
Summary
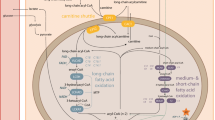
Skin fibroblast carnitine uptake studies may identify and differentiate primary and secondary carnitine deficiency disorders. To confirm the specificity of these studies in differentiating primary from secondary carnitine deficiency disorders, we have studied carnitine uptake in the cultured skin fibroblasts from 5 children who have various enzymatic defects in intramitochondrial β-oxidation including short-chain, medium-chain and long-chain acyl-CoA dehydrogenase and short-chainl-3-hydroxyacyl-CoA dehydrogenase deficiencies, and in 4 children with cytochrome oxidase deficiency. Carnitine uptake was normal in the intramitochondrial β-oxidation cases, suggesting other mechanisms for their carnitine deficiency. Therefore, intramitochondrial β-oxidation defects associated with carnitine deficiency can be differentiated from primary carnitine deficiency not only by the presence of an abnormal dicarboxylic aciduria but by normal skin fibroblast carnitine uptake. In contrast to these findings, carnitine uptake in the cultured skin fibroblasts of four children with secondary carnitine deficiency due to cytochrome oxidase deficiency demonstrated a partial decrease in the maximal velocity of uptake (20–47% controlV max), similar to that observed in the primary carnitine deficiency heterozygotes. We propose that this observation may be due to a generalized decrease in intracellular ATP, thus decreasing the efficiency of the energy- and sodium-dependent carnitine transporter. We conclude that carnitine uptake studies in cultured skin fibroblasts will contribute to an understanding of the mechanisms of carnitine depletion in the primary and secondary carnitine deficiency disorders.
Similar content being viewed by others


References
Angelini C, Trevisan C, Isaya G, Pegolo G, Vergani L (1987) Clinical varieties of carnitine and carnitine palmitoyltransferase deficiency.Clin Biochem 20: 1–7.
Arts WFM, Scholte HR, Loonen MCB et al (1987) Cytochromec oxidase deficiency in subacute necrotizing encephalomyelopathy.J Neurol Sci 77: 103–115.
Bremer J (1983) Carnitine-metabolism and functions.Physiol Rev 63: 1420–1480.
Chapoy PR, Angelini C, Brown WJ, Stiff JE, Shug AL, Cederbaum SD (1980) Systemic carnitine deficiency — a treatable inherited lipid-storage disease presenting as Reye's syndrome.N Engl J Med 303: 1389–1394.
Coates PM, Hale DE, Stanley CA, Glasgow AM (1984) Systemic carnitine deficiency simulating Reye syndrome.J Pediatr 105: 679.
Coates PM, Hale DE, Stanley CA, Corkey BE, Cortner JA (1985) Genetic deficiency of medium-chain acyl coenzyme A dehydrogenase: studies in cultured skin fibroblasts and peripheral mononuclear leukocytes.Pediatr Res 19: 671–676.
Coates PM, Hale DE, Finocchiaro G, Tanaka K, Winter SC (1988) Genetic deficiency of short-chain acyl-coenzyme A dehydrogenase deficiency in cultured fibroblasts from a patient with muscle carnitine deficiency and severe skeletal muscle weakness.J Clin Invest 81: 171–175.
DiMauro S (1979) Metabolic myopathies. In Vinken PJ, Bruyn GW, eds.Handbook of Clinical Neurology, vol 41, Diseases of Muscle, Part II. New York: North Holland, 175–234.
DiMauro S, Mendell JR, Sahenk Z et al (1980) Fatal infantile mitochondrial myopathy and renal dysfunction due to cytochrome-c-oxidase deficiency.Neurology 30: 795–804.
Engel AG, Angelini C (1973) Carnitine deficiency of human skeletal muscle with associated lipid storage myopathy: a new syndrome.Science 179: 899–902.
Engel AG, Rebouche CJ, Wilson DM, Glasgow AM, Romshe CA, Cruse RP (1981) Primary systemic carnitine deficiency. II. Renal handling of carnitine.Neurology 31: 819–825.
Eriksson BO, Lindstedt S, Nordin I (1988) Hereditary defect in carnitine membrane transport is expressed in skin fibroblast. [Letter]Eur J Pediatr 147: 662–663.
Fong JC, Schulz H (1978) On the rate determining step of fatty acid oxidation in heart. Inhibition of fatty acid oxidation by 4-pentenoic acid.J Biol Chem 253: 6917–6922.
Frerman FE, Goodman SI (1985) Fluorometric assay of acyl-CoA dehydrogenases in normal and mutant human fibroblasts.Biochem Med 33: 38–44.
Hale DE, Cruse RP, Engel AG (1985) Familial systemic carnitine deficiency. [Letter]Arch Neurol 42: 1133.
Hale DE, Stanley CA, Coates PM (1990) Genetic defects of acyl-CoA dehydrogenases: Studies using an electron transfer flavoprotein reduction assay. In Tanaka K, Coates PM, eds.Fatty Acid Oxidation: Clinical, Biochemical and Molecular Aspects. New York: Alan R. Liss, 333–348.
Holme E, Greter J, Jacobson CE et al (1989) Carnitine deficiency induced by pivampicillin and pivmecillinam therapy.Lancet 2: 469–473.
Holme E, Greter J, Jacobson CE et al (1990) Studies on secondary carnitine deficiency induced by pivaloyloximethyl-esterified antibiotics.Proceedings V International Congress Inborn Errors of Metabolism, Asilomar California June 1–5, W2.1.
Husain M, Steenkamp DJ (1983) Electron transfer flavoprotein from pig liver mitochondria. A simple purification and re-evaluation of some of the molecular properties.Biochem J 209: 541–545.
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the folin phenol reagent.J Biol Chem 193: 265–275.
McGarry JD, Foster DW (1976) An improved and simplified radioisotopic assay for the determination of free and esterified carnitine.J Lipid Res 17: 277–281.
Müller-Höcker J, Pongratz D, Deufel T, Trijbels, JMF, Endres W, Hübner G (1983) Fatal lipid storage myopathy with deficiency of cytochrome-c-oxidase and carnitine. A contribution to the combined cytochemical-finestructural identification of cytochrome-c-oxidase in longterm frozen muscle.Virchows Arch [A]399: 11–23.
Rebouche CJ, Engel AG (1983) Carnitine metabolism and deficiency syndromes.Mayo Clin Proc 58: 533–540.
Rebouche CJ, Mack DL (1984) Sodium gradient-stimulated transport ofl-carnitine into renal brush border membrane vesicles: kinetics, specificity and regulation by dietary carnitine.Arch Biochem Biophys 235: 393–402.
Reichmann H, Srihari T, Pette D (1983) Ipsi and contralateral fibre transformations by cross-reinnervation. A principle of symmetry.Pflugers Arch 397: 202–208.
Schulz H (1974) Long-chain enoyl coenzyme A hydratase from pig heart.J Biol Chem 249: 2704–2709.
Srere PA (1969) Citrate synthase.Methods Enzymol 13: 3–11.
Stanley CA (1987) New genetic defects in mitochondrial fatty acid oxidation and carnitine deficiency.Adv Pediatr 34: 59–88.
Stanley CA, Berry GT, Treem WR (1990) Differences in the evolution of carnitine deficiency among secondary carnitine deficiency disorders.Proceedings V International Congress Inborn Errors of Metabolism, Asilomar, California June 1–5, W17.10.
Tein I, De Vivo DC, Bierman F et al (1990) Impaired skin fibroblast carnitine uptake in primary systemic carnitine deficiency manifested by childhood carnitine-responsive cardiomyopathy.Pediatr Res 28: 247–255.
Tein I, De Vivo DC, Hale DE et al (1991) Short-chainl-3-hydroxyacyl-CoA dehydrogenase deficiency in muscle: A new cause for recurrent myoglobinuria and encephalopathy.Ann Neurol 30: 415–419
Treem WR, Stanley CA, Finegold DN, Hale DE, Coates PM (1988) Primary carnitine deficiency due to a failure of carnitine transport in kidney, muscle and fibroblasts.N Engl J Med 319: 1331–1336.
Tripp ME, Katcher ML, Peters HA et al (1981) Systemic carnitine deficiency presenting as familial endocardial fibroelastosis: a treatable cardiomyopathy.N Engl J Med 305: 385–390.
Turnbull DM, Bartlett K, Stevens DL et al (1984) Short-chain acyl-CoA dehydrogenase deficiency associated with a lipid-storage myopathy and secondary carnitine deficiency.N Engl J Med 311: 1232–1236.
Waber LJ, Valle D, Neill C, DiMauro S, Shug A (1982) Carnitine deficiency presenting as familial cardiomyopathy: a treatable defect in carnitine transport.J Pediatr 101: 700–705.
Wharton DC, Tzagaloff A (1967) Cytochrome oxidase from beef heart mitochondria.Methods Enzymol 20: 245–250.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Tein, I., De Vivo, D.C., Ranucci, D. et al. Skin fibroblast carnitine uptake in secondary carnitine deficiency disorders. J Inherit Metab Dis 16, 135–146 (1993). https://doi.org/10.1007/BF00711327
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/BF00711327