
Summary
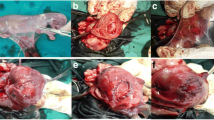
Amnion membrane implantation has been proposed as an approach to enzyme replacement in mucopolysaccharidoses. Human amnion membranes have been subcutaneously implanted in the abdominal wall in 19 patients with mucopolysaccharidoses (MPS I, II and III). A protocol was developed for the objective evaluation of experimental treatments of these patients. Systematic evaluation of the clinical status before and 6 months after amnion membrane implantation reveals no change in function except improvement in joint mobility. The sum of all joint movements showed improvement from baseline values to 6 months after implantation by ANOVA followed bypost-hoc analysis (p<0.056). The only specific joint movements to significantly improve after 6 months were shoulder extension (p<0.01) and hip internal rotation (p<0.05). Serial measurements of the deficient lysosomal enzyme activity in serum and white blood cells did not increase in any patient after amnion membrane implantation. Urinary glycosaminoglycan excretion decreased transiently in 2 of 10 patients after implantation, but a second amnion membrane implantation did not result in any change. Biopsy of the implantation site in 10 patients 6 months after amnion membrane implantation revealed a foreign-body reaction with giant cell formation and fibrosis and no recognizable amnion membrane tissue. We conclude that human amnion membrane implantation is not an effective therapy in mucopolysaccharidoses.
Similar content being viewed by others


References
Adinolfi, M. and Brown, S. Strategies for the correction of enzymatic deficiencies in patients with mucopolysaccharidoses.Dev. Med. Child. Neurol. 26 (1984) 404–408
Adinolfi, M., Akle, C. A., McColl, I., Fensom, A. H., Tansley, L., Connolly, P., Hsi, B. L., Faulk, W. P., Travers, P. and Bodmer, W. F. Expression of HLA antigens beta 2-microglobulin and enzymes by human amniotic epithelial cells.Nature 295 (1982) 325–327
Akle, C. A., Adinolfi, M., Welsh, K. I., Leibowitz, S. and McColl, I. Immunogenicity of human amniotic epithelial cells after transplantation into volunteers.Lancet 2 (1981) 1003–1005
Akle, C., McColl, I., Dean, M., Adinolfi, M., Brown, S., Fensom, A. H., Marsh, J. and Welsh, K. Transplantation of amniotic epithelial membranes in patients with mucopolysaccharidoses.Exp. Clin. Immunogenet. 2 (1985) 43–48
Bartsocas, C. S., Papasotiriou, N., Karageorga, M., Moser, H. W. and Moser, H. W. Hunter's syndrome and Cooley's anaemia in the same patient. Effect of multiple transfusions.Acta Pediatr. Scand. 62 (1973) 66–68
Blumenkrantz, N. and Asboe-Hansen, G. New method for quantitative determination of uronic acids.Anal. Biochem. 54 (1973) 484–489
Booth, C. W. and Nadler, H. L. Plasma infusions in an infant with Hurler's syndrome.J. Pediatr. 82 (1973) 273–278
Brown, F. R. III, Hall, C. W., Neufeld, E. F., Munoz, L. L., Braine, H., Andrzejewski, S., Camargo, E. E., Mark, S. A., Richard, J. M. and Moser, H. W. Administration of iduronate sulfatase by plasma exchange to patients with Hunter syndrome: a clinical study.Am. J. Med. Genet. 13 (1982) 309–318
Caruso, R. C., Kaiser-Kupfer, M. I., Muenzer, J., Ludwig, I. H., Zasloff, M. A. and Mercer, P. A. Electroretinographic findings in the mucopolysaccharidoses.Ophthalmology 93 (1986) 1612–1616
Danes, B. S., Degnan, M., Salk, L. and Flynn, F. J. Plasma infusions in the Hurler syndrome. Influence during the first year of life.Am. J. Dis. Child. 125 (1973) 533–535
Dean, M. F., Muir, H., Benson, P. F., Button, L. R., Batchelor, J. R. and Bewick, M. Increased breakdown of glycosaminoglycans and appearance of corrective enzyme after skin transplants in Hunter syndrome.Nature 257 (1975) 609–612
Dean, M. F., Stevens, R. L., Muir, H., Benson, P. F., Button, L. R., Anderson, R. L., Boylston, A. and Mowbray, J. Enzyme replacement therapy by fibroblast transplantation: long-term biochemical study in three cases of Hunter's syndrome.J. Clin. Invest. 63 (1979) 138–145
Dekaban, A. S., Holden, K. R. and Constantopoulos, G. Effects of fresh plasma or whole blood transfusions on patients with various types of mucopolysaccharidosis.Pediatrics 50 (1972) 688–692
Di Ferrante, N., Nichols, B. L., Donnelly, P. V., Neri, G., Hrgovcic, R. and Berglund, R. K. Induced degradation of glycosaminoglycans in Hurler's and Hunter's syndromes by plasma infusion.Proc. Natl. Acad. Sci. USA 68 (1971) 303–307
Erickson, R. P., Sandman, R., Robertson, W. V. B. and Epstein, C. J. Inefficacy of fresh frozen plasma therapy of mucopolysaccharidosis II.Pediatrics 50 (1972) 693–701
Gibbs, D. A., Spellacy, E., Tompkins, R., Watts, R. W. and Mowbray, J. F. A clinical trial of fibroblast transplantation for the treatment of mucopolysaccharidoses.J. Inher. Metab. Dis. 6 (1983) 62–81
Guise, K. S., Korneluk, R. G., Waye, J., Lamhonwah, A. M., Quan, F., Palmer, R., Ganschow, R. E., Sly, W. S. and Gravel, R. A. Isolation and expression in Escherichia coli of a cDNA clone encoding human beta-glucuronidase.Gene 34 (1985) 105–110
Hall, C. W., Liebaers, I., DiNatale, P. and Neufeld, E. F. Enzymatic diagnosis of the genetic mucopolysaccharide storage disorders.Methods Enzymol. 50 (1978) 439–456
Harris, R. and Ukaejiofo, E. O. Tissue typing using a routine one-step lymphocyte separation procedure.Br. J. Haematol. 18 (1970) 229–235
Hobbs, J. R., Hugh-Jones, K., Barrett, A. J., Byrom, N., Chambers, D., Henry, K., James, D. C., Lucas, C. F., Rogers, T. R., Benson, P. F., Tansley, L. R., Patrick, A. D., Mossman, J. and Young, E. P. Reversal of clinical features of Hurler's disease and biochemical improvement after treatment by bone marrow transplantation.Lancet 2 (1981) 709–712
Hugh-Jones, K., Hobbs, J. R., Chambers, D., White, S., Byrom, N., Williamson, S., Barrett, J. and Henry, K. Bone marrow transplantation in mucopolysaccharidoses. In Barranger, J. A., Brady, R. O. (eds.)Molecular Basis of Lysosomal Storage Disorders, Academic Press, New York, 1984, pp. 411–421
Hunt, J. S., Andrews, G. K., Fishback, J. L., Feess, M. and Wood, G. W., Amnion membrane epithelial cells express class I HLA and contain class I HLA mRNA.J. Immunol. 140 (1988) 2790–2795
Hussels, I. E., Eikman, E. A., Kenyon, K. R. and McKusick, V. A. Treatment of mucopolysaccharidoses.Birth Defects 10 (1974) 212–225
Kaplan, A., Achord, D. T. and Sly, W. S. Phosphohexosyl components of a lysosomal enzyme are recognized by pinocytosis receptors on human fibroblasts.Proc. Natl. Acad. Sci. USA 74 (1977) 2026–2030
Knudson, A. G., Di Ferrante, N. and Curtis, J. E. Effect of leukocyte transfusion in a child with type II mucopolysaccharidosis.Proc. Natl. Acad. Sci. USA, 68 (1971) 1738–1741
Krivit, W., Pierpont, M. E., Ayaz, K., Tsai, M., Ramsay, N. K., Kersey, J. H., Weisdorf, S., Sibley, R., Snover, D. and McGovern, M. M. Bone marrow transplantation in the Maroteaux-Lamy syndrome (mucopolysaccharidosis type VI). Biochemical and clinical status 24-months after transplantation.N. Engl. J. Med. 311 (1984) 1606–1611
Krivit, W., Whitley, C. B., Chang, P., Shapiro, E., Belani, K. G., Snover, D., Summers, C. G. and Blazar, B. Lysosomal storage diseases treated by bone marrow transplantation: review of 21 patients. In Johnson, F. J. and Pochedly, C. (eds.)Bone Marrow Transplantation in Children, Raven Press, New York, 1990, pp. 261–287
Lakatos, M. and DiFerrante, N. Simple measurement of urinary glycosaminoglycans and degradation products.Biochem. Med. 9 (1974) 256–260
Lowry, O. H., Rosebrough, N. J., Farr, A. L. and Randall, R. J.J. Biol. Chem. 193 (1951) 265–275
Muenzer, J. Mucopolysaccharidoses. In Barness, L. (ed.)Advances in Pediatrics, 33, Year Book Medical Publishers, Chicago, (1986) pp. 269–302
Neufeld, E. F. The biochemical basis for mucopolysaccharidoses and mucolipidoses. In Steinberg, A. G. and Bearn, A. G. (eds.)Progress in Medical Genetics, Vol. X, Grune and Stratton, New York, 1974, pp. 81–101
Neufeld, E. F. and Muenzer, J. The mucopolysaccharidoses. In Scriver, C. R., Beaudet, A. L., Sly, W. S. and Valle, D. (eds.)The Metabolic Basis of Inherited Disease, 6th edn., McGraw-Hill, New York, 1989, pp. 1565–1588
Orii, T., Yamaguchi, M., Minami, R., Ito, H. and Nakao, T. Influence of fresh plasma infusions on patients with genetic mucopolysaccharidosis.Tohoku J. Exp. Med. 114 (1974) 385–392
Robertson, D. A., Freeman, C., Nelson, P. V. and Hopwood, J. J. Human glucosamine-6-sulfatase cDNA reveals homology with steroid sulfatase.Biochem. Biophys. Res. Commun. 30 (1988) 218–224
Rome, L.H., Weissman, B. and Neufeld, E. F. Direct demonstration of binding of a lysosomal enzyme, α-l-iduronidase, to receptors on cultured fibroblasts.Proc. Natl. Acad. Sci. USA 76 (1979) 2331–2334
Scaggiante, B., Pineschi, A., Sustersich, M., Andolina, M., Agosti, E. and Romeo, D. Successful therapy of Niemann-Pick disease by implantation of human amniotic membrane.Transplantation 44 (1987) 59–61
Schuchman, E. H., Jackson, C. E. and Desnick, R. J. Human arylsulfatase B: MOPAC cloning, nucleotide sequence of a full-length cDNA, and regions of amino acid identity with arylsulfatases A and C.Genomics 6 (1990) 149–158
Shull, R. M., Hastings, N. E., Selcer, R. R., Jones, J. B., Smith, J. R. and Cullen, W. C. Bone marrow transplantation in canine mucopolysaccharidosis I. Effects within the nervous system.J. Clin. Invest. 79 (1987) 435–443
Sly, W. S., Fischer, H. D., Gonzalez-Noriega, A., Grubb, J. H. and Natowicz, M. Role of the 6-phosphomannosyl-enzyme receptor in intracellular transport and adsorptive pinocytosis of lysosomal enzymes.Methods Cell. Biol. 23 (1981) 191–214
Thompson, J. N., Roden, L. and Reynertson, R., Oligosaccharide substrates for heparin sulfamidase.Anal. Biochem. 152 (1986) 412–422
Tylki-Szymanska, A., Maciejko, D., Kidawa, M., Jablonska-Budaj, U. and Czartoryska, B. Amniotic tissue transplantation as a trial of treatment in some lysosomal storage diseases.J. Inher. Metab. Dis. 8 (1985) 101–104
Wasteson, A. and Neufeld, E. F. Iduronate sulfatase from human plasma.Methods Enzymol. 83 (1982) 573–578
Wenger, D. A., Gasper, P. W., Thrall, M. A., Dial, S. M., LeCouteur, R. A. and Hoover, E.A. Bone marrow transplantation in the feline model for arylsulfatase B deficiency.Birth Defects 22 (1986) 177–186
Whitley, C. B., Kersey, J. H., Tsai, M. Y., Sharp, H. L., Sibley, R. K., Snover, D. C., Filipovich, A. H., Ramsay, N. K. C. and Krivit, W. Reversal of hepatic glycosaminoglycans accumulation in Hurler syndrome after bone marrow transplantation.Am. J. Hum. Genet. 36 (1984) 81s
Whitley, C. B., Belani, K. G., Ramsay, N. K. C., Kersey, J. H. and Krivit, W. Progressive elevation of CSF glycosaminoglycan in neuropathic mucopolysaccharidosis and normal levels after bone marrow transplantation.Am. J. Hum. Genet. 39 (1986) A87
Wilson, P. J., Morris, C. P., Anson, D. S., Occhiodoro, T., Bielicki, J., Clements, P. R. and Hopwood, J. J. Hunter syndrome: Isolation of an iduronate-2-sulfatase cDNA clone and analysis of patient DNA.Proc. Natl. Acad. Sci. USA 87 (1990) 8531–8535
Yatziv, S., Statter, M., Abeliuk, P., Meshulam, M. and Russel, A. A therapeutic trial of fresh plasma infusions over a period of 22 months in two siblings with Hunter syndrome.Isr. J. Med. Sci. 11 (1975) 802–808
Yeager, A. M., Singer, H. S., Buck, J. R., Matalon, R., Brennan, S., O'Toole, S. O. and Moser, H. W. A therapeutic trial of amniotic epithelial cell implantation in patients with lysosomal storage diseases.Am. J. Med. Genet. 22 (1985) 347–355
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Muenzer, J., Neufeld, E.F., Constantopoulos, G. et al. Attempted enzyme replacement using human amnion membane implantations in mucopolysaccharidoses. J Inherit Metab Dis 15, 25–37 (1992). https://doi.org/10.1007/BF01800340
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF01800340