
Summary
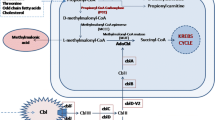
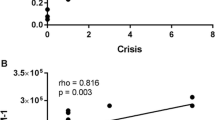
Nine patients with maple syrup urine disease (MSUD), of whom eight were detected by mass-screening of neonates for inherited metabolic desease, were studied to determine possible relationships between clinical features and properties of the branched-chain α-keto acid dehydrogenase complex (BCKDH) in cultured lymphoblastoid cells. Based on their tolerance for leucine and on the clinical manifestations observed after 2 years of age, most could be classified into three types; classical (tolerate less than 600 mg of leucine per day, N=2), intermediate (N=3) and intermittent (N=3) types. In the other patient two of these three phenotypes were present. The BCKDH activities measured at a lower α-ketoisovaleric acid concentration (0.054 mM) were 0.026±0.015 in classical, 0.118±0.016 in intermediate and 0.625±0.139 in intermittent types and 7.052±0.779 (nmol/h per milligram of protein) in two controls, respectively; the differences being statistically significant (P<0.01, classical vs intermediate types; P<0.01, intermediate vs intermittent types; P<0.01, intermittent vs control). Kinetic and immunochemical analyses of the BCKDH revealed that, although there are a few exceptions, classical, intermediate and intermittent types correspond to the enzyme properties of sigmoidal kinetics with E1β subunit deficiency, near-sigmoidal kinetics with E1β subunit deficiency and hyperbolic kinetics with E2 subunit deficiency of the BCKDH, respectively.
Similar content being viewed by others

References
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chuang DT, Niu W-L, Cox RP (1981) Activities of branched-chain 2-oxo acid dehydrogenase and its components in skin fibroblasts from normal and classical-maple-syrup-urine-disease subjects. Biochem J 200:59–67
Dancis J, Levitz M, Miller S, Westall RG (1959) Maple syrup urine disease. Br Med J 1:91–93
Dancis J, Hutzler J, Rokkones T (1967) Intermittent branched-chain ketonuria. Variant of maple-syrup-urine disease. N Engl J Med 276:84–89
Dancis J, Hutzler J, Snyderman SE, Cox RP (1972) Enzyme activities in classical and variant forms of maple syrup urine disease. J Pediatr 81:312–320
Danner DJ, Armstrong N, Heffelfinger SC, Seqell ET, Priest JH, Elsas LJ (1985) Absence of branched chain acyl-transferase as a cause of maple syrup urine disease. J Clin Invest 75:858–860
Harper AE, Miller RH, Block KP (1984) Branched-chain amino acid metabolism. Annu Rev Nutr 4:409–454
Heffelfinger SC, Sewell ET, Danner DJ (1983) Identification of specific subunits of highly purified bovine liver branched-chain ketoacid dehydrogenase. Biochemistry 22:5519–5522
Hutson SM, Harper AE (1981) Blood and tissue branched-chain amino and α-keto acid concentrations: effects of diet, starvation, and disease. Am J Clin Nutr 34:173–183
Ide S, Hayakawa T, Okabe K, Koike M (1967) Lipoamide dehydrogenase from human liver. J Biol Chem 242:54–60
Indo Y, Kitano A, Endo F, Akaboshi I, Matsuda I (1987) Altered kinetic properties of the branched-chain α-keto acid dehydrogenase complex due to mutation of the β-subunit of the branchedchain α-keto acid decarboxylase (E1) component in lymphoblastoid cells derived from patients with maple syrup urine disease. J Clin Invest 80:63–70
Jinno Y, Akaboshi I, Katsuki T, Matsuda I (1984a) Study on established lymphoid cells in maple syrup urine disease. Correlation with clinical heterogeneity. Hum Genet 65:358–361
Jinno Y, Akaboshi I, Matsuda I (1984b) Complementation analysis in lymphoid cells from five patients with different forms of maple syrup urine disease. Hum Genet 68:54–56
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Livesey G, Lund P (1980) Enzymic determination of branched-chain amino acids and 2-oxo acids in rat tissues. Biochem J 188:705–713
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265–275
Lyons LB, Cox RP, Dancis J (1973) Complementation analysis of maple syrup urine disease in heterokaryons derived from cultured human fibroblasts. Nature 243:533–535
Matsuda I, Yamamoto J, Nagata N, Ninomiya N, Akaboshi I, Ohtsuka H, Katsuki T (1977) Lysosomal enzyme activities in cultured lymphoid cell lines. Clin Chim Acta 80:483–486
Menkes JH, Hurst PL, Craig JM (1954) A new syndrome progressive familial infantile cerebral dysfunction associated with an unusual urinary substance. Pediatrics 14:462–466
Morris MD, Lewis BD, Doolan PD, Harper HA (1961) Clinical and biochemical observations on an apparently nonfatal variant of branched-chain keto aciduria (maple syrup urine disease). Pediatrics 28:918–923
Pettit FH, Yeaman SJ, Reed LJ (1978) Purification and characterization of branched chain α-keto acid dehydrogenase complex of bovine kidney. Proc Natl Acad Sci USA 75:4881–5522
Reed LJ, Damuni Z, Merryfield ML (1985) Regulation of mammalian pyruvate and branched-chain α-keto acid dehydrogenase complexes by phosphorylation-dephosphorylation. Curr Top Cell Regul 27: 41–49
Rosenberg LE, Scriver CR (1980) Disorders of amino acid metabolism. In: Bondy PK, Rosenberg LE (eds) Metabolic control and disease. Saunders, Philadelphia, pp 583–776
Schulman JD, Lustberg TJ, Kennedy JL, Museles M, Seegmiller JE (1970) A new variant of maple syrup urine disease (branched chain ketoaciduria). Am J Med 49:118–124
Scriver CR, Mackenzie S, Clow CL, Delvin E (1971) Thiamino-responsive maple-syrup-urine disease. Lancet I:310–312
Singh S, Willers I, Goedde HW (1977) Heterogeneity in maple syrup urine disease: aspects of cofactor requirement and complementation in cultured fibroblasts. Clin Genet 11:277–284
Tanaka K, Rosenberg LE (1983) Disorders of branched chain amino acid and organic acid metabolism. In: Stanbury JB, Wyngaarden JB, Fredrickson DS, Goldstein JL, Brown MS (eds) The metabolic basis of inherited disease. McGraw-Hill, New York, pp 440–473
Wendel U, Langenbeck U (1984) Intracellular levels and metabolism of leucine and α-ketoisocaproate in normal and maple syrup urine disease fibroblasts. Biochem Med 31:294–302
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Indo, Y., Akaboshi, I., Nobukuni, Y. et al. Maple syrup urine disease: a possible biochemical basis for the clinical heterogeneity. Hum Genet 80, 6–10 (1988). https://doi.org/10.1007/BF00451447
Received:
Revised:
Issue Date:
DOI: https://doi.org/10.1007/BF00451447