
Abstract
Information integration and workflow technologies for data analysis have always been major fields of investigation in bioinformatics. A range of popular workflow suites are available to support analyses in computational biology. Commercial providers tend to offer prepared applications remote to their clients. However, for most academic environments with local expertise, novel data collection techniques or novel data analysis, it is essential to have all the flexibility of open-source tools and open-source workflow descriptions. Workflows in data-driven science such as computational biology have considerably gained in complexity. New tools or new releases with additional features arrive at an enormous pace, and new reference data or concepts for quality control are emerging. A well-abstracted workflow and the exchange of the same across work groups have an enormous impact on the efficiency of research and the further development of the field. High-throughput sequencing adds to the avalanche of data available in the field; efficient computation and, in particular, parallel execution motivate the transition from traditional scripts and Makefiles to workflows. We here review the extant software development and distribution model with a focus on the role of integration testing and discuss the effect of common workflow language on distributions of open-source scientific software to swiftly and reliably provide the tools demanded for the execution of such formally described workflows. It is contended that, alleviated from technical differences for the execution on local machines, clusters or the cloud, communities also gain the technical means to test workflow-driven interaction across several software packages.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
An enormous amount of data is available in public databases, institutional data archives or generated locally. This remote wealth is immediately downloadable, but its interpretation is hampered by the variation of samples and their biomedical condition, the technological preparation of the sample and data formats. In general, all sciences are challenged with data management, and particle physics, astronomy, medicine and biology are particularly known for data-driven research.
The influx of data further increases with more technical advances and higher acceptance in the community. With more data, the pressure raises on researchers and service groups to perform analyses quickly. Local compute facilities grow and have become extensible by public clouds, which all need to be maintained and the scientific execution environment be prepared.
Software performing the analyses steadily gains functionality, both for the core analyses and for quality control. New protocols emerge that need to be integrated in existing pipelines for researchers to keep up with the advances in knowledge and technology. Small research groups with a focus on biomedical research sensibly avoid the overhead entailed in developing full solutions to the analysis problem or becoming experts in all details of these long workflows, instead concentrating on the development of a single software tool for a single step in the process. Best practices emerge, are evaluated in accompanying papers and are then shared with the community in the most efficient way [37].
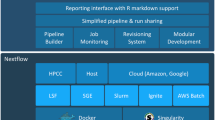
Over the past 5 years, with the avalanche of high-throughput sequencing data in particular, the pressure on research groups has risen drastically to establish routines in non-trivial data processing. Companies like Illumina offer hosted bioinformatics services (http://basespace.illumina.com/) which developed into a platform in its own right. This paper suggests workflow engines to take the position of a core interface between the users and a series of communities to facilitate the exchange, verification and efficient execution of scientific workflows [32]. Prominent examples of workflow and workbench applications are Apache Taverna [38], with its seeds in the orchestration of web interfaces; Galaxy [1, 11], which is popular for allowing the end-users’ modulation of workflows in nucleic acid sequencing analysis and other fields; the lightweight highly compatible Nextflow [8]; and KNIME [5] which has emerged from machine learning and cheminformatics environments and is now also established for bioinformatics routine sequence analyses [13].Footnote 1
Should all these workflow frameworks bring the software for the execution environment with them (Fig. 1) or should they depend on system administrators to provide the executables and data they need?

Workflow specifications comprise formal references to a range of packages to be installed without specifying the execution environment. The separation between problem-specific software, and the packages a Linux distribution provides, is fluid
2 Methods
The foundations of functional workflows are sources for readily usable software. The software tools of interest are normally curated by maintainers into installable units that we will generically call “packages”. We reference the following package managers that can be installed on one single system and, together, represent all bioinformatics open-source software packages in use today:
-
Debian GNU/Linux (http://www.debian.org) is a software distribution that encompasses the kernel (Linux) plus a large body of other user software including graphical desktop environments, server software and specialist software for scientific data processing. Debian and its derivatives share the deb package format with a long history of community support for bioinformatics packages [26, 27].
-
GNU Guix (https://www.gnu.org/software/guix/) is a package manager of the GNU project that can be installed on top of other Linux distributions and represents the most rigorous approach towards dependency management. GNU Guix packages are uniquely isolated by a hash value computed over all inputs, including the source package, the configuration and all dependencies. This means that it is possible to have multiple versions of the same software and even different combinations of software, e.g. Apache with ssl and without ssl compiled in on a single system.
-
Conda (https://conda.io/docs/) is a package installation tool that, while popularised by the Anaconda Python distribution, can be used to manage software written in any language. Coupled with Bioconda [7] (https://bioconda.github.io/), its software catalogue provides immediate access to many common bioinformatics packages.
-
Brew (https://brew.sh) is a package manager that distributes rules to compile source packages, originally designed to work on macOS but capable of managing software on Linux environments as well.
The common workflow language (CWL) [3] is a set of open-community-made standards with a reference implementation for maintaining workflows with a particular strength for the execution of command-line tools. CWL will be adopted also by already established workflow suites like Galaxy and Apache Taverna. It is also of interest as an abstraction layer to reduce complexity in current hard-coded pipelines. The CWL standards provide
-
formalisms to derive command-line invocations to software
-
modular, executable descriptions of workflows with auto-fulfillable specifications of run-time dependencies
The community embracing the CWL standards provides
-
tools to execute workflows in different technical environments, i.e. local clusters and remote cloudsFootnote 2
-
auto-installable run-time environments using lightweight isolation from the underlying operating system
For isolation from an operating system (Fig. 1), it is now popular to adopt software container technologies such as Docker (https://www.docker.com/). Increasingly, high-performance computing sites turn to the compatible Singularity [21] (http://singularity.lbl.gov/), which is considered to be well suited for research containerisation, as it is also for non-privileged users. The Open Container Initiative’s Open Container Format and Open Container Interface configuration files (https://www.opencontainers.org/) specify the contents of these containers (such as via Dockerfiles), representing an interface to the bespoke packages of the Bioconda community and the underpinning base of a GNU/Linux distribution (such as that produced by the Debian project). In high-performance compute environments, the acceptance rate of Docker is relatively low due to its technical overhead and demand for special privileges (https://thehftguy.com/2016/11/01/docker-in-production-an-history-of-failure). With the focus of this article being on the provisioning of software, we use the availability of auto-configured Docker images as an example that has a low barrier to enter for new users; other alternative configuration and software management engines can also be used to create setups of the same packages, e.g. Puppet, Chef or Ansible (https://docs.ansible.com/ansible/intro_installation.html). Automated or programmable deployment technologies are also enabling for collaborative computational environments, for example, by sharing folders at an Infrastructure-as-a-Service cloud provider (e.g. [35] or https://aws.amazon.com/health/genomics/). Incompatible versions of interacting tools would disrupt the workflow as a whole. This motivated Dockstore to shift from dynamic image creation with Ansible to ready-prepared Docker images [30]. The exact specification of versions from snapshot.debian.org, Bioconda, or unspecified versions of two tools together in the same release of a distribution is expected to overcome this difficulty.
The Debian GNU/Linux distribution provides base systems for the Docker images and has a rich repository of scientific software with special interest groups for science in general, and additional efforts, for example Astronomy, Bioinformatics and Chemistry (https://blends.debian.org/med/tasks/) [27]. The Bioconda community provides additional packages homogeneously for all Linux distributions and macOS. Differences between these communities and consequences for the specification of workflows are described below.
3 Results and Discussion
The scientific method needs scientific software to be inspectable, that is, it should be open source (https://www.heise.de/tp/features/Open-Science-and-Open-Source-3443973.html). Openness is increasingly a requirement from funding agencies or the policy of institutes—either way, it is good scientific practice. Bioinformatics is no exception, which would not be noteworthy if there was not the vicinity to the pharmaceutical industry and medical technology. It is likely that the past dispute over the public accessibility of the human genome has manifested the open-source principles in this community ([19] and Ewan Birney, 2002 at http://archive.oreilly.com/pub/a/network/2002/01/28/bioday1.html). Beyond inspectability, for the exchange and collaborative development of workflows, software licences must allow redistribution of the software. While well-maintained, pre-compiled, non-inspectable, black-box tools may also be redistributable, this is also potentially undesirable for the technical reason that the pre-compiled binary will not use the latest processor features and optimised external libraries to their full potential. Targeted optimisation is not achievable with any centralised distribution of software since the end-users’ hardware is too diverse to optimise for them all; however, only the most CPU-intensive packages need to be optimised and this can be done centrally for various common cloud platforms. With scientific software distributed as source code, local recompilations are relatively easy and the automated compilation recipes that are provided by Linux distributions facilitate that.
3.1 Distribution: Getting Software from Its Developers to Its Users
A software distribution may be small. Common on traditional closed-source operating systems like macOS or Windows, the developers themselves are likely to offer a readily installable package to download. Binaries of the executable and also binaries of the libraries the software uses are bundled. The version of the self-developed software is the latest, but the versions of the libraries are whatever the author knows to be compatible with a particular release.
In the philosophy of Linux distributions, common functions should be broken out into libraries, and libraries should only be installed once with all tools depending on one single installation. This philosophy developed, in part, as a reaction to the problems that were experienced in dealing with monolithic software systems where big, expensive computers (mainframes, minicomputers) required considerable effort from local system administrators to build software from source (from tape, possibly from the vendor), or by installing binary versions directly from vendors. Inflexibility of solutions and a “look but don’t touch” policy from vendors made local tailoring and improvement in software problematic in many instances. A key step was the creation of pre-compiled, Internet-distributed Linux distributions which saved the local system administrator from the tiresome task of compiling everything by hand. An important stage in the development of the distribution was the standardisation of which compiler was used, the versions of libraries that would be included, the file locations on disk and the removal of pointless variations between software packages [37]. Such requirements are codified in documents such as the Filesystem Hierarchy Standard (FHS, http://www.pathname.com/fhs/) and Debian Policy (https://www.debian.org/doc/debian-policy/).
Standardisation made shared libraries the norm. The shared library model of maintenance only works when the application programming interfaces (APIs) are stable or at least not changing in backwards-incompatible ways on a frequent basis. Such stability permits the use of the same library across software both new and old. Difficulties due to incompatibilities between versions are possible and need to be fixed—in the library or the calling code—as part of regular software maintenance, and the requisite changes are communicated back to the respective authors of the software. In mature and well-designed code bases, incompatible changes are rare and for some languages such as C, there is a formal way by which they can be tracked and described in the shared object name and version (soname and sover, https://www.gnu.org/software/libtool/manual/html_node/Updating-version-info.html). Further, peer pressure and code review help avoid incompatible changes. In less mature software, in code bases that have grown organically, without the benefit of team design, refinement, regular code review, and with different pressures on the development, keeping backwards compatibility during regular development and maintenance is harder and breaking changes are an unwelcome companion. Notably, much scientific software possesses these attributes. Maintainers of software packages in Linux distributions (the largest of which have more than 2000 contributors maintaining packages) use the distribution’s infrastructure to notify difficulties, report them to the developers of the software or prepare a respective fix themselves.
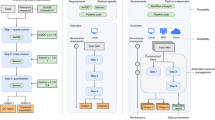
Contributors to Linux distributions see a fluid transition from the code written by the program’s authors to a perpetual maintenance effort to keep the program working for all its users within the distribution and with the current versions of any dependent libraries, as illustrated in Fig. 2. The shared library maintenance model avoids keeping additional, redundant, separate copies of the same or only slightly different versions of a particular library. All tools in the same distribution benefit from the latest advancements of that library, including security-related bug fixes. Through wider testing, problems are found earlier and for a larger fraction of routines in the library. Except for the possibility of newly introduced problems, this increases performance for everyone. With eyeballs concentrating on the same latest version, this also helps the early detection of new concerns.
There is a consensus that in an ideal world the authors of many tools and libraries indeed collaborate closely to ensure that shared library resources are performant, flexible and suitable for all tools that use them. In Linux distributions, the shared library development model indeed works well for the packages very close to the core of the distribution, such as the ones required to get the machine to boot and show the graphical user interface or based on suitably mature APIs. However, use of the shared library maintenance model comes at the cost of an increased time from software being released by its author to the time that the software is released in a stable release of a Linux distribution.
New versions of a distribution are released periodically, with different user groups seeking different release cadences: desktop users might be happy with relatively short cycles every 6 months (Ubuntu, Fedora), or perhaps more stable cycles of 24 months (Debian, Ubuntu LTS), while infrastructure and large Linux installations (e.g. a compute cluster) are likely to run older releases with even longer release cycles (Red Hat family, 4–5 years). Compiling new software to older releases of the distribution (“backporting”) is typically not automated and, due to the extensive dependency trees of the shared library model, can make this effort quite difficult.
For a scientist, there are competing forces for their tooling. It is the science that they care about, not the finer details of distribution maintenance, and so the convenience and reproducibility of the analytical environment created by the Linux distribution should not be underestimated. At the same time, the prospect of a delay in obtaining for new research tools is not acceptable as they will not benefit from new optimisations, new features or, most importantly, improved accuracy. From the perspective of the scientist, the scientific software is special in its need to be up to date, while the base system functionality (which may be just sufficiently recent to provide compatible compilers and core libraries) is a mere cumbersome consequence of the computational demands. Through the lens of looking for the latest tools to address a scientific problem, the Linux distribution and the delays in process and QA it entails become an unwelcome barrier to code delivery. Container-based deployment of tools thus offers an alluring possibility: a stable base distribution running on the hardware with containers of bespoke tools deployed on top. Within the container, a domain-specific (or language-specific) package manager can be used to install and upgrade the tooling.
With a focus on problem-tailored workflows that are comprised of multiple tools, scientists have a desire for a repository that contains recent releases. However, they still want the benefits of a curated software library in which the software is known to work. The fundamental dichotomy of software management is that both “recent” and “well-tested” are difficult to achieve at the same time. The maintenance effort is better distributed across a community, to avoid a perpetual investment of time for updates that is unrewarding (both scientifically and in terms of career advancement) for the individual scientist. Thus, to allow for a focus on workflows with better reproducibility across installations, one wants recent scientific software nonetheless readily installable as packages.
The Gentoo Linux distribution popularised the concept of storing readily executable commands for downloading and installing packages in a public code repository (https://wiki.gentoo.org/wiki/Project:Science/Overlay). The approach of distributing compilation recipes rather than compiled code found broad acceptance within the Homebrew initiative for macOS (https://brew.sh) together with its Linuxbrew companion (https://linuxbrew.sh). While technically similar, for bioinformatics, the Bioconda initiative (https://bioconda.github.io/) propelled itself to the heart of the community. Since command-line instructions are mostly the same across Linux distributions, these build instructions can be shared across different execution environments. There is no lock-in to one particular community. Further, these build instructions are often easily adapted to new versions of the software and are technically easy to improve or extend for anyone familiar with software code maintenance with git (http://git-scm.com). By reducing the scope of the integration problem to a smaller software domain, there is less of the overhead that delays traditional Linux distributions. The software is readily installable and suitable for automated installation into pristine environments.
The downside is that a maintenance and installation procedure that works nicely across distributions cannot be deeply integrated with any distribution because the integration and QA work of the package maintainer has not been performed; when the API changes in ways that break compatibility, someone must do the integration work to create the coherent software stack, and someone must be prepared to apply the polish and ensure standards conformance of the package. Yet again a forerunner is the Gentoo distribution with the introduction of its Prefix concept (https://wiki.gentoo.org/wiki/Project:Prefix) to allow for user-defined installation at non-privileged locations [2].
The method of having an “alternative root” prefix with a FHS-like file hierarchy, e.g. /alt/bin, /alt/lib, /alt/var, allows software distribution tools like Brew to have binaries and libraries installed side-by-side with the operating system’s own libraries; in effect being a secondary software distribution, bypassing library version incompatibility issues. One can consider this approach to be pioneered by the StoreAdm system [6] (http://storeadm.sf.net/), which used a file hierarchy of symbolic links to individually captured software installations, relying on the rpath mechanism (https://en.wikipedia.org/wiki/Rpath) to modify the search path for dynamic libraries.
Poor integration can manifest itself in a lack of documentation (manual pages are missing from Bioconda), missing resources or unusual on-disk locations for the included files. While documentation may be provided instead by command-line help in modern tools, integration problems are harder for the user to ameliorate. In Bioconda, Python modules are not installed in a system-accessible fashion but instead with every package in a separate directory and not available to the Python interpreter without further action by the user. While this permits co-installability of package versions, access to provided libraries is less immediate and convenient as when the package is centrally managed such as in Debian. Further, the ability to install multiple versions of the same module does not permit the one Python process to use these multiple versions, even if that might be required by the overall software stack. The container has not removed the need for integration tests and QA work on the stack, merely attempted to reduce the size of the problem domain.
Simple, automated deployment of bespoke analysis tools is synergistic with the wide deployment of container technologies and the dynamic deployment of cloud instances. Both provide almost immediate access to single tools or complete workflows. For large projects, because of the then increasing likelihood of a failure, deployment of the same packages or containers shall be performed in an environment that detects and reacts to such outages [34].

The software distribution process for packages in a Linux distribution such as Debian (upper) and in a smaller immediate-availability catalogue such as Conda/Bioconda (lower). The Conda system renders packages immediately available across many releases of many Linux distributions and the macOS. The arrival in Debian’s “unstable” distribution can be equally fast, then with build and QA tests, grants users time to comment prior to the transition to testing. In the Linux distribution, the new release will not be automatically made available (“backported”) to the current stable release
3.2 Shipping Confidence: A Workflow Perspective
The confidence in the correct execution of a workflow has its foundation in the confidence in all its component tools and their integration. The right workflow must have been selected for the right kind of properly formatted data. A new software installation needs to perform correctly and that should be testable by the local user and also by the package maintainer and integrator through QA tests.
Modern software ships with self-tests including unit tests of functions, functional tests (for a fixed input, the output should not change across installations), interfacing tests (errors in the input should throw the expected error messages) and integration tests with other parts of the ecosystem. The Debian Project, in particular, invests a considerable effort to test every new submission to its distribution:
-
Can it be built? (https://buildd.debian.org/)
-
Can it be installed, upgraded and removed? (https://piuparts.debian.org/)
-
Does its test suite still pass? (https://ci.debian.net/)
-
How does that new version differ? (https://snapshot.debian.org)
Additionally, packages that depend on a newly uploaded library are tested in the same way to prevent problems cascading through the software stack. Packages with self-tests conducted as part of the build process will have these tests executed at build time by the build system, and after the build the piuparts system will test if the package installs, upgrades and removes as it should. The continuous integration (CI) system will install the package and run the defined self-tests to verify that the packages work as expected when installed. Such testing is a key feature of the distribution’s QA work and is important to verify that newer dependencies than those the authors of the software may have had available at the time the software was released, work with the package as built.
A workflow also needs to be tested as a whole, since the exact combination of tools that determines the input of a tool cannot be foreseen by the individual authors. Build dependencies for packages should also be minimal which will constrain interface tests with other tools to static textual representations—an external tool’s update with a change to its default file format will not have an immediate effect. Like regular applications, workflow pipelines may also ship with a test suite [12]. However, there is still a need to develop ways to perform tests in a package-independent manner. We may see a transition from testing for technical completion of a workflow towards the finding of regressions in the performance of the tool. The Genome in the Bottle consortium provides a gold standard [39] for sequencing and variant calling to support respective benchmarking [22]. Others have independently evaluated tools for molecular docking [15], and, generally, every new competing method will have to prove its performance in some way. The local confirmation of such an evaluation would yield the highest possible confidence in a workflow.
The common workflow language (CWL) project has published a standard to describe command-line tools for the integration in workflows. The CWL community provides its own workflow engine for reference and development use. This reference executor can be used as an interface from workflows to command-line tools, e.g. for the Apache Taverna workflow engine, it may substitute an earlier tool wrapper [20] and is also finding acceptance by the Galaxy community. It could also be the means to exchange ways to evaluate the performance of tools and whole workflows independently of any distribution.
Successful transfer of experience from one community to another needs a mapping of software packages across distributions. Source package matching can be performed based on the package name, which should be very similar if not identical, and the home page at which the tool is presented. Each way is not without difficulty, both for very young and very old tools. To the rescue come registries of software like Bio.Tools [17], and the resource identification initiative (https://scicrunch.org/resources) expands the same concept well beyond software. For sharing whole workflows, myExperiment (http://myexperiment.org/) is a well-established repository used by multiple workflow systems and research communities [10], complementing more specific workflow repositories like CWL Viewer [33] (https://view.commonwl.org/), Galaxy’s ToolShed (https://galaxyproject.org/toolshed/workflow-sharing/), and Dockstore [30].
3.3 How Distributions Meet
Recipes for the execution of data analysis tools and workflows refer to a basic image of a Linux distribution plus a series of additional packages to install. When combined with lightweight containers such as Docker or Singularity, the recipe becomes directly installable (Fig. 2) [28]. It needs to be left to the users’ opinion where to draw the line between traditional distribution-provided packages that are already used to communicate with the kernel and the end-user packages of the given scientific discipline, which are often redundantly available from the Linux distribution and a Linuxbrew/Bioconda community, or other repositories like Bioconductor [9] (http://bioconductor.org). There are advantages to each approach, as shown in Table 1.
By way of example, Debian already provides many packages for the R statistical analysis suite. The setup time for the container could be much reduced by pre-installing those into the container prior to cloning and use; the flexibility to do so is left to the user’s discretion. On every computer, local or remote, users have the choice to use software that is directly retrieved from the developers, with all its redundancies, or instead the coherent and curated presentation via their Linux distribution. A Linux distribution cannot and should not provide all possible software for all user communities. However, the software engineering lessons learned in maintaining large distributions should be adopted as much as possible and the effort towards minimal redundancy and maximal testing can be shared within the community.
Software catalogues and registries have a key role in facilitating cooperation and synchronisation of communities, in that these refer to workflows using a particular tool and propose means to install the software, with all the user feedback known from regular “app stores”. Resource identifiers (RRID) [4] offer to act as a common reference point also for scientific software which further strengthens cross-platform activities and reproducibility, albeit not without semantic deficiencies: Sharing the same RRID is both (1) a early version of the tool Bowtie that is still maintained for aligning short reads and (2) a newer version of Bowtie used for new technologies that provide longer reads. (However, this situation is not a problem when the tool identifier (like RRID) is combined with a desired version.) The registries OMICtools [14] and Bio.Tools [17] have begun to integrate Debian’s curated package descriptions into their catalogues. With Debian, all control over the packaging is with the individual package maintainers, but the scientific packages are commonly team-maintained, which facilitates mass changes like the introduction of references to catalogues from Debian in analogy to references to publications that are already offered today. Bulk retrieval of any such edited annotation is possible via the Debian database and its API (https://udd.debian.org/) to bidirectionally maintain links. Bio.Tools is also collaborating with the community of SEQanswers.com [24], and it can be reasonably predicted that further coordination will develop. Once catalogues also serve workflows and best practices with example data, pan-package testing can become routine, which will be of interest to all users of any of the tools involved.
Another aspect of such registries is their approach to describe their collections of computational tools, and the implications that such characterisations entail. While the OMICtools registry leverages a tailor-made taxonomy that tags each tool and enables researchers finding the most pertinent tool for their data analysis task, the Bio.Tools registry employs the EDAM ontology [16]. Terms from EDAM (a collaboratively and openly maintained ontology) can describe a tool in terms of its topic, operation, data, and format, thus providing a multifaceted characterisation: tools can be grouped by functionality, and compatible data formats. Within the scope of sharing workflows, and enhancing testing and reproducibility through workflow modularity, such features can make the difference between enabling users to manually build their own computational pipelines, and assisting users in recommending them pertinent tools to automate constructing them. A controlled vocabulary of terms or a structured inference-ready ontology can already provide ground for automated or semi-automated decision-making system.
The CWL CommandLineTool standard describes tools at a very syntactical level. While these CWL-based tools descriptions may be optionally earmarked with EDAM annotations, there is no generic direct transfer possible from the command-line interface to any such semantic annotation.
The Docker configuration shown in Table 2 illustrates how a base system from a Linux distribution can be prepared, and then further code can be deployed within the container by adding-in external resources. The recipe described by Table 2 demonstrates vividly that users may not explicitly care about the particular version that is installed, either of the operating system or the installed library, with the only specification being that it should be the “latest reliable”. It is not uncommon to just refer to the latest version released by a trusted maintainer, but this trust can only be earned by solid testing against a good reference, and at best any such test environment is available locally to confirm the installation. Tables 3 and 4 show a readily usable implementation from the EBI Metagenomics [25] workflow. It was initially prepared for Docker but was adjusting to allow for a regular distributions package via the SciCrunch Research Resource ID of the tool infernal and the CWL’s SoftwareRequirement specification (https://w3id.org/cwl/v1.0/CommandLineTool.html#SoftwareRequirement). Debian packages reference the same catalogues to support the matching.
The installation procedure of Bioconductor in Table 2 is trusted, and the rest performs in an automated manner. Version information can be retrieved at run-time. For the user who desires to always have the latest released version of a bespoke tool, the distribution providing backported versions is a significant advantage. While technically and socially difficult to undertake en masse, automated backporting of much scientific software is possible, and efforts to provide such packages either officially within the distribution (http://backports.debian.org) or through external repositories (http://neuro.debian.net/) will continue to grow in importance.
A yet unresolved issue with Bioconductor and other tools and frameworks that provide their own packages is that they have their own release scheme of highly interconnected packages which do not synchronise with an underlying Linux distribution. This issue is, albeit to a lesser degree, shared with the rolling Conda and GNU Guix distributions. GNU Guix does allow multi-versioning of packages and their dependencies, but the main software distribution typically only includes recent releases of software. Currently, no software packaging system distributes the same software with many versions, e.g. compatibility with R version 3.4 and with a particular earlier version of this established statistics environment as requested for a Bioconductor release. The community has not yet found an answer to this problem though Conda channels can be used to achieve this. For GNU Guix a similar “channels” system is being considered for supporting older software packages.
4 Conclusion
We have discussed the many efforts at different levels that are contributed by volunteers. User-installability in HPC environments is provided by containers like Singularity or the Prefix concept of Gentoo. Every distribution needs to provide proofs for the reliability of their packages by themselves. Conceptional differences remain in the degree of manual curation. Via cross-distributional efforts like AppStream, a good part of this curation may be shifting to the upstream source tree, to be equally used by all distributions. The EDAM annotations are a prime candidate to make this transition from Debian or software catalogues into the source tree.
We have presented Conda-based packages as a cross-distributional resource of readily usable software packages. There is an ongoing need for a traditional Linux distribution underneath, of which the focus in this article lies on the Debian distribution as a point of comparison with Conda. Philosophical differences between these efforts persist, especially towards the avoidance of redundancy between packages. We did not explore here other package managers such as GNU Guix and Brew. These four package managers together represent the full range of free and open-source software in use today and can be installed on one system without interfering with each other, each providing some level of convenience, robustness and reproducibility.
By placing a high value on standardisation, policy compliance (https://www.debian.org/doc/debian-policy/) and quality assurance, it is understood that immediate participation of newcomers in Debian maintainership is rendered more difficult; contributions are undoubtedly more difficult than a pull request on GitHub which the Conda initiatives requests. The Debian community is moving towards technologies with lower barriers to entry, but its focus on using free software tools to develop a free operating system [29, 31], and a strict adherence to correctness and policy will keep this barrier relatively high for the foreseeable future.
With a focus on the exchange of workflows, we need to find ways to eliminate hurdles for an exchange of experiences between distributions of scientific software. Formally, this can be performed by an exchange of tests/benchmarks of tools and complete workflows alike. This is a likely challenge for upcoming informal meetings like a Codefest [26].
Workflow testing and modularisation can highly benefit from a more homogeneous characterisation of all available software tools. The EDAM ontology provides such descriptors, but its terms have to be associated manually, and their precise attribution highly depends on the very knowledge of the ontology itself. The lack of a protocol formalising whether the developer or the package maintainer has to provide them brings, however, ground for both communities to decide whether to channel their efforts towards a better tool description enrichment.
We have experimented with packages of the Bioconda software infrastructure for Debian to reduce the overhead for users of Debian and derivative distributions, e.g., Ubuntu, to add Bioconda-provided packages to their workflows. Conversely, it would be feasible to add a bit of extra logic to the Conda infrastructure to install system packages if those are available and the user has the permission to install to system directories. Similar points can be made for GNU Guix and Brew. We also note that containerisation technologies, such as Docker and Singularity, allow for easy deployment of software tools with their dependencies inside workflows. All mentioned software packaging technologies play well with containers.
A bioinformatics pipeline can be expressed as a workflow plus data plus (containerized) software packages which is a first step towards reproducible analysis. Once a research project is completed and the results are published, the analysis still needs to be reproducible, both for researchers in the community and for oneself when going back to past projects. Institutional data archives are now commonplace, and researchers deposit data in them as part of the publication process. What is needed to complete the reproducibility is the analysis tooling. This holds for any size of data, including local findings of a pre-clinical study or remote big data such as from physics or astronomy. Hence, researchers need well-established transitions between arbitrary research environments that render our research the most productive, and an environment that may be reliably installed across many clinical environments. This is a long-term goal that we had better all start working towards now. We are approaching it with the CWL, and seeing the CWL as an integral part of a Linux Distribution with all the distributions’ established policies and infrastructure to assess correctness and reliability, will be beneficial for all.
Notes
For a growing list of alternative workflow engines and formalisms, see https://github.com/common-workflow-language/common-workflow-language/wiki/Existing-Workflow-systems and https://github.com/pditommaso/awesome-pipeline. An overview of workbench applications in bioinformatics is in [23, 36] and [18] pp. 35–39, http://bora.uib.no/bitstream/handle/1956/10658/thesis.pdf#page=35.
References
Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Eberhard C, Grüning B, Guerler A, Hillman-Jackson J, Von Kuster G, Rasche E, Soranzo N, Turaga N, Taylor J, Nekrutenko A, Goecks J (2016) The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res 44(W1):W3. https://doi.org/10.1093/nar/gkw343
Amadio G, Xu B (2016) Portage: bringing hackers’ wisdom to science. CoRR arXiv:1610.02742
Amstutz P, Crusoe MR, Tijanić N, Chapman B, Chilton J, Heuer M, Kartashov A, Leehr D, Ménager H, Nedeljkovich M, Scales M, Soiland-Reyes S, Stojanovic L (2016) Common workflow language, v1.0. figshare. https://doi.org/10.6084/m9.figshare.3115156.v2
Bandrowski A, Brush M, Grethe JS, Haendel MA, Kennedy DN, Hill S, Hof PR, Martone ME, Pols M, Tan S, Washington N, Zudilova-Seinstra E, Vasilevsky N (2015) The resource identification initiative: a cultural shift in publishing [version 2; referees: 2 approved]. F1000Research 6(ISCB Comm J):1075. https://doi.org/10.12688/f1000research.6555.2 (Poster)
Berthold MR, Cebron N, Dill F, Gabriel TR, Kötter T, Meinl T, Ohl P, Sieb C, Thiel K, Wiswedel B (2008) KNIME: the Konstanz information miner. Springer, Berlin, pp 319–326. https://doi.org/10.1007/978-3-540-78246-9_38
Christensen A, Egge T (1995) Store—a system for handling third-party applications in a heterogeneous computer environment. In: Estublier J (ed) Software configuration management: ICSE SCM-4 and SCM-5 workshops selected papers. Springer, Berlin, pp 263–276. https://doi.org/10.1007/3-540-60578-9_22
Grüning B, Dale R, Sjödin A, Rowe J, Chapman BA, Tomkins-Tinch CH, Valieris R, The Bioconda Team, Köster J (2017) Bioconda: a sustainable and comprehensive software distribution for the life sciences. bioRxiv. https://doi.org/10.1101/207092. https://www.biorxiv.org/content/early/2017/10/21/207092 (Preprint)
Di Tommaso P, Chatzou M, Floden EW, Barja PP, Palumbo E, Notredame C (2017) Nextflow enables reproducible computational workflows. Nat Biotechnol 35(4):316–319. https://doi.org/10.1038/nbt.3820
Gentleman R, Carey V, Bates D, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini A, Sawitzki G, Smith C, Smyth G, Tierney L, Yang J, Zhang J (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5(10):R80. https://doi.org/10.1186/gb-2004-5-10-r80
Goble CA, Bhagat J, Aleksejevs S, Cruickshank D, Michaelides D, Newman D, Borkum M, Bechhofer S, Roos M, Li P, De Roure D (2010) myExperiment: a repository and social network for the sharing of bioinformatics workflows. Nucleic Acids Res 38(suppl. 2):W677–W682. https://doi.org/10.1093/nar/gkq429
Goecks J, Nekrutenko A, Taylor J, Galaxy Team (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11:R86. https://doi.org/10.1186/gb-2010-11-8-r86
Guimera RV, Chapman B (2012) Bcbio-nextgen: automated, distributed next-gen sequencing pipeline. EMBnet J 17:30. https://doi.org/10.14806/ej.17.b.286
Hastreiter M, Jeske T, Hoser J, Kluge M, Ahomaa K, Friedl MS, Kopetzky SJ, Quell JD, Mewes HW, Küffner R (2017) KNIME4NGS: a comprehensive toolbox for next generation sequencing analysis. Bioinformatics 33(10):1565–1567. https://doi.org/10.1093/bioinformatics/btx003
Henry VJ, Bandrowski AE, Pepin AS, Gonzalez BJ, Desfeux A (2014) OMICtools: an informative directory for multi-omic data analysis. Database 2014:bau069. https://doi.org/10.1093/database/bau069
Irwin J (2008) Community benchmarks for virtual screening. J Comput Aided Mol Des 22(3–4):193–199. https://doi.org/10.1007/s10822-008-9189-4
Ison J, Kalaš M, Jonassen I, Bolser D, Uludag M, McWilliam H, Lopez JMR, Pettifer S, Rice P (2013) EDAM: an ontology of bioinformatics operations, types of data and identifiers, topics and formats. Bioinformatics 29(10):1325–1332. https://doi.org/10.1093/bioinformatics/btt113
Ison J, Rapacki K, Ménager H, Kalaš M, Rydza E, Chmura P, Anthon C, Beard N, Berka K, Bolser D, Booth T, Bretaudeau A, Brezovsky J, Casadio R, Cesareni G, Coppens F, Cornell M, Cuccuru G, Davidsen K, Vedova GD, Dogan T, Doppelt-Azeroual O, Emery L, Gasteiger E, Gatter T, Goldberg T, Grosjean M, Grüning B, Helmer-Citterich M, Ienasescu H, Ioannidis V, Jespersen MC, Jimenez R, Juty N, Juvan P, Koch M, Laibe C, Li JW, Licata L, Mareuil F, Mičetić I, Friborg RM, Moretti S, Morris C, Möller S, Nenadic A, Peterson H, Profiti G, Rice P, Romano P, Roncaglia P, Saidi R, Schafferhans A, Schwämmle V, Smith C, Sperotto MM, Stockinger H, Vařeková RS, Tosatto SC, delaTorre V, Uva P, Via A, Yachdav G, Zambelli F, Vriend G, Rost B, Parkinson H, Løngreen P, Brunak S (2016) Tools and data services registry: a community effort to document bioinformatics resources. Nucleic Acids Res 44(D1):D38. https://doi.org/10.1093/nar/gkv1116
Kalaš M (2015) Efforts towards accessible and reliable bioinformatics. Ph.D. thesis, University of Bergen, Norway. https://doi.org/10.5281/zenodo.33715. http://hdl.handle.net/1956/10658
Kent WJ, Haussler D (2001) Assembly of the working draft of the human genome with GigAssembler. Genome Res 11(9):1541–1548. https://doi.org/10.1101/gr.183201
Krabbenhöft HN, Möller S, Bayer D (2008) Integrating ARC grid middleware with Taverna workflows. Bioinformatics 24(9):1221–1222. https://doi.org/10.1093/bioinformatics/btn095
Kurtzer G, Sochat V, Bauer M (2017) Singularity: scientific containers for mobility of compute. PLoS ONE 12(5):e0177,459. https://doi.org/10.1371/journal.pone.0177459
Laurie S, Fernandez-Callejo M, Marco-Sola S, Trotta J, Camps J, Chacón A, Espinosa A, Gut M, Gut I, Heath S, Beltran S (2016) From wet-lab to variations: concordance and speed of bioinformatics pipelines for whole genome and whole exome sequencing. Hum Mutat 37(12):1263–1271. https://doi.org/10.1002/humu.23114
Leipzig J (2017) A review of bioinformatic pipeline frameworks. Brief Bioinform 18(3):530–536. https://doi.org/10.1093/bib/bbw020
Li JW, Robison K, Martin M, Sjödin A, Usadel B, Young M, Olivares EC, Bolser DM (2012) The SEQanswers wiki: a wiki database of tools for high-throughput sequencing analysis. Nucleic Acids Res 40(suppl 1,D1):D1313–D1317. https://doi.org/10.1093/nar/gkr1058
Mitchell A, Bucchini F, Cochrane G, Denise H, Hoopen Pt, Fraser M, Pesseat S, Potter S, Scheremetjew M, Sterk P, Finn RD (2015) EBI metagenomics in 2016 - an expanding and evolving resource for the analysis and archiving of metagenomic data. Nucleic Acids Res 44(D1):D595–D603. https://doi.org/10.1093/nar/gkv1195
Möller S, Afgan E, Banck M, Bonnal R, Booth T, Chilton J, Cock P, Gumbel M, Harris N, Holland R, Kalaš M, Kaján L, Kibukawa E, Powel D, Prins P, Quinn J, Sallou O, Strozzi F, Seemann T, Sloggett C, Soiland-Reyes S, Spooner W, Steinbiss S, Tille A, Travis A, Guimera R, Katayama T, Chapman B (2014) Community-driven development for computational biology at Sprints, Hackathons and Codefests. BMC Bioinform 15(Suppl. 14):S7. https://doi.org/10.1186/1471-2105-15-S14-S7
Möller S, Krabbenhöft H, Tille A, Paleino D, Williams A, Wolstencroft K, Goble C, Holland R, Belhachemi D, Plessy C (2010) Community-driven computational biology with Debian Linux. BMC Bioinform 11(Suppl 12):S5. https://doi.org/10.1186/1471-2105-11-s12-s5
Moreews F, Sallou O, Ménager H, Le Bras Y, Monjeaud C, Blanchet C, Collin O (2015) BioShaDock: a community driven bioinformatics shared Docker-based tools registry [version 1; referees: 2 approved]. F1000Research 4:1443. https://doi.org/10.12688/f1000research.7536.1
Murdock IA (1994) The Debian Linux Manifesto. http://www.ibiblio.org/pub/historic-linux/distributions/debian-0.91/info/Manifesto. Included in the release of Debian version 0.91
O’Connor BD, Yuen D, Chung V, Duncan AG, Liu XK, Patricia J, Paten B, Stein L, Ferretti V (2017) The Dockstore: enabling modular, community-focused sharing of Docker-based genomics tools and workflows [version 1; referees: 2 approved]. F1000Research 6:52. https://doi.org/10.12688/f1000research.10137.1
Perens B (1997) Debian’s “Social Contract” with the free software community. debian-announce@lists.debian.org (msg00017). https://lists.debian.org/debian-announce/1997/msg00017.html. Re-published as Debian Social Contract, Version 1.0
Prins P, de Ligt J, Tarasov A, Jansen RC, Cuppen E, Bourne PE (2015) Toward effective software solutions for big biology. Nat Biotechnol 33:686–687. https://doi.org/10.1038/nbt.3240
Robinson M, Soiland-Reyes S, Crusoe M, Goble C (2017) CWL Viewer: the common workflow language viewer [version 1; not peer reviewed]. F1000Research stuff. https://doi.org/10.7490/f1000research.1114375.1
Schulz W, Durant T, Siddon A, Torres R (2016) Use of application containers and workflows for genomic data analysis. J Pathol Inform 7(1):53. https://doi.org/10.4103/2153-3539.197197
Shanahan HP, Owen AM, Harrison AP (2014) Bioinformatics on the cloud computing platform Azure. PLoS ONE 9(7):1–9. https://doi.org/10.1371/journal.pone.0102642
Spjuth O, Bongcam-Rudloff E, Hernández GC, Forer L, Giovacchini M, Guimera RV, Kallio A, Korpelainen E, Kańduła MM, Krachunov M, Kreil DP, Kulev O, Łabaj PP, Lampa S, Pireddu L, Schönherr S, Siretskiy A, Vassilev D (2015) Experiences with workflows for automating data-intensive bioinformatics. Biol Direct 10(1):43. https://doi.org/10.1186/s13062-015-0071-8
Taschuk M, Wilson G (2017) Ten simple rules for making research software more robust. PLoS Comput Biol 13(4):e1005,412. https://doi.org/10.1371/journal.pcbi.1005412
Wolstencroft K, Haines R, Fellows D, Williams A, Withers D, Owen S, Soiland-Reyes S, Dunlop I, Nenadic A, Fisher P, Bhagat J, Belhajjame K, Bacall F, Hardisty A, Nieva de la Hidalga A, Balcazar Vargas MP, Sufi S, Goble C (2013) The Taverna workflow suite: designing and executing workflows of Web Services on the desktop, web or in the cloud. Nucleic Acids Res 41(W1):W557–W561. https://doi.org/10.1093/nar/gkt328
Zook J, Chapman B, Wang J, Mittelman D, Hofmann O, Hide W, Salit M (2014) Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat Biotechnol 32(3):246–251. https://doi.org/10.1038/nbt.2835
Acknowledgements
The authors thank Fabien Pichon, Stephan Struckmann and Georg Fuellen for their review and comments to the manuscript. This work has been supported by funding from the European Union’s Horizon 2020 research and innovation programme under H2020-PHC-2014-two-stage Grant Agreement 633589 “Ageing with Elegans” and H2020-EINFRA-2015-1 Grant Agreement 675728 “BioExcel CoE”. In addition, the project has benefited from EU ICT COST Action “cHiPSet” (IC1406). This publication reflects only the authors’ views, and the commission is not responsible for any use that may be made of the information it contains.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
AT, MRC, SM and PR received travel grants by Debian for participating in a Debian Med Sprint. MRC and SSR are members of the CWL Leadership Team at the Software Freedom Conservancy.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Möller, S., Prescott, S.W., Wirzenius, L. et al. Robust Cross-Platform Workflows: How Technical and Scientific Communities Collaborate to Develop, Test and Share Best Practices for Data Analysis. Data Sci. Eng. 2, 232–244 (2017). https://doi.org/10.1007/s41019-017-0050-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41019-017-0050-4