
Summary
A 10 year old boy from an ethnically German family developed severe hemolysis after ingestion of fava beans. Extremely low activity of G-6-PD (1–4% of normal) was found in the patient's red cells. The enzyme deficiency was also observed in the red cells of two other members of this family.
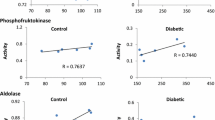
This G-6-PD variant was partially purified and biochemically characterized. The defect enzyme has an increased affinity for its substrate G-6-P. The Km for G-6-P is 26–28 µM (normal: 40–60 µM). The Km for NADP is within the normal range (4.7 µM). The enzyme variant shows an abnormal biphasic pH optimum curve and a marked thermal instability. The rate of utilization of the substrate analogues 2-deoxyglucose-6-phosphate and galactose-6-phosphate is elevated.
Some characteristics of the investigated G-6-PD variant resemble those of the Mediterranean variety of G-6-PD, but there is no identity. In addition, the electrophoretic behaviour of the patient's leucocyte G-6-PD shows no similarity with the Mediterranean type of the enzyme.
The differences between the known types of variants and the type described here are sufficient to postulate the existence of a new G-6-PD variant.
Zusammenfassung
Ein 10jähriger Junge einer deutschstämmigen Familie erlitt eine schwere Hämolyse nach dem Genuß von Fava-Bohnen. In den Erythrocyten des Patienten wurde eine sehr niedrige Aktivität der G-6-PD gefunden (1–4% der normalen Aktivität). Der Enzymmangel wurde auch in den Erythrocyten von 2 weiteren Familienangehörigen beobachtet.
Die G-6-PD-Variante des Patienten wurde teilweise gereinigt und biochemisch charakterisiert. Das defekte Enzym hat eine erhöhte Affinität zu seinem Substrat G-6-P. Die Km für G-6-P beträgt 26–28 µM (normal 40–60 µM). Die Km für NADP liegt im Normalbereich (4,7 µM). Die Enzymvariante hat eine abnormale biphasische pH-Abhängigkeitskurve und eine erhebliche Thermolabilität. Die Umsatzrate der Substratanalogen 2-Desoxyglucose-6-Phosphat und Galactose-6-Phosphat ist erhöht.
In einigen Eigenschaften gleicht die untersuchte G-6-PD-Variante dem mediterranen Typ der G-6-PD. Es besteht aber keine Identität. Auch das elektrophoretische Verhalten der G-6-PD aus den Leukocyten des Patienten weicht von dem der mediterranen G-6-PD ab.
Auf Grund der unterschiedlichen Eigenschaften des hier beschriebenen Enzym-Typs und der bisher bekannten Varianten wird die Existenz einer neuen G-6-PD-Variante mit Favismus angenommen.
Similar content being viewed by others


References
Bannert, N., Thal, W., Lubas, E.: Glucose-6-Phosphat-dehydrogenase-Mangel als Ursache des hämolytischen Ikterus bei einer deutschen Familie. Mschr. Kinderheilk.117, 675 (1969)
Benöhr, H. Chr., Usener, H., Wagner, H., Kleihauer, E., Waller, H. D.: Spontane Hämolyse bei einem Kranken mit Glukose-6-P-Dehydrogenase-Mangel (mediterraner Typ) und heterozygoter Beta-Thalassämie. Med. Welt24, 1091 (1970).
—— Waller, H. D.: Eigenschaften der Glucose-6-P-Dehydrogenase Typ „Tübingen“. Klin. Wschr.48, 71 (1970).
Beutler, E., Mathai, C. K., Smith, J. E.: Biochemical variants of glucose-6-phosphate dehydrogenase giving rise to congenital nonspherocytic hemolytic disease. Blood31, 131 (1968).
—— L-dopa and favism. Blood36, 523 (1970).
Busch, D., Boie, K.: Glucose-6-Phosphat-Dehydrogenase-Defekt in Deutschland. II. Eigenschaften des Enzyms (Typ Freiburg). Klin. Wschr.48, 74 (1970).
Cremer, Hj., Witt, I.: Kongenitale nichtsphärozytäre hämolytische Anämie infolge G-6-PD-Inaktivität. Arch. Kinderheilk.182, 75 (1970).
Cohen, P., Rosemeyer, M. A.: Human glucose-6-phosphate dehydrogenase: purification of the erythrocyte enzyme and the influence of ions on its activity. Europ. J. Biochem.8, 1 (1969).
Davis, B. J.: Disc electrophoresis. II. Method and application to human serum proteins. Ann. N. Y. Acad. Sci.121, 404 (1964).
Gehrmann, G., Sturm, A., Amelung, D.: Favismus in Deutschland. Dtsch. med. Wschr.88, 1865 (1963).
Helge, H., Borner, K.: Kongenitale nichtsphärozytäre hämolytische Anämie, Katarakt und Glukose-6-Phosphat- Dehydrogenase-Mangel. Dtsch. med. Wschr.91, 1584 (1966).
Hennemann, H. H., Esser, U., Stecher, G.: Chronischer, nichtsphärozytärer hämolytischer Ikterus bei völligem Fehlen der Glukose-6-Phosphatdehydrogenase-Aktivität der Erythrocyten. Klin. Wschr.42, 1250 (1964).
Johannsen, L. P., Witt, I., Künzer, W.: Favismus bei einer deutschen Familie. Dtsch. med. Wschr.93, 2463 (1968).
Schröter, W., Drescher, J., Fischer, K.: Über eine seltene Form des Glukose-6-Phosphatdehydrogenase-Mangels mit kongenitaler nichtsphärozytärer hämolytischer Anämie. Klin. Wschr.45, 355 (1967).
Sitzmann, F. C., Kornhuber, B., Stephan, U., Truckenbrodt, H.: Enzymopenische hämolytische Anämie infolge Glukose-6-Phosphatdehydrogenasemangel bei einem Kind deutscher Abstammung. Z. Kinderheilk.93, 189 (1965).
Waller, H. D., Löhr, G. W., Gayer, J.: Hereditäre, nichtsphärozytäre hämolytische Anämie durch Glukose-6-Phosphatdehydrogenase-Mangel. Klin. Wschr.44, 122 (1966).
Weidtman, V., Tönz. O.: Glukose-6-Phosphat-Dehydrogenase-Mangel der Erythrocyten bei 3 deutschen Kindern. Nachweis einer frischen Mutation. Klin. Wschr.44, 446 (1966).
Witt, I., Witz, D.: Reinigung und Charakterisierung von Phosphopyruvat-Hydratase (=Enolase, EC 4.2.1.11) aus Neugeborenen- und Erwachsenen-Erythrocyten. Hoppe-Seylers Z. physiol. Chem.351, 1232 (1970).
World Health Organization Technical Report Series: Standardization of procedures for the study of glucose-6-phosphate dehydrogenase. Report of a WHO Scientific Group. Wld Hlth Org. tech. Rep. Ser. 366 (1967).
Author information
Authors and Affiliations
Additional information
This paper is dedicated to Professor Dr. Joachim Kühnau for his 70th birthday.
S. Yoshioka wishes to thank the Alexander von Humboldt-Stiftung for a research grant.
Rights and permissions
About this article
Cite this article
Witt, I., Yoshioka, S. Biochemical characterization of a glucose-6-phosphate dehydrogenase variant with favism: G-6-PD Zähringen. Klin Wochenschr 50, 205–209 (1972). https://doi.org/10.1007/BF01487145
Issue Date:
DOI: https://doi.org/10.1007/BF01487145