
Summary
Acid maltase deficiency (glycogenosis type II; M. Pompe) exists in three different clinical forms:
-
1.
Infantile course with rapid progressive myopathy and cardiac involvement. This form presents as a rule a fatal disease with death in early infancy.
-
2.
Juvenile form characterized by progressive proximal myopathy beginning in the adolescence associated with only mild cardiomyopath.
-
3.
Very rare adult cases showing a late onse myopathy without cardiac symptoms, but with development of a respiratory deficiency.
Two cases of the last mentioned type are described and compaired with the typical course of a juvenile form.
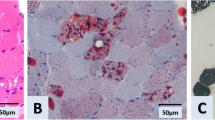
From the morphological point of view there are striking differences between these two forms of the disease. While in the case of juvenile onset myopathy marked vacuoles in most muscle fibers are detectable and an excessive accumulation of glycogen can be demonstrated, structural changes in late onset cases are slight and as a rule can be shown only in scattered fibers, whereas great areas of muscle tissue appear normal.
Biochemically, the juvenile form is characterized by a clearly elevated glycogen content in combination with a significantly decreased activity of acid maltase. Adult forms in contrast show only decreased enzyme activity without detectable glycogen elevation.
Possible causes for the different expression of the disease are discussed and diagnostic difficulties in the detection of late onset forms pointed out.
Similar content being viewed by others


Literatur
Engel AG (1970) Acid maltase deficiency in adults: Studies in four cases of a syndrome which may mimic muscular dystrophy or other myopathies. Brain 93:599–615
Engel AG, Gomez MR (1970) Acid maltase levels in muscle in heterozygous acid maltase deficiency and in non-weak and neuromuscular disease controls. J Neurol Neurosurg Psychiat 33:801–804
Engel AG, Gomez MR, Marjoric E, Seybold ME, Lamert EH (1973) The spectrum and diagnosis of acid maltase deficiency. Neurology 23:95–106
Hers HG (1964) Glycogen storage disease. Adv Metab Disord 1:1–44 and 335–336
Hers HG, Verhue W, Hoof F van (1967) The Determination of Amylo-1,6-Glucosidase. Eur J Biochem 2:257–264
Keppler D, Decker K (1970) Glykogen. Bestimmung mit Amylo-1,6-Glucosidase. In: Bergmeyer HU (Hrsg) Methoden der enzymatischen Analyse. Verlag Chemie, Weinhcim
Kölmel HW, Assmus H, Seiler D (1974) Myopathie bei Saure-Maltase-Mangel. Die Pompesche Erkrankung im Jugend- und Erwachsenenalter. Arch Psychiat Nervenkr 218:93–106
Lilienthal JL, Zierler KL, Folk BP, Buka R, Riley MJ (1950) A reference base and system for analysis of muscle constituents. J Biol Chem 183:501–508
Lowry OH, Rosebrough NJ, Farr AL (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193:265–275
Loonen MCB (1979) The variability of Pompe's disease. A clinical, biochemical and genetic study of glyocgen storage disease type 2, or acid maltase deficiency. Profeschrift, University Hospital Rotterdam, The Netherlands
Mehler M, DiMauro S (1977) Residual acid maltase activity in late-onset acid maltase deficiency. Neurology 27:178–184
Pompc JC (1933) Hypertrophie idiopathique du coeur. Ann Anat Pathol 10:23
Pongratz D (1976) Differentialdiagnose der Erkrankungen der Skelettmuskulatur anhand von Muskelbiopsien. Thieme, Stuttart
Pongratz D, Hübner G, Deufel Th, Wieland OH, Pongratz E, Liphardt E (1979) Klinische, morphologische und biochemische Befunde bei Carnitinmangelmyopathien. Klin Wochenschr 57:927–936
Steckel F, Gieselmann V, Waheed A, Hasilik A, Figura K v, Elferink RO, Kalsbeek R, Tager JM (1982) Biosynthsis of acidα-glucosidase in late onset forms of glycogenosis type II (Pompe's discase). FEBS-Letters 150:69–76
Author information
Authors and Affiliations
Additional information
Mit Unterstützung der Friedrich Baur-Stiftung, München und der Deutschen Forschungsgemeinschaft
Rights and permissions
About this article
Cite this article
Pongratz, D., Hübner, G., Deufel, T. et al. Zur Kenntnis mitigierter adulter Formen des Mangels an saurer Maltase (Morbus Pompe). Klin Wochenschr 61, 743–750 (1983). https://doi.org/10.1007/BF01497401
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF01497401