
Summary
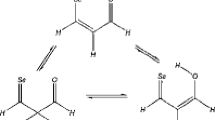
The relative stabilities of syn- and anti-isomers of 2-substituted furan and thiophene carbonyl derivatives are investigated by theab initio MO method. The energy differences between the rotamers are 1–3 kcal mol−1 but the barriers to rotation are ca. 10 kcal mol−1 so that free rotational mode is predicted to be a rather difficult process. Application of the self-consistent reaction field (SCRF) method to account for the solvent effect indicates that the isomer with a higher dipole moment (syn) is favored in solution. An electron withdrawing 2-substituent favors syn-isomers for furan carbonyls in contrast to thiophene carbonyls for which anti-isomers are favored. These trends are ascribable to a decrease in electrostatic repulsive and attractive interactions, respectively, in the syn forms of furan and thiophene carbonyls. Contribution of non-bonded repulsive interaction in the anti-isomer is important for the relative stability of the syn-isomer of furan carbonyl derivative. Solvent effects due to higher dielectric continuum are small on the absolute values of energy differences but can reverse the order of stability of the two isomers due to a greater stability acquired by an isomer (syn) with higher dipole moment in solution. The major factor determining stability, or instability, of syn-isomer is a repulsive electrostatic interaction between the two oxygen atoms for furan carbonyls and an attractive electrostatic interaction between the sulfur and oxygen atoms for thiophene carbonyls.
Similar content being viewed by others


References
Determination of reactivity by MO theory, Part 96. Part 95., Lee D, Kim CK, Lee BS, Lee BC, Lee I (submitted for publication)
Roques B, Combrisson S, Riche C, Pascard-Billy C (1970) Tetrahedron 26:3555;
Larsen RB, Nicolaisen F, Nielsen JT (1972) Acta Chem Scand 26:1736;
Abraham RJ, Siverns TM (1972) Tetrahedron 28:3015;
Monnig F, Dreizler H, Rudolph HD (1965) Z Naturforsch 20a:1323;
Arlinger L, Dahlquist K, Forser S (1970) Acta Chem Scand 24:662
Abraham RJ, Bretschneider E (1974) In: Orville-Thomas WJ (ed.) Internal rotation in molecules, Chap. 13 Wiley, London
Allen G, Bernstein HJ (1955) Can J Chem 33:1055
Pople JA, Beveridge DL (1970) Approximate molecular orbital theory. McGraw-Hill, New York;
Wong MW, Frisch MJ, Wiberg KB (1991) J Am Chem Soc 113:4776
Jorgensen WL (1983) J Phys Chem 87:5304;
Jorgensen WL (1989) Acc Chem Res 22:184
Kirkwood JG (1934) J Chem Phys 2: 351;
Onsager L (1936) J Am Chem Soc 58:1486
Rivail JL, Terryn B, Rinaldi D, Ruiz-Lapez MF (1985) THEOCHEM 120:387;
Tapia O, Gascinski O (1975) Mol Phys 29:1653;
Karelson MW, Tamm T, Katritzky AR, Szefran M, Zerner MC (1990) Int J Quantum Chem 37:1;
Karelson MM, Katritzky AR, Zerner MC (1986) Int J Quantum Chem Symp 20:521
Frisch MJ, Trucks GW, Head-Gordon M, Gill PMW, Wong MW, Foresman JB, Johnson BG, Schlegel HB, Robb MA, Replogle ES, Gomperts R, Andress JL, Raghavachari K, Binkley JS, Gonzalez C, Martin RL, Fox DJ, Defrees DJ, Baker J, Stewart JJP, Pople JA (1992) Gaussian 92, Revision C. Gaussian Inc, Pittsburgh, PA
Moller C, Plesset MS (1934) Plup Rev 46:618;
Hehre WJ, Radom L, Schleyer PvR, Pople JA (1986)Ab initio molecular orbital theory. Wiley-Interscience, New York
Ref [10b, p 470];
Pople JA, Krishnan R, Schlegl HB, Binkley JS (1979) Int J Quant Chem s13:225
Taken from Tables of Interatomic Distances and Configuration in Molecule and Ions. Sutton LE (ed) (1965), Sp. Pub. no. 18, The Chem Soc, London
Carsky P, Urban M (1980)Ab initio calculations methods and applications in chemistry, Lecture Notes in Chem, No 16. Springer Berlin, p 74
Taft RW (1956) In: Newman M (ed) Steric effects in organic chemistry, New York, Chap. 13, Wiley
The inductive (σ1) and resonance (σR) substituent constants are taken from Klumpp GW (1982) Reactivity in organic chemistry. Wiley, New York, p 104
Fleming I (1976) Frontier orbitals and organic chemical reactions, Chap. 4. Wiley, London
Lee I, Cheun YG, Yang K (1982) J Comput Chem 3:565
[Ref 10c, p 266]
Dahlquist K-I, Forsen S (1965) J Phys Chem 69:4062;
Dahlquist K-I, Forsen S (1970) Acta Chem Scand 24:662
Payne PW, Allen LC (1977) Modern theoretical chemistry, Vol 4. Schaeffer HF (ed) Plenum, New York, p 29
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Han, IS., Kim, C.K., Jung, H.J. et al. Ab Initio studies on the rotational equilibria of 2-substituted furan and thiophene carbonyl derivatives. Theoret. Chim. Acta 93, 199–210 (1996). https://doi.org/10.1007/BF01113417
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF01113417