardiomyopathy in long-chain 3-hydroxy-acyl-CoA dehydrogenase deficiency. Eur J Pediatr 151:121–126
Carpenter K, Pollitt RJ, Middelton B (1992) Human liver long-chain 3-hydroxyacyl-coenzyme A dehydrogenase is a multifunctional membrane-bound beta-oxidation enzyme of mitochondria. Biochem Biophys Res Commun: 183:443–448
Corr PB, Creer MH, Yamada KA, Saffitz JE, Sobel BE (1989) Prophylaxis of early ventricular fibrillation by inhibition of acylcarnitine accumulation. J Clin Invest 83:927–936
Demaugre F, Bonnefont J-P, Brivet M, Cepancec C, Pollitt RJ, Priestly BL, Saudubray J-M, Leroux J-P (1992) Pathophysiological approach to carnitine palmitoyltransferase II deficiencies. In: Coates PM, Tanaka K (eds). Progress in clinical and biological research, vol 375: new developments in fatty acid oxidation. Wiley-Liss, New York, pp 301–309
Dionisi-Vici C, Burlina AB, Bertini E, Bachmann C, Mazziotta MRM, Zacchello F, Sabetta G, Hale DE (1991) Progressive neuropathy and recurrent myoglobinuria in a child with longchain 3-hydroxyacyl-coenzyme A deficiency. J Pediatr 118:744–746
Duran M, Wanders RJA, Jager JP de, Dorland L, bruinvis L, Ketting D, Iljist L, Sprang van FJ (1991) 3-Hydroxydicarboxylic aciduria due to long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency associated with sudden neonatal death: protective effect of medium-chain triglyceride treatment. Eur J Pediatr 150:190–195
El-Fakhiri M, Middleton B (1979) The existence of two different L-3-hydroxyacyl-coenzyme A dehydrogenases in rat tissues. Biochem Soc Trans 7: 392–393
Fischbach PS, Corr PB, Yamada KA (1992) Long-chain acylcarnitine increases intracellular Ca++ and induces after depolarisation in adult ventricular myocytes. Circulation 96:748
Greter J, Lindstedt S, Seeman H, Steen G (1980) 3-Hydroxydecanedioic acid and related homologues: urinary metabolites in ketoacidosis. Clin Chem 26:261–265
Hagenfeldt L, Döblen U von, Holme E, Alm J, Brandberg G, Enocksson E, Lindberg L (1990) 3-Hydroxydicarboxylic aciduria — a fatty acid oxidation defect with severe prognosis. J Pediatr 116:387–392
Hale DE, Bennett MJ (1992) Fatty acid oxidation disorders: a new class of metabolic diseases. J Pediatr 121:1–11
Hale DE, Batshaw ML, Coates PM, Frerman FE, Goodman SI, Singh I, Stanley CA (1985) Long-chain acyl coenzyme A dehydrogenase deficiency. An inherited cause of nonketotic hypoglycemia. Pediatr Res 19: 666–671
Hale DE, Thorpe C, Braat K, Wright JH, Roe CR, Coates PM, Hashimoto T, Glasgow AM (1990) The L-3-hydroxyacyl-CoA dehydrogenase deficiency. In: Tanaka K, Coates PM (eds) Fatty acid oxidation: clinical, biochemical and molecular aspects. Alan R. Liss, New York, pp 503–510
Ijlst L, Wanders RJA, Ushikubo S, Kamijo T, Hashimoto T (1993) Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: new information on the mutant protein and first results of mutation analysis. Abstracts of the 31st Symposium SSIEM, Manchester P061
Jackson S, Bartlett K, Land J, Moxon ER, Pollitt RJ, Leonard JV, Turnbull DM (1991) Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Pediatr Res 29:406–411
Moore R, Glasgow JFT, Bingham MA, Dodge JA, Pollitt RJ, Olpin SE, Middleton B, Carpenter K (1993) Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency — diagnosis, plasma carnitine fractions and management in a further patient. Eur J Pediatr 152:433–436
Olpin SE, Manning NJ, Carpenter K, Middleton B, Pollitt RJ (1992) Differential diagnosis of hydroxydicarboxylic aciduria based on release of3H2O from [9, 10-3H] myristic and [9, 10-3H] palmitic acids by intact cultured fibroblasts. J Inherited Metab Dis 15:883–891
Pollitt RJ (1990) Clinical and biochemical presentations in 20 cases of hydroxydicarboxylic aciduria. In: Tanaka K, Coates PM (eds) Fatty acid oxidation: clinical, biochemical and molecular aspects. Alan R. Liss, New York, pp 495–502
Pollitt RJ, Losty H, Westwood A (1987) 3-Hydroxydicarboxylic aciduria: a distinctive type of intermittent dicarboxylic aciduria of possible diagnostic significance. J Inherited Metab Dis 10 [Suppl 2]:266–269
Poll-The BT, Bonnefont J-P, Ogier H, Charpentier C, Pelet A, Le Fur JM, Jakobs C, Kok RM, Duran M, Divry P, Scotto J, Saudubray J-M (1988) Familial hypoketotic hypoglycaemia associated with peripheral neuropathy, pigmentary retinopathy and C6–C14 hydroxydicarboxylic aciduria. A new defect in fatty acid oxidation? J Inherited Metab Dis 11 [suppl 2]:183–185
Ribes A, Riudor E, Navarro C, Boronat M, Marti M, Hale DE (1992) Fatal outcome in a patient with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J Inherited metab Dis 15: 278–279
Rocchiccioli F, Aubourg P, Bougneres PF (1986) Medium- and long-chain dicarboxylic aciduria in patients with Zellweger syndrome and neonatal adrenoleukodystrophy. Pediatr Res 20: 62–66
Rocchiccioli F, Wanders RJA, Aubourg P, Viancy-Liaud C, Ijlst L, Fabre M, Cartier N, Bougneres P-F (1990) Deficiency of long-chain 3-hydroxyacyl-CoA dehydrogenase: a cause of lethal myopathy and cardiomyopathy in early childhood. Pediatr Res 28:657–662
Sewell AC, Böhles HJ (1991) 4-Hydroxycyclohexanecarboxylic acid: a rare compound in urinary organic acid analysis. Clin Chem 37:1301–1302
Sewell AC, Herwig J, Böhles HJ, Rinaldo P, Bhala A, Hale DE (1993) A new case of short-chain acyl-CoA dehydrogenase deficiency with isolated ethylmalonic aciduria. Eur J Pediatr 152:922–924
Stanley CA (1992) Plasma and mitochondrial membrane carnitine transport defects. In: Coates PM, Tanaka K (eds) Progress in clinical and biological research, Vol 375: new developments in fatty acid oxidation. Wiley-Liss, New York, pp 289–301
Stanley CA, Hale DE, Coates PM, Hall CL, Corkey BE, Yang W, Kelley RI, Gonzales EL, Williamson JR, Baker L (1983) Medium-chain acyl-CoA dehydrogenase deficiency in children with non-ketotic hypoglycaemia and low carnitine levels. Pediatr Res 17: 877–884
Treem WR, Witzleben Ca, Piccoli DA, Stanley CA, Hale DE, Coates PM, Watkins JB (1986) Medium-chain and long-chain acyl CoA dehydrogenase deficiency: clinical, pathologic and ultrastructural differentiation from Reye's syndrome. Hepatology 6: 1270–1278
Wanders RJA, Ijlst L (1992a) Fatty acid β-oxidation in leukocytes from control subjects and medium-chain acyl-CoA dehydrogenase deficient patients. Biochim Biophys Acta 1138: 80–84
Wanders RJA, Ijlst L (1992b) Long-chain 3-hydroxyacyl-CoA dehydrogenase in leukocytes and chorionic villus fibroblasts: potential for pre- and postnatal diagnosis. J Inherited Metab Dis 15:356–358
Wanders RJA, Ijlst L, Gennip AH van, Jakobs C, Jager JP de, Dorland L, Sprang FJ van, Duran M (1990) Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: identification of a new inborn error of mitochondrial fatty acid β-oxidation. J Inherited metab Dis 13: 311–314
Wanders RJA, Ijlst L, Duran M, Jakobs C, Klerk JBC de, Przyrembel H, Rocchiccioli F, Aubourg P (1991) Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: different clinical expression in three unrelated patients. J Inherited Metab Dis 14:325–328
Wanders RJA, Ijlst L, Poggi F, Bonnefont J-P, Munnich A, Brivet M, Rabier D, Saudubray J-M (1992) Human trifunctional protein deficiency: a new disorder of mitochondrial fatty acid oxidation. Biochem Biophys Res Commun 180:1139–1145
Vamecq J, Draye J-P (1989) Comparison between the formation and the oxidation of dicarboxylcarnitine esters in rat liver and skeletal muscle: possible implications for human inborn disorders of mitochondrial β-oxidation. J Inherited Metab Dis 12:58–63
Yamaguchi S, Indo Y, Coates PM, Hashimoto T, Tanaka K (1993) Identification of very-long-chain acyl-CoA dehydrogenase deficiency in three patients previously diagnosed with long-chain acyl-CoA dehydrogenase deficiency. Eur J Pediatr 34:111–113
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Sewell, A.C., Bender, S.W., Wirth, S. et al. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: A severe fatty acid oxidation disorder. Eur J Pediatr 153, 745–750 (1994). https://doi.org/10.1007/BF01954492
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF01954492