
Abstract
Purpose
This review provides an update on the current clinical, epidemiological and pathophysiological evidence alongside the diagnostic, prevention and treatment approach to chemotherapy-induced peripheral neuropathy (CIPN).
Findings
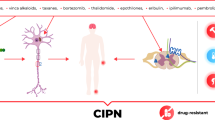
The incidence of cancer and long-term survival after treatment is increasing. CIPN affects sensory, motor and autonomic nerves and is one of the most common adverse events caused by chemotherapeutic agents, which in severe cases leads to dose reduction or treatment cessation, with increased mortality. The primary classes of chemotherapeutic agents associated with CIPN are platinum-based drugs, taxanes, vinca alkaloids, bortezomib and thalidomide. Platinum agents are the most neurotoxic, with oxaliplatin causing the highest prevalence of CIPN. CIPN can progress from acute to chronic, may deteriorate even after treatment cessation (a phenomenon known as coasting) or only partially attenuate. Different chemotherapeutic agents share both similarities and key differences in pathophysiology and clinical presentation. The diagnosis of CIPN relies heavily on identifying symptoms, with limited objective diagnostic approaches targeting the class of affected nerve fibres. Studies have consistently failed to identify at-risk cohorts, and there are no proven strategies or interventions to prevent or limit the development of CIPN. Furthermore, multiple treatments developed to relieve symptoms and to modify the underlying disease in CIPN have failed.
Implications
The increasing prevalence of CIPN demands an objective approach to identify at-risk patients in order to prevent or limit progression and effectively alleviate the symptoms associated with CIPN. An evidence base for novel targets and both pharmacological and non-pharmacological treatments is beginning to emerge and has been recognised recently in publications by the American Society of Clinical Oncology and analgesic trial design expert groups such as ACTTION.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Chemotherapy-induced peripheral neuropathy is a common adverse event which affects the sensory, motor and autonomic nerves. |
The diagnosis of chemotherapy-induced peripheral neuropathy lacks a gold standard. |
There are currently no proven strategies or interventions to prevent or limit the development of chemotherapy-induced peripheral neuropathy. |
A mechanistic approach is needed to address strategies for prevention and treatment of chemotherapy-induced peripheral neuropathy. |
Introduction
The most recent estimation for all-cause cancer incidence is 18.1 million new cases per year [1]. With more effective targeted cancer treatments, long-term cancer survival is increasing in high-income countries [2], as evidenced by the 27% drop in the overall cancer death rate in the United States between 1991 and 2016 [3, 4]. However, chemotherapy-induced peripheral neuropathy (CIPN) is a common and challenging complication of several frequently administered antineoplastic agents [5]. The development of CIPN may result in prolonged infusion times, dose reduction or premature cessation of chemotherapy [6–8], which may negatively impact both treatment efficacy and patient survival [9, 10]. A meta-analysis of randomised controlled trials and cohort studies showed that around half of all patients develop CIPN during treatment [10].
There is currently no gold standard for the assessment of CIPN, with a variety of clinical tools utilised in studies with heterogeneous primary outcome measures [11–21]. Indeed, subclinical nerve damage and motor involvement are poorly defined when using current standardised clinical instruments [15]. Accurate comparisons of the prevalence, incidence, prevention and treatment of CIPN are therefore problematic (Table 1). Additionally, there are considerable disparities in patient- and clinician-reported neurotoxicity. For example, in the ICON7 trial, clinicians reported CIPN in 28% of patients, while 67% of patients reported ‘quite a bit’ or ‘very much’ tingling or numbness, with poor agreement between patients and clinicians (κ = 0.236, 95% confidence interval, 0.177–0.296, p < 0.001) [22].
Chemotherapeutic agents result in neurotoxicity through a variety of mechanisms, culminating in a predominantly symmetrical sensory or sensorimotor, length-dependent neuropathy and autonomic dysfunction [23–28]. Neuropathic syndromes specific to chemotherapeutic agents can be observed, each with their own presentation and natural history [29–33] (Table 2). CIPN can develop, or continue to worsen, several months after treatment has stopped, in a phenomenon termed “coasting”. The prevalence of CIPN one month after finishing chemotherapy approaches 68%, and persists in approximately one third of patients beyond 6 months [10]. Risk factors for CIPN include the agent used, cumulative dose, number of cycles, treatment duration, combination therapies, genetic predisposition, age, existing nerve damage, severity of acute symptoms and chronic alcohol consumption amongst others [15]. The ageing population and more efficacious chemotherapeutic regimens will continue to increase cancer cure rates and long-term cancer survival [34], together with CIPN [35]. It is therefore imperative to develop effective strategies for the early identification with prevention and more efficacious management of CIPN.
Literature Search Methodology
Electronic database searches were undertaken in EMBASE, PubMed, OVID and Cochrane CENTRAL to identify included articles. The reference lists of relevant articles were searched, and in addition, studies were identified by authors with expertise in CIPN. Studies published from initial curation of the electronic database to March 2021 were identified, and those felt not relevant by authors were excluded with the guidance of the senior author (U.A.). This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Toxicity Versus Benefit
Balancing the risk of different manifestations of chemotoxicity and the potential benefit of reduced disease burden/remission is a demanding aspect of oncological practice. For instance, there is an increased risk of CIPN with oxaliplatin compared to cisplatin, but the risk of thromboembolism is greater for cisplatin than for oxaliplatin [36], with a small survival benefit with oxaliplatin compared to cisplatin. Accordingly, there are many options to try to limit CIPN by reducing the number of doses and cumulative toxicity, especially in older individuals or those more likely to have pre-existing neuropathy such as diabetes [37]. Patients are less likely to continue chemotherapy if they develop serious complications. In a population of older patients in a non-curative setting, lower doses of oxaliplatin and capecitabine were better tolerated, resulting in patients receiving a greater number of cycles and a small survival benefit [37]. Al-Batran et al. [38] reported that the rate of CIPN in patients treated with epirubicin, cisplatin and fluoropyrimidine was half that observed in patients treated with oxaliplatin, docetaxel and fluoropyrimidine. Similarly, Cunningham et al. [36] reported that the rate of thromboembolism doubled in patients treated with epirubicin, cisplatin and fluoropyrimidine [38]. Ultimately, there are trade-offs when treating patients with cancer, especially those with limited therapeutic options, worse prognosis or pre-existing conditions that may predispose them to chemotherapy-related complications.
Oxaliplatin-Induced Peripheral Neuropathy
Platinum-based chemotherapeutics (oxaliplatin, cisplatin and carboplatin) are used in the treatment of solid tumours of the gut, bladder, testes, ovary, uterus, lung, head and neck [39, 40]. Platinum chemotherapeutic agents have the highest prevalence rates of CIPN, affecting ~ 70% of patients, often complicated by coasting [29, 41]. The main anatomical structure injured by platinum agents is the dorsal root ganglion, and manifests as a sensory neuropathy with prominent pain accompanied by cold-induced allodynia and muscle cramps due to peripheral nerve hyperexcitability or neuromyotonia. Acute oxaliplatin-induced peripheral neuropathy (OIPN) can result in prolonged infusion times (~ 22%), dose reduction (15–43%) and treatment cessation (6–21.4%) [42–46]. A systematic analysis of studies including 6211 participants undergoing oxaliplatin treatment found acute OIPN with an incidence of 4–98% [42]. The wide range of incidence may be attributed to heterogeneous dosing regimens, drug combinations, dosing intervals and screening instruments used to identify acute OIPN [42]. A longitudinal study following 346 participants undergoing FOLFOX chemotherapy demonstrated a 3-day peak in acute OIPN, with sensory symptoms including cold-induced hypersensitivity (71%), sensitivity to swallowing cold food and drink (71%), throat discomfort (63%) and muscle cramps (42%) [25]. Symptoms often persist between treatments and increase in severity with subsequent doses [25, 26]. The initial severity of acute OIPN also predicts progression to chronic sensory OIPN [25, 47], which can be identified in 84% of patients after 25 months, with long-term impact on functionality and quality of life.
Pathogenesis of Platinum-Induced Peripheral Neuropathy
The dorsal root ganglion (DRG) is particularly susceptible to chemotherapeutic agents, as it lies outside the central nervous system and is not protected by the blood–brain barrier [48]. In an animal model of CIPN, the accumulation of oxaliplatin in DRG neurons was associated with intracellular overexpression of Octn1/2 and Mate1 transporters [49]. Oxaliplatin also interferes with DNA cross-linking, resulting in direct neurotoxicity [50] and early p38 and ERK1/2 activation, reduced mitochondrial respiration, increased oxidative stress and dose-dependent apoptosis of DRG neurons [51]. Cell culture studies have shown greater neuronal cell body atrophy and apoptosis when exposed to oxaliplatin compared to both paclitaxel and controls, promoting a sensory neuronopathy (neuronal cell body) as opposed to an axonopathy that is phenotypic of other chemotherapeutic agents. OIPN also correlates with mitochondrial morphological artefacts, decreased adenosine triphosphate generation and depressed respiration rates in mitochondrial complexes I and II [52–54] 55. Indeed, platinum agents and their metabolites form adducts with mitochondrial DNA (mtDNA), disrupting replication and transcription, with a reduction in neuronal cell body mitochondrial populations [56]. Oxidative stress leads to oxidation of intracellular moieties of neurons, diminishing neuronal energy status and increasing apoptosis [55, 57–61]. Reduction of oxidative stress with phenyl N-tert-butylnitrone has been shown to decrease oxaliplatin-induced mechanical hyperalgesia and cold allodynia [62, 63]. Oxaliplatin also interacts with voltage-gated potassium channels (VGKC) expressed on peripheral motor neurons and is implicated in the acute phase of OIPN in which patients exhibit nerve hyperexcitability, prolonged depolarisation, increased neurotransmission and muscle contraction similar to that seen in neuromyotonia [44]. Notably, the excitability of Aδ and C-type fibres of the maxillary branch of the trigeminal nerve are controlled by VGKCs. Further, VGKC isotype 4.3 channels had slower deactivation after administration of oxaliplatin, and this may underlie cold-induced orofacial allodynia [64]. Intramuscular injections at the base of the tail of mice with oxaliplatin were shown to cause acute transient dose-dependent changes in excitability of both motor and sensory axons and evoked ectopic activity in these fibres [65]. Moreover, mathematical modelling indicates that oxaliplatin causes slow inactivation of voltage-gated sodium (NaV) channels and reduces the resting membrane potential of nerve fibres through the reduction of fast potassium conductance in the acute phase of OIPN [65]. Indeed, in preclinical studies, NaV channel-blocking drugs such as topiramate have recently been shown to have a neuroprotective effect in the prevention of both the acute and chronic phase of OIPN, with no interactions with the antineoplastic activity of oxaliplatin [66].
Cold hyperalgesia is a major feature of OIPN and is thought to be driven by TRPA1 and p38 MAPK activation in DRG neurons and increased activity of NaV channel isoforms NaV1.6 and NaV1.9 in nociceptive subpopulations of peripheral and DRG neurons. Further, there is a potential role played by transient receptor potential melastatin 8 (TRPM8) in acute cold-induced allodynia [67]. Altered expression of pain receptor-associated TREK-1, TRAAK, TRPA1 NaV channel isoforms and hyperpolarisation-activated cyclic nucleotide–gated (HCN1) channels in sensory neurons contribute to the prominent neuropathic pain associated with this condition [68]. Oxalate chelates Ca2+ ions, contributing to neuronal excitability and increasing spontaneous pain signalling [69]. There is also increased expression of pro-inflammatory cytokines including tumour necrosis factor-α (TNF-α) and interleukin (IL)-1β and decreased expression of the neuroprotective cytokines IL-10 and IL-4 [70–72] through the activation of astrocytes by platinum-based chemotherapy agents. A summary figure of these processes is shown in Fig. 1.

The current hypothesis for the pathogenesis of OIPN. Accumulation of oxaliplatin occurs in dorsal root ganglion neurons, where it interferes with DNA and mtDNA cross-linking. This results in a direct dose-dependent toxicity of DRG neurons and neuronal mitochondria. There is a subsequent decrease in mitochondrial respiration and ATP. The resultant oxidative stress contributes to disruption in DNA and mtDNA replication and transcription, leading to diminished energy status and increased neuronal apoptosis. Increased production of ROS together with activation of astrocytes causes the release of pro-inflammatory mediators TNF-α and IL-1β and decreased expression of cytokines IL-10 and IL-4 with a neuroprotective function. Subsequently, leucocytes are activated and travel down a chemotactic gradient to the dorsal root ganglion and peripheral nerves, leading to neuroinflammation. Neuroinflammation and ROS cause damage to dorsal root ganglion neurons, leading to apoptosis, which contributes to calcium dysregulation, axonal energy depletion and damage to neuronal organelles. Both ROS and neuroinflammation are implicated in nociceptor sensitisation, mechanical hyperalgesia and cold allodynia in preclinical models. Oxaliplatin interacts directly with VGKC, NaV channel, TRPM isoforms in sensory neurons contributing to cold hyperalgesia/allodynia and hyperexcitability of peripheral neurons. Further, a metabolite of oxaliplatin, oxalate chelates Ca2+ ions in the acute phase, contributing to neuronal excitability and increasing spontaneous activity of neurons. ATP: adenosine triphosphate, Ca2+: calcium, DNA: deoxyribonucleic acid, DRG: dorsal root ganglion, NaV: voltage-gated sodium, OIPN: oxaliplatin-induced peripheral neuropathy, mtDNA: mitochondrial DNA, ROS: reactive oxygen species, IL-1B: interleukin 1B, IL-4: interleukin 4, IL-10: interleukin 10, TRPM: transient receptor potential melastatin, TNF-α: tumour necrosis factor-α, VGKC: voltage-gated potassium channel
Taxane-Induced Peripheral Neuropathy
The taxanes (paclitaxel, docetaxel and cabazitaxel) are currently first-line treatments for breast, ovarian, lung, bladder, prostate and other solid tumour cancers [34, 73, 74]. Taxane-induced peripheral neuropathy (TIPN) is the most common non-haematological adverse event of treatment, which may result in dose reduction and cessation of treatment, impacting patient survival [75]. TIPN primarily causes an acute, length-dependent distal sensory neuropathy, accompanied by neuropathic pain, which may progress proximally in more severe cases. Aβ fibres and to a lesser extent Aδ and C-fibres are affected in a glove-and-stocking distribution [31, 76, 77]. Patients report tingling, numbness, paraesthesia, neuropathic pain, cold-induced dysaesthesia and muscle cramps [26, 78], which typically worsen with treatment and gradually improve with cessation [26], although 31–44% of patients treated with docetaxel or paclitaxel report symptoms after up to 6 years of follow-up [79–81]. TIPN incidence in non-small cell lung cancer (NSCLC) in phase III trials occurred in 13–62% of patients [82]. Severe TIPN (FACT-Lung grade ≥ 3) occurred in 21–40% of patients, with worse outcomes after receiving paclitaxel as opposed to docetaxel-based chemotherapy regimens [83]. Thus, docetaxel is generally considered to be less neurotoxic than paclitaxel [84].
Pathogenesis of TIPN
Studies have identified an increase in the incidence of abnormal axonal mitochondria in C-fibres when compared to controls after ~ 1 month of paclitaxel treatment [85]. Paclitaxel interacts with the mitochondrial permeability transition pore, leading to mitochondrial dysfunction, decreased mitochondrial respiration and disruption of neuronal ATP generation [86, 87], with disruption of the axonal microtubule network [88]. Taxane treatment of rat DRG neuronal stem cells increased ROS production and oxidative stress, simultaneously decreasing mitochondrial metabolic activity, membrane potential and antioxidant bioavailability [62, 89]. Similarly, taxane treatment of rat and human DRG neurons lowered the resting and threshold membrane potential and increased the frequency of ectopic spontaneous activity [90]. In experimental models, paclitaxel increased the expression of voltage-gated calcium channels (Cav) 3.2 and calcium current amplitude and decreased the excitability threshold of dorsal root sensory neurons, which when inhibited decreased mechanical hypersensitivity [87, 91, 92]. Further, toll-like receptor (TLR) 4 is also upregulated, resulting in increased intracellular calcium mediated by the co-located protein Cav3.2. Moreover, paclitaxel increases the expression of Nav 1.7 channels in a dose-dependent manner in human DRG neurons in culture, leading to increased ectopic spontaneous activity [92, 93]. Notably, paclitaxel can bind and activate TLR4 on macrophages, engaging signalling pathways that lead to increased gene expression and release of nuclear factor kappa B (NF-kB), initiating inflammatory and cytokine cascades [94]. TLR4, MyD88 and ERK1/2 expression is increased in IB4− and CGRP+ DRG neurons [94–96]. Inflammatory mediators IL-6, IL-8, IL-10, monocyte chemoattractant protein-1 (MCP-1) and activated Langerhans cells are upregulated, where they propagate the further release of pro-inflammatory cytokines [97–99]. Furthermore, there is increased expression of stress and inflammatory markers in Schwann cells and lumbar DRG neurons [100, 101]. Activation and migration of CD68+ macrophages, CD8+ T cells and CD11b+ leucocytes towards the DRG and peripheral nerves has been identified [99–101]. Thus, sensitisation of C-fibres, net energy loss, neuroinflammation and hyperexcitability contribute to paclitaxel-induced peripheral neuropathy. The hypotheses of the pathomechanism of TIPN is summarised in Fig. 2.

The current hypothesis for the pathogenesis of TIPN. Taxanes such as paclitaxel directly interact with TLR-4 on macrophages. This interaction upregulates the expression of TLR-4 and activates macrophages leading to the release of NF-kB, leading to further downstream pro-inflammatory cascades. Activated Langerhans cells release IL-6, IL-8, IL-10 and MCP-1. Subsequently, there is activation and migration of macrophages, cytotoxic T-cells, monocytes and neutrophils towards the DRG and peripheral nerves. DRG neurons and IB4-/GCRP+ peripheral fibres increase expression of inflammatory associated markers such as TLR4, MyD88 and ERK1/2. Similarly, inflammatory signalling is increased in Schwann cells, microglia and DRG neurons together with markers of cellular stress. Oxidative stress and the generation of ROS further impacts upon mitochondrial performance, limits intracellular energy stores of peripheral neurons and contributes to inflammation and intracellular damage. Further, taxanes such as paclitaxel interact with the MPTP, which culminates in a reduction in ATP generation and mitochondrial generation. Taxanes disrupt microtubule polymerisation and impair the function of the axonal microtubule network. The expression of CaV 3.2 and NaV 1.7 are upregulated after treatment with taxanes, resulting in changes to the excitability threshold of peripheral neurons. The sensitisation of peripheral neurons and subsequent changes in neuronal excitability result in mechanical hypersensitivity and ectopic spontaneous activity which contribute to the development of TIPN. ATP: adenosine triphosphate, CaV: low voltage-activated T-type calcium channel, CGRP: calcitonin gene-related peptide, DAMP: damage-associated molecular pattern, DRG: dorsal root ganglion, ERK1/2: extracellular signal-regulated kinase, IB4: isolectin B4-binding glycoprotein, IL-6: interleukin 6, IL-8: interleukin 8, IL-10: interleukin 10, NaV: voltage-gated sodium, NF-kB: nuclear factor kappa B, MCP-1: monocyte chemoattractant protein-1, MEK: mitogen-activated protein kinase kinase, ROS: reactive oxygen species, TIPN: taxane-induced peripheral neuropathy, TLR-4: toll-like receptor 4
Vinca Alkaloids
Vinca alkaloids are natural (vincristine and vinblastine) and semi-synthetic (vindesine and vinorelbine) chemotherapeutics derived from the periwinkle plant and are used either alone or in combination therapy to treat haematological malignancies, testicular cancer, myeloma, sarcoma, non-small cell lung cancer and tumours of the kidney, liver, lung, brain and breast [102]. Vincristine is arguably the most neurotoxic vinca alkaloid, with a majority of patients developing vincristine-induced neuropathy (VIPN) [10, 103], the severity of which is dose-dependent [104]105. Genetic polymorphisms in genes associated with Charcot-Marie-Tooth (CMT) disease appear to increase the risk of VIPN [106]. The incidence of VIPN or vinorelbine-induced neuropathy leading to sensory neuropathy is ~ 20%, with motor impairment in 17.5% of adult patients [107, 108]. The most common presentation of VIPN is a length-dependent sensory neuropathy, with significant motor impairment and occasional cranial nerve involvement [107]. Surprisingly, 91% of patients reported continuing symptoms 12 months after cessation of treatment [109], and there is evidence for long-term distal sensory [107, 110] and motor deficits in vincristine-treated cancer survivors [30].
Pathogenesis of VIPN
Anterograde transport of organelles and membrane proteins and retrograde transport of signalling molecules depends on microtubule-based transport [88]. Vinca alkaloids interfere with and disrupt microtubule assembly and mitotic spindle formation [111, 112]. They also increase the stability of microtubules, which impacts negatively on the ability of the cell to dynamically alter the structure of the cytoskeleton affecting axonal transport [88] 113. Additionally, vincristine is mitotoxic and can impair the mitochondrial electron transport chain, resulting in defective energy production [114]. Axonal degeneration requires both sterile alpha and TIR motif-containing proteins SARM1 and MAPK, and the deletion of SARM1 protects mice from developing VIPN [115]. Other intracellular targets include the NF-E2-related factor and haeme oxygenase 1/carbon monoxide system (Nrf2/HO-1/CO) which modulates the expression of connexin 43 (Cx43), protecting against nerve damage and reducing vincristine-induced neuroinflammation [116]. Increased expression of inflammatory markers TNF-α and IL-1β and increased expression of TRPA1 were recently identified in models of VIPN [117]. Moreover, mRNA gene ontology has identified the inflammatory role of vincristine on microglia and upregulation of pro-inflammatory genes including frizzled-related protein 2 (SFRP2) and C-X-C motif chemokines (CXCL) 10 and 9 [118]. The current available data on the hypothesised mechanism of VIPN is shown in Fig. 3A.

The current hypothesis for the pathogenesis of VIPN (A), ThiPN (B) and BIPN (C). A Vinca alkaloids such as vincristine activate leucocytes and microglia, causing the attraction and activation of downstream pro-inflammatory cytokines, leading to neuroinflammation. Vinca alkaloids inhibit the polymerisation of microtubules and therefore the formation of mitotic spindles causing disruption to axonal transport. This, together with mitotoxicity, causes net energy loss by impairing the electron transport chain. These mechanisms culminate in a distal sensorimotor axonal neuropathy. B Thalidomide inhibits VEGF, b-FGF, NF-kB and TNF-a, leading to dysregulation of neurotrophins. This impedes signalling responsible for the survival and proliferation of neurons. Further, antiangiogenic properties of thalidomide cause secondary ischaemia and hypoxia of small nerve fibres, leading to damage to sensory nerve fibres. C Bortezomib causes the release of intracellular calcium from the endoplasmic reticulum in sensory neurons, leading to caspase activation and subsequent cellular apoptosis. Pro-inflammatory mediators are upregulated after treatment with bortezomib, leading to further cytokine signalling cascades and neuroinflammation. Bortezomib is mitotoxic, leading to damage to neuronal mitochondria, diminished respiration and reduced ATP production, culminating in neuronal energy failure. Further, oxidative stress and ROS contribute to intracellular damage to neuronal organelles (including mitochondria) and apoptotic mechanisms. Ultrastructural changes are seen in the myelin sheath of neurons, although the contribution of these changes warrants further investigation. AT: adenosine triphosphate, b-FGF: basic fibroblast growth factor, Ca2+ calcium, CCL21 -CXCL-9-C-X-C motif chemokine 9, CXCL-10-C-X-C motif chemokine 10, C-X-C motif chemokine 21, IL-1B: interleukin 1B, NF-kB: nuclear factor kappa B, SARM1: sterile alpha and TIR motif-containing 1, SFRP2: frizzled-related protein 2, TNF-α: tumour necrosis factor α, VEGF: vascular endothelial growth factor
Thalidomide-Induced Peripheral Neuropathy
Thalidomide is a US Food and Drug Administration (FDA)-approved treatment for multiple myeloma (MM) [119]. Patients treated with thalidomide for MM, glioblastoma, renal cell carcinoma, colorectal and lung, melanoma, and breast and prostate cancer can develop thalidomide-induced peripheral neuropathy (ThiPN) [32, 102, 120–122]. Symptoms include symmetrical numbness, tingling, burning pain and sensitivity to touch and heat, with hyperaesthesia, hypoaesthesia and paraesthesia in a glove-and-stocking distribution [32] with tremor, muscle cramps, distal muscle weakness, areflexia, loss of proprioception, gait ataxia and/or a lack of coordination [32, 123–128]. The incidence of ThiPN ranges from 11 to 75% and is dependent on dose [120, 122, 129–134] and duration of exposure [125]. As such, the results of phase I studies giving thalidomide to the maximum tolerable dose are not representative of patients who are receiving this medication over a longer duration. Peripheral neuropathy induced by the thalidomide analogues lenalidomide and pomalidomide are less severe and occur at a lower incidence [135–137], making them the agents of choice in those with pre-existing neuropathy. MM is currently incurable and requires long durations of exposure to thalidomide and its analogues, which results in accumulative chemotoxicity [138]. This is especially relevant as the 5-year relative survival rate of M has increased in recent years [139]. Barlogie et al. [140] reported that 90% of participants with a National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) ≥ 2 grade ThiPN improved to a grade ≤ 2 within 3–4 months of thalidomide dose attenuation. However, complete clinical recovery is limited to approximately one quarter of patients [124, 128, 141–143].
Pathogenesis of ThiPN
The exact pathomechanism of thalidomide is yet to be fully elucidated, but antiangiogenic properties [144] may lead to hypoxia of small nerve fibres [145]. Additionally, the immunomodulatory action of thalidomide inhibits basic fibroblast growth factor (b-FGF), vascular endothelial growth factor (VEGF), TNF-α and NF-kB and dysregulates neurotrophins; the signalling molecules responsible for the proliferation, survival and function of neurons are shown in Fig. 3B [146]. Further, preclinical ThiPN animal models demonstrate improvement of NCS following the injection of VEGF [147].
Bortezomib
The proteasome inhibitors bortezomib, ixazomib and carfilzomib are FDA-approved treatments for MM [148, 149] and are used in the treatment of progressive, relapsed or refractory MM and mantle cell lymphoma [150, 151]. Bortezomib-induced peripheral neuropathy (BIPN) is a distal, symmetrical, length-dependent axonal sensorimotor neuropathy characterised by mild to moderate sensory loss, mild to severe neuropathic pain and mild motor weakness of the distal lower extremities [[33, 152]. Phase II trials have identified a BIPN incidence of 31–37%, with grade ≥ 2 neuropathy present in 28% of participants [153–155]. Although ixazomib and carfilzomib have a lower incidence of CIPN [156–158], long-term treatment [159] with the addition of other chemotherapeutic agents [160] is required to maintain remission.
Pathogenesis of BIPN
Bortezomib initiates apoptosis through the release of intracellular Ca2+ in the endoplasmic reticulum, leading to activation of caspase, a protease enzyme essential for programmed cell death [161]. A study showed vacuolation of DRG-associated mitochondria [162], although these findings could not be replicated [163]. Bortezomib treatment increased the number of swollen and vacuolated mitochondria in A-fibres and C-fibres compared to controls, and mitochondrial respiration and adenosine triphosphate production were reduced, indicating cumulative energy failure as a pathogenic mechanism of BIPN [164]. In a recent study, bortezomib exhibited neurotoxicity in PC12 neuroblastoma cells through the induction of apoptosis which was ameliorated with antioxidants, implicating oxidative stress in the pathogenesis of BIPN [165]. Ultrastructural features of myelin sheath degeneration of large nerve fibres and axonal degeneration of C- fibres have been identified [162, 163]. Inhibition of NF-kB and TNF-α attenuates the severity of BIPN in preclinical models [166, 167]. Indeed, bortezomib treatment increases the expression of GATA-binding protein (GATA3), a transcription factor implicated in the regulation of inflammatory signalling cascades and upregulation of the T-cell chemoattractant chemokine C–C motif ligand 21 (CCL21) in dorsal horn neurons, which when silenced attenuates mechanical allodynia in Sprague Dawley rats [168]. The current hypothesis for the pathomechanism of BIPN is summarised in Fig. 3C.
Diagnostic Methods
Electrodiagnostic methods are considered the reference standard for the functional assessment of large sensory and motor fibres which drive paraesthesia, numbness and weakness seen in people with CIPN. Although sensory testing used in composite scoring systems is often deployed in the clinical setting, a rigorous, lengthy battery of standardised sensory tests is required to reliably identify a patient’s sensory phenotype. Further, these tests are subjective and cannot discriminate between a central or peripheral disease process of the somatosensory nervous system, and benefit from the addition of a structural measure of peripheral nerve fibres. In light of this, we include an overview of self-reported outcome measures, composite scoring systems, functional tests of large fibres, structural measure of small fibres such as skin biopsy, and highlight the novel, reiterative method of corneal confocal microscopy. This method is of particular interest, as the early detection of CIPN may enable health care professionals to determine subclinical nerve damage and assist in changes to dosing strategies before the neuropathy becomes irreversible. In this section we highlight the methods used to quantify CIPN in both clinical and research settings.
Identification of CIPN and Grading
The methods used in both clinical trials and medical practice to identify and grade the severity of CIPN can be broadly separated into instruments which utilise patient-reported outcomes, composite scoring systems with a functional assessment component, and quality-of-life tools [169]. Most commonly used is the clinician-led patient-reported tool, National Cancer Institute-Common Terminology Criteria for Adverse Events (NCI-CTCAE), although other instruments such as the Eastern Cooperative Oncology Group (ECOG) criteria and the World Health Organization (WHO) neurotoxicity scale [170] are also used. The latest version of the NCI-CTCAE (version 5.0) (Table 3) grades both motor and sensory neuropathy according to asymptomatic (grade 1), moderate (grade 2), severe (grade 3) or life-threatening (grade 4) neurotoxicity. Composite scoring systems such as the Total Neuropathy Score (TNS) use patient-reported symptoms, physical examination, vibration perception threshold and nerve conduction studies to grade CIPN, although there are versions which omit vibration perception threshold (TNSr) and nerve conduction studies (TNSc) [169]. Further, the TNS clinical (TNSc) and nurse-administered TNS (TNSn) have been shown to correlate well with the emergence of sensory and motor symptoms after the completion of chemotherapy, identifying 88% of participants who developed CIPN [171].
Functional assessments are self-reported questionnaires measuring both the quality of life and symptoms specific to how neurotoxicity impairs activity. These measures are often tailored to the primary cancer such as the Functional Assessment of Cancer Therapy/Gynecologic Oncology Group-neurotoxicity (FACT/COG-Ntx) tool, which has been shown to correlate well with the TNSc and TNSn [171]. Other examples of functional assessments include the European Organisation for Research and Treatment of Cancer (EORTC) and the chemotherapy-induced peripheral neuropathy questionnaire (CIPN20). These instruments have been reviewed extensively by Cavaletti et al. [170] and Park et al. [169].
Nerve Conduction Studies (NCS)
NCS provide an objective measure of large fibre function and are considered the reference standard for the diagnosis of large fibre involvement in CIPN [172]. Peripheral nerve demyelination is accompanied by conduction slowing and latency prolongation, and axonal loss is accompanied by a reduction in amplitude.
In patients treated with paclitaxel and oxaliplatin, NCS can be used to confirm a symmetric, length-dependent, predominantly sensory distal neuropathy [173–177]. However, in acute OIPN there are rarely significant changes in NCS, although motor axons can develop increased refractoriness resulting in repetitive motor discharges [173, 178, 179]. Further, a change in sensory excitability in acute OIPN predicts the development of chronic OIPN, a purely sensory neuropathy with a reduction in sensory sural nerve action potential (SNAP) and nerve conduction velocity (SNCV) without motor NCS involvement [180]. In a longitudinal study of ten participants, the phenomenon of coasting was evidenced by worsening median and sural sensory amplitudes at least 3 months after completing oxaliplatin-based chemotherapy [181]. The typical presentation of TIPN is that of a predominantly distal sensory axonal neuropathy with some motor involvement [172, 182]. A recent longitudinal study identified significantly reduced SNAP amplitudes predominantly in the upper limbs, but to a lesser extent in the lower limbs, 12 months after completion of taxane chemotherapy, arguing for a non-length-dependent effect [15]. Both acute and chronic thalidomide neurotoxicity are characterised by attenuation of median, radial and sural SNAPs and compound muscle action potentials (cMAPs) of the peroneal and tibial nerves [126]. NCS in patients with BIPN largely indicate a predominantly small fibre sensory axonal neuropathy, with less frequent motor neuropathy [129]. Bortezomib and thalidomide combination therapy is associated with a significant reduction in sural SNAP, peroneal motor nerve action potential (PMNAP) and peroneal motor nerve conduction velocity (PMNCV) [33, 127]. NCS in people with VIPN is characterised by a distal sensorimotor axonal neuropathy and motor involvement [172], with prolongation of distal latencies but preserved conduction velocities [183]. Furthermore, NCS parameters may deteriorate before or after the development of neuropathic symptoms [47, 127, 129, 172, 184–187].
Quantitative Sensory Testing (QST)
QST provides an extensively validated mechanism-based and symptom-orientated approach to neuropathic pain. The loss of nerve fibre sensitivity or deafferentation can be detected using quantitative sensory testing for different nerve fibre populations. The loss of Aβ-fibre sensitivity is indicated by impaired vibration perception, light touch or mechanical detection thresholds. C-fibre dysfunction is reflected by abnormal heat detection and heat pain thresholds, whilst Aδ-fibre dysfunction is indicated by abnormal thresholds to pinprick stimuli, mechanical pain and cold detection [188–190]. The majority of patients with CIPN from a range of drugs exhibit reduced or absent pinprick and vibration perception thresholds and impaired proprioception [191]. Early impairment of vibration detection and cold detection thresholds have been identified from week 12 of treatment with oxaliplatin, with an increase in mechanical detection thresholds 6 months after finishing treatment [180, 192]. Cold pain threshold can be used to dichotomise participants with acute OIPN and change over time [193]. People with TIPN exhibit diminished tactile perception in the upper and lower extremities, with worsening VPT in the lower limbs [194]. Participants with VIPN and BIPN exhibit widespread abnormalities in touch detection, pinprick detection and heat detection thresholds both within and outside self-reported areas of involvement [195, 196].
Skin Biopsy
The accepted gold standard for diagnosing small fibre pathology is skin biopsy [197, 198]. Normative age- and sex-related values for intraepidermal nerve fibre density have been published for clinical use [199]. In preclinical models of paclitaxel- and vincristine-induced peripheral neuropathy, there is a significant reduction in intraepidermal nerve fibres [91, 98]. Indeed, a significant decrease in intraepidermal nerve fibre density (IENFD) at the distal leg was identified in eight patients 6 months after oxaliplatin treatment had been stopped [200]. Notably, a recent study found a significant time-dependent decrease in IENFD 6 months after treatment had been stopped [180]. In patients with BIPN, whilst epidermal nerve density did not differ, there was a reduction in subepidermal nerve fibre density [201]. Further work is needed to characterise the differential effect of different chemotherapy drugs on small nerve fibres in the skin.
Corneal Confocal Microscopy
Corneal confocal microscopy (CCM) is a non-invasive, reiterative ophthalmic imaging technique that detects small nerve fibre abnormalities in the subbasal nerve plexus in a range of peripheral neuropathies [202–210]. A large body of published data shows that CCM has good diagnostic [211] and prognostic [212] utility in diabetic neuropathy. Recently, CCM has been proposed to have utility in the diagnosis and follow-up of patients with CIPN [213].
In an early study of 15 patients with colorectal cancer treated with oxaliplatin, 10 patients developed a significant worsening of TNSc and 8 patients developed NCV evidence of a sensory axonal neuropathy [214]. CCM demonstrated a significant abnormality in 10/15 patients characterised by a reduction in corneal nerve fibre density (40%) and length (37%). Interestingly, after receiving the final cycle of chemotherapy, two patients with normal clinical and neurophysiological findings had evidence of severe corneal nerve loss, and 3 weeks later they developed neuropathic symptoms, indicative of coasting [214]. In 21 patients with gastro-oesophageal cancer without neuropathic symptoms there was evidence of corneal nerve loss which correlated with the stage of cancer. After three cycles of platinum-based chemotherapy, 61.5% of patients developed grade 1 symptomatic paraesthesia on CTCAE criteria; however, all patients except those with metastatic liver disease showed an increase in corneal nerve fibre length [205]. CCM has also shown a significant reduction in corneal nerve fibre density, length and beading in patients with MM undergoing treatment with bortezomib, despite clinically evident neuropathy being present in only 38.5% of patients [215]. More recently, of 63 patients who had received docetaxel for breast cancer (n = 28) or oxaliplatin for colorectal cancer (n = 35) 5 years prior to detailed neuropathy phenotyping, 41.3% still had evidence of CIPN, of whom 58% had pure large fibre neuropathy based on NCS [216]. Detailed QST revealed increased cold, warm, mechanical and vibration detection thresholds with no evidence of pinprick hyperalgesia or dynamic mechanical, cold or warm allodynia. CCM demonstrated no significant difference in corneal nerve fiber length, density or branch density between controls and patients with CIPN with and without small fibre neuropathy [216]. In a study comparing CCM in different peripheral neuropathies, patients with CIPN had evidence of corneal nerve fibre loss in a distinct pattern based on the corneal nerve fractal dimension, which differed from patients with diabetic neuropathy or chronic inflammatory demyelinating neuropathy [217]. A study of 70 patients with breast, colorectal, upper gastrointestinal and gynaecological cancer having received either paclitaxel (n = 40) or oxaliplatin (n = 30) within the past 3 to 24 months showed evidence of a significant reduction in the corneal nerve fibre and inferior whorl lengths [218]. Furthermore, corneal nerve fiber length, inferior whorl length, average nerve fiber length and corneal nerve fiber density were significantly lower in patients with neuropathy compared to those without neuropathy based on the correlation of TNSr and inferior whorl length with hand dexterity [218]. These data suggest that CCM may have diagnostic and prognostic value in CIPN.
Chemotherapy and Neuropathic Pain
A large meta-analysis of 13,683 people with CIPN estimated the prevalence of neuropathic pain to be as high as 40% [219]. A recent international study of 2003 patients with CIPN has found a similar prevalence of neuropathic pain, which significantly impacted upon quality of life and daily functioning [220]. CIPN is predominantly a sensory neuropathy, as summarised in Table 2, with pain being the most bothersome symptom [221]. Indeed, the symptom burden of CIPN including sensory disturbances and neuropathic pain profoundly impacts on the quality of life of survivors of cancer [84, 191, 222–227]. CIPN also affects functionality and the capacity to work both during and after treatment, fuelling unemployment and loss of working time [228]. Moreover, a recent US administrative claims analysis by Song et al. [229] found that individuals with painful CIPN incur a significant economic burden driven by costs of analgesic drug prescriptions, increased rates of hospitalisation, emergency department visits and outpatient hospital visits compared to participants treated for cancer who did not develop CIPN. Pike et al. [230] showed that painful CIPN was associated with higher average costs of $17,344 compared to patients without CIPN. Notably, oxaliplatin- or paclitaxel-based chemotherapy regimens are more likely to result in neuropathic pain, and the pain associated with OIPN and TIPN is more severe and protracted [15]. The treatment of chronic neuropathic pain is often inadequate and may be poorly tolerated [231].
Preventative Treatment
A Cochrane systematic review of interventions and an expert group systematic review by the American Society of Clinical Oncology (ASCO) recommended against the use of a range of interventions (acupuncture, cryotherapy, exercise therapy or ganglioside-monosialic acid (GM-1), retinoic acid, amifostine, amitriptyline, calcium magnesium infusion (Ca/Mg), calmangafodipir, cannabinoids, carbamazepine, l-carnosine, diethyldithiocarbamate (DDTC), gabapentin, pregabalin, glutamate, glutathione, goshajinkigan (GJG), metformin minocycline, N-acetylcysteine, nimodipine, omega-3 fatty acids, ORG 2766, oxcarbazepine, recombinant human leukemia inhibitory factor, venlafaxine, vitamin B or vitamin E) in CIPN [232, 233]. Moreover, acetyl-l-carnitine is strongly advised against due to high-quality evidence indicating worsening neuropathy [232, 234].
The ACTTION [Analgesic, Anesthetic, and Addiction Clinical Trial Translations, Innovations, Opportunities, and Networks]/CONCEPPT [Clinical Endpoints and Procedures for Peripheral Neuropathy Trials] consortia developed recommendations for CIPN prevention studies [235]. These included the selection of outcome measures and endpoints, eligibility criteria, potential effects of the investigational therapy on the efficacy of chemotherapy and accurate sample size estimation [235]. Summaries of studies evaluating putative preventative therapies are detailed in Table 4.
Nutraceuticals
Nutraceuticals as neuroprotective agents have not yielded strong evidence for the prevention of neurotoxicity. For instance, α-lipoic acid [236], OPERA [237], curcumin [238] and Neuronorm [239] have shown no benefit in randomised controlled trials despite positive findings in preclinical studies. Similarly, despite positive pilot studies, a large phase III trial did not demonstrate a significant neuroprotective effect of vitamin E and glutathione supplementation [240–243, 244, 245] [246]. However, in two randomised, standard-of-care-controlled trials and a smaller non-randomised standard-of-care-controlled trial, glutamine was associated with reduced incidence and severity of dysaesthesias, nerve conduction impairment and interference with daily functioning [247–249]. Amifostine demonstrated a clinically meaningful benefit for the prevention of sensory and auditory CIPN but was associated with worsening nausea and vomiting [250–252]. Patients administered diethyldithiocarbamate (DDTC), with lower cumulative doses of cisplatin, were more likely to withdraw from treatment due to CisPN-related adverse events [253]. Similarly, the hexapeptide analogue of ACTH, ORG 2766, increased the incidence of CIPN in a smaller cohort study [252]. Caution is advised with nutraceuticals and supplements with unproven efficacy.
Calcium and Magnesium Infusion
Retrospective studies of patients with advanced colorectal cancer treated with oxaliplatin found that calcium and magnesium infusion (Ca/Mg) significantly reduced the incidence of all-grade OIPN compared to 5-fluorouracil and leucovorin [254, 255]. Notably, a meta-analysis found that Ca/Mg treatment reduced the incidence of severe chronic OIPN (grade ≥ 2) (0.44 (95% CI 0.23–0.85; p = 0.01)) but does not reduce the incidence of acute OIPN (0.41 (95% CI 0.11–1.49; p = 0.18)) [256]. The reduction in acute OIPN incidence with Ca/Mg infusions has not been replicated in a phase I RCT and a large phase III RCT (Table 4) [257–259].
Symptomatic Treatments
Recently published ASCO guidelines indicate that duloxetine is the only currently recommended treatment; however, due to a lack of definitive efficacy, no recommendations can be made for exercise therapy, acupuncture, scrambler therapy, gabapentin, pregabalin, topical gel treatment (containing baclofen/amitriptyline plus/minus ketamine), tricyclic antidepressants or oral cannabinoids in the treatment of symptomatic CIPN [232]. Based on current clinical trial data (Table 5), larger, high-quality studies are needed to confirm efficacy and identify risks of treatment [9, 232, 235, 260, 261].
Acupuncture
In a systematic review of acupuncture for the treatment of CIPN, two out of three trials found acupuncture to be effective in improving self-reported CIPN measures [262–264], but one trial found no benefit [265]. A recent systematic review identified 19 RCTs with 1174 patients and showed that acupuncture significantly improved not only pain but also, surprisingly, nerve conduction velocity [266]. Pilot studies of electro-acupuncture and laser acupuncture have shown improvements in self-reported measures and sensory testing in patients with chronic CIPN [267, 268].
Exercise
A secondary analysis of a large phase III randomised controlled trial of non-pharmaceutical interventions in cancer patients found that exercise reduced sensory symptoms in participants with OIPN, TIPN or VIPN, especially in participants who were older, male or had breast cancer [269]. A recent systematic review and meta-analysis indicated that exercise interventions significantly improve CIPN symptoms, and a sensorimotor-based exercise intervention reduced CIPN-induced loss of postural stability [270].
Topical therapies
A large phase III randomised, placebo-controlled trial of participants with CIPN treated with topical 2% ketamine plus 4% amitriptyline showed no benefit on mean pain, numbness or tingling scores when compared to placebo (p = 0.363) [271].However, a pilot study of 44 participants with CIPN treated with topical 10% amitriptyline showed a five-point reduction in mean pain scores after 4 weeks (p < 0.0001) [272].
High-Strength Capsaicin Patch
An in vitro study showed that oxaliplatin modulates the sensitivity of the capsaicin receptor (TRPV1) response through a secondary intracellular messenger [273]. In a single-centre study, the high-dose capsaicin 8% patch reduced pain by 84% in 18 participants with OIPN, 12 weeks after the patch was applied [274]. Similarly, a single-centre, open-label, longitudinal study showed that the capsaicin 8% patch ameliorated neuropathic pain in 16 participants with chronic CIPN, with evidence of regeneration of intraepidermal nerve fibres, suggestive of initial degeneration due to capsaicin [275]. Indeed, the latest ASCO guidelines indicate that the efficacy of the high-dose 8% capsaicin patch should be further explored [232].
Oxycodone
In a multicentre, phase IV study, oxycodone and naloxone taken together with gabapentin (≥ 900 mg/day) was found to decrease mean numeric rating scale pain scores from 6.0 ± 1.3 to 4.7 ± 2.1, after 4 weeks (p = < 0.0001) [276]. Similarly, treatment with controlled-release oxycodone reduced mean pain intensity from 7.6 to 1.3 at day 14 (p < 0.002) [277]. However, close monitoring of long-term opioid therapy, particularly in combination with gabapentinoids, is advised [278].
Gabapentinoids
A double-blind, randomised, placebo-controlled trial found pregabalin to be more effective than both gabapentin and amitriptyline in decreasing pain scores, with a morphine-sparing effect associated with pregabalin monotherapy [279]. A pilot study and a cohort study identified gabapentin as a potential treatment with improved self-reported measures of CIPN [280, 281]. Nevertheless, a randomised, double-blind, placebo-controlled, crossover, phase III trial (n = 115) failed to show a significant change in the pain score with gabapentin in patients with CIPN [282]. Further, the pre-emptive administration of pregabalin did not decrease the risk of painful OIPN [283].
Tricyclic Antidepressants
A phase III randomised, double-blind, placebo-controlled, crossover trial of nortriptyline in participants with CisPN showed no benefit on paraesthesia or neuropathic symptoms, although there was an improvement in sleep (p < 0.02) [284]. Amitriptyline has shown no efficacy for the improvement or prevention of CIPN symptoms in two double-blind, randomised, placebo-controlled trials [285, 286].
Selective Serotonin Reuptake Inhibitors (SSRI)
There is limited evidence for the use of SSRIs in painful CIPN [232].
Selective Serotonin and Norepinephrine Reuptake Inhibitor (SNRI)
Studies in experimental models of painful neuropathy have demonstrated a superior antinociceptive effect of norepinephrine compared to serotonin [287], and combined increases in both serotonin and norepinephrine result in a better analgesic effect than an increase in either one alone [288]. A multicentre randomised, double-blind, placebo-controlled crossover trial demonstrated the efficacy of duloxetine in participants undergoing platinum or taxane chemotherapy regimens [289]. At 5 weeks, participants receiving duloxetine reported a greater mean decrease in pain score compared to placebo (1.06 [95% CI 0.72–1.40] vs 0.34 [95% CI 0.01–0.66] p = 0.00)3 [290]. Similar results have been reported in other smaller studies [291, 292]. Further, participants with painful OIPN are more likely to respond to duloxetine that those with TIPN [293]. Duloxetine has a greater effect than venlafaxine on pain scores [294]. Recent ASCO guidelines advise a moderate recommendation for the use of duloxetine in CIPN [232]. A randomised, double-blind, placebo-controlled phase III trial found greater improvements in pain relief by ≥ 50% in participants receiving venlafaxine compared to placebo (p = 0.02) [295]. A retrospective cohort study of participants with painful TIPN or OIPN found that venlafaxine achieved relief of paraesthesia in over half the participants for up to 9 weeks (p < 0.001) [296]. However, a small pilot randomised, placebo-controlled, double-blind study found no significant effect of venlafaxine in the prevention of OIPN [297].
Conclusion
CIPN is a major dose-limiting side effect of chemotherapy, and the burden of CIPN continues to increase with increasing cancer-survivorship. Clinical guidance for the treatment of CIPN highlights the paucity of preventative strategies and symptom management. The diagnosis and assessment of CIPN lacks a reference standard, with studies utilising heterogeneous CIPN assessment tools dependent on self-reported outcome measures. The recent ACTTION recommendations endorse a pathomechanism-driven treatment discovery approach to CIPN. CCM may provide an adjunct to NCS in natural history studies and trials of disease-modifying therapies. Detailed mechanistic research in CIPN and CIPN-related neuropathic pain is needed to address the substantial burden on the patient, families and society.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. 2018;68(6):394–424.
Arnold M, Rutherford MJ, Bardot A, Ferlay J, Andersson TML, Myklebust TÅ, et al. Progress in cancer survival, mortality, and incidence in seven high-income countries 1995–2014 (ICBP SURVMARK-2): a population-based study. Lancet Oncol. 2019;20(11):1493–505.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA. 2019;69(1):7–34.
Henley SJ, Singh SD, King J, Wilson RJ, O’Neil ME, Ryerson AB. Invasive cancer incidence and survival-United States, 2012. MMWR. 2015;64(49):1353–8.
Argyriou AA, Bruna J, Marmiroli P, Cavaletti G. Chemotherapy-induced peripheral neurotoxicity (CIPN): an update. Crit Rev Oncol Hematol. 2012;82(1):51–77.
Addington J, Freimer M (2106) Chemotherapy-induced peripheral neuropathy an update on the current understanding. Crit Rev 5:10
Cavaletti G, Marmiroli P. Chemotherapy-induced peripheral neurotoxicity. Nature reviews. Neurology. 2010;6:657.
Cavaletti G, Grp CIPS. Chemotherapy-induced peripheral neurotoxicity (CIPN): the dilemma of proper assessment. Nat Rev Neurol. 2018;12:657–66.
Colvin LA. Chemotherapy-induced peripheral neuropathy: where are we now? Pain. 2019;160(Suppl 1):S1–10.
Seretny M, Currie GL, Sena ES, Ramnarine S, Grant R, MacLeod MR, et al. Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: a systematic review and meta-analysis. Pain. 2014;155(12):2461–70.
Trotti A, Colevas AD, Setser A, Rusch V, Jaques D, Budach V, et al. CTCAE v3.0: development of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol. 2003;13(3):176–81.
Smith EML, Knoerl R, Yang JJ, Kanzawa-Lee G, Lee D, Bridges CM. In search of a gold standard patient-reported outcome measure for use in chemotherapy-induced peripheral neuropathy clinical trials. Cancer Control. 2018;25(1):1073274818756608.
Smith EML, Cohen JA, Pett MA, Beck SL. The reliability and validity of a modified total neuropathy score-reduced and neuropathic pain severity items when used to measure chemotherapy-induced peripheral neuropathy in patients receiving taxanes and platinums. Cancer Nurs. 2010;33(3):173–83.
Pachman DR, Barton DL, Watson JC, Loprinzi CL. Chemotherapy-induced peripheral neuropathy: prevention and treatment. Clin Pharmacol Ther. 2011;90(3):377–87.
Molassiotis A, Cheng HL, Lopez V, Au JSK, Chan A, Bandla A, et al. Are we mis-estimating chemotherapy-induced peripheral neuropathy? Analysis of assessment methodologies from a prospective, multinational, longitudinal cohort study of patients receiving neurotoxic chemotherapy. BMC Cancer. 2019;19(1):132.
Mendoza TR, Wang XS, Williams LA, Shi Q, Vichaya EG, Dougherty PM, et al. Measuring therapy-induced peripheral neuropathy: preliminary development and validation of the treatment-induced neuropathy assessment scale. J Pain. 2015;16(10):1032–43.
Kautio AL, Saarto T, Haanpää M, Leminen A, Kalso E, Kautiainen H. Oxaliplatin scale and National Cancer Institute-common toxicity criteria in the assessment of chemotherapy-induced peripheral neuropathy. Anticancer Res. 2011;31(10):3493–6.
Cavaletti G, Frigeni B, Lanzani F, Piatti M, Rota S, Briani C, et al. The Total Neuropathy Score as an assessment tool for grading the course of chemotherapy-induced peripheral neurotoxicity: comparison with the National Cancer Institute-Common Toxicity Scale. J Peripher Nerv Syst. 2007;12(3):210–5.
Alberti P, Rossi E, Cornblath DR, Merkies ISJ, Postma TJ, Frigeni B, et al. Physician-assessed and patient-reported outcome measures in chemotherapy-induced sensory peripheral neurotoxicity: two sides of the same coin. Ann Oncol. 2014;25:257–64.
Alberti P. Chemotherapy-induced peripheral neurotoxicity—outcome measures: the issue. Expert Opin Drug Metab Toxicol. 2017;13(3):241–3.
Abdi S, Dougherty PM. Chemotherapy-induced peripheral neuropathy: a challenge for clinicians. Oncology. 2016;30(11):1030.
Park SB, Kwok JB, Asher R, Lee CK, Beale P, Selle F, et al. Clinical and genetic predictors of paclitaxel neurotoxicity based on patient-versus clinician-reported incidence and severity of neurotoxicity in the ICON7 trial. Ann Oncol. 2017;28(11):2733–40.
Yeo F, Ng CC, Loh KWJ, Molassiotis A, Cheng HL, Au JSK, et al. Minimal clinically important difference of the EORTC QLQ-CIPN20 for worsening peripheral neuropathy in patients receiving neurotoxic chemotherapy. Support Care Cancer. 2019;27(12):4753–62.
Pachman DR, Qin R, Seisler DK, Smith EML, Beutler AS, Ta LE, et al. Clinical course of patients with oxaliplatin-associated neuropathy: N08CB (Alliance). J Clin Oncol. 2014;32(15):3595.
Pachman DR, Qin R, Seisler DK, Smith EM, Beutler AS, Ta LE, et al. Clinical course of oxaliplatin-induced neuropathy: results from the randomized phase III Trial N08CB (Alliance). J Clin Oncol. 2015;33(30):3416–22.
Pachman DR, Qin R, Seisler D, Smith EM, Kaggal S, Novotny P, et al. Comparison of oxaliplatin and paclitaxel-induced neuropathy (Alliance A151505). Support Care Cancer. 2016;24(12):5059–68.
Le-Rademacher J, Kanwar R, Seisler D, Pachman DR, Qin R, Abyzov A, et al. Patient-reported (EORTC QLQ-CIPN20) versus physician-reported (CTCAE) quantification of oxaliplatin- and paclitaxel/carboplatin-induced peripheral neuropathy in NCCTG/Alliance clinical trials. Support Care Cancer. 2017;25(11):3537–44.
Kaiser K, Lyleroehr M, Shaunfield S, Lacson L, Corona M, Kircher S, et al. Neuropathy experienced by colorectal cancer patients receiving oxaliplatin: a qualitative study to validate the Functional Assessment of Cancer Therapy/Gynecologic Oncology Group-Neurotoxicity scale. World J Gastrointest Oncol. 2020;12(2):205–18.
Soveri LM, Lamminmaki A, Hanninen UA, Karhunen M, Bono P, Osterlund P. Long-term neuropathy and quality of life in colorectal cancer patients treated with oxaliplatin containing adjuvant chemotherapy. Acta Oncol. 2019;58(4):398–406.
Grisold A, Ackerl M, Surböck B, Giometto B, Grisold W. Multifocal neuropathy in vinorelbine treatment for breast cancer (P6.186). Neurology. 2017;88(16 Suppl):186.
Tamburin S, Park SB, Alberti P, Demichelis C, Schenone A, Argyriou AA. Taxane and epothilone-induced peripheral neurotoxicity: from pathogenesis to treatment. J Peripher Nerv Syst. 2019;24(Suppl 2):S40-s51.
Grammatico S, Cesini L, Petrucci MT. Managing treatment-related peripheral neuropathy in patients with multiple myeloma. Blood Lymphat Cancer. 2016;6:37–47.
Thawani SP, Tanji K, De Sousa EA, Weimer LH, Brannagan TH 3rd. Bortezomib-associated demyelinating neuropathy—clinical and pathologic features. J Clin Neuromuscul Dis. 2015;16(4):202–9.
World Health Organization (2019) International Agency for Research on Cancer. WHO-IARC, Global Cancer Obsevatory Lyon, France. https://gco.iarc.fr/
Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, et al. Cancer treatment and survivorship statistics. CA. 2016;66(4):271–89.
Cunningham D, Starling N, Rao S, Iveson T, Nicolson M, Coxon F, et al. Capecitabine and oxaliplatin for advanced esophagogastric cancer. N Engl J Med. 2008;358(1):36–46.
Hall PS, Swinson D, Waters JS, Wadsley J, Falk S, Roy R, et al. Optimizing chemotherapy for frail and elderly patients (pts) with advanced gastroesophageal cancer (aGOAC): the GO2 phase III trial. J Clin Oncol. 2019;37(15 Suppl):4006.
Al-Batran S-E, Homann N, Pauligk C, Goetze TO, Meiler J, Kasper S, et al. Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): a randomised, phase 2/3 trial. Lancet. 2019;393(10184):1948–57.
Zajączkowska R, Kocot-Kępska M, Leppert W, Wrzosek A, Mika J, Wordliczek J. Mechanisms of chemotherapy-induced peripheral neuropathy. Int J Mol Sci. 2019;20(6):1451.
Hana S, Irina V. Pathophysiology of Chemotherapy-Induced Peripheral Neuropathy. Front Mol Neurosci. 2017;10:174.
Kroigard T, Schroder HD, Qvortrup C, Eckhoff L, Pfeiffer P, Gaist D, et al. Characterization and diagnostic evaluation of chronic polyneuropathies induced by oxaliplatin and docetaxel comparing skin biopsy to quantitative sensory testing and nerve conduction studies. Eur J Neurol. 2014;21(4):623–9.
Gebremedhn EG, Shortland PJ, Mahns DA. The incidence of acute oxaliplatin-induced neuropathy and its impact on treatment in the first cycle: a systematic review. BMC Cancer. 2018;18(1):410.
Park SB, Lin CSY, Krishnan AV, Goldstein D, Friedlander ML, Kiernan MC. Long-term neuropathy after oxaliplatin treatment: challenging the dictum of reversibility. Oncologist. 2011;16(5):708–16.
Lehky TJ, Leonard GD, Wilson RH, Grem JL, Floeter MK. Oxaliplatin-induced neurotoxicity: acute hyperexcitability and chronic neuropathy. Muscle Nerve. 2004;29(3):387–92.
Land SR, Kopec JA, Cecchini RS, Ganz PA, Wieand HS, Colangelo LH, et al. Neurotoxicity from oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for stage II and III colon cancer: NSABP C-07. J Clin Oncol. 2007;25(16):2205–11.
de Gramont A, Figer A, Seymour M, Homerin M, Hmissi A, Cassidy J, et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18(16):2938–47.
Briani C, Argyriou AA, Izquierdo C, Velasco R, Campagnolo M, Alberti P, et al. Long-term course of oxaliplatin-induced polyneuropathy: a prospective 2-year follow-up study. J Peripher Nerv Syst. 2014;19(4):299–306.
Branca JJV, Morucci G, Paternostro F, Gulisano M, Pacini A, Maresca M, et al. Oxaliplatin-induced blood brain barrier loosening: a new point of view on chemotherapy-induced neurotoxicity. Oncotarget. 2018;9(34):23426–38.
Fujita S, Hirota T, Sakiyama R, Baba M, Ieiri I. Identification of drug transporters contributing to oxaliplatin-induced peripheral neuropathy. J Neurochem. 2019;148(3):373–85.
Banach M, Juranek JK, Zygulska AL. Chemotherapy-induced neuropathies-a growing problem for patients and health care providers. Brain Behav. 2017;7:e00558.
Scuteri A, Galimberti A, Maggioni D, Ravasi M, Pasini S, Nicolini G, et al. Role of MAPKs in platinum-induced neuronal apoptosis. Neurotoxicology. 2009;30:312–9.
Melli G, Taiana M, Camozzi F, Triolo D, Podini P, Quattrini A, et al. Alpha-lipoic acid prevents mitochondrial damage and neurotoxicity in experimental chemotherapy neuropathy. Exp Neurol. 2008;214:276.
Podratz JL, Knight AM, Ta LE, Staff NP, Gass JM, Genelin K, et al. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol Dis. 2011;41(3):661–8.
Flatters SJL, Bennett GJ. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain. 2006;122(3):245–57.
Zheng H, Xiao WH, Bennett GJ. Functional deficits in peripheral nerve mitochondria in rats with paclitaxel- and oxaliplatin-evoked painful peripheral neuropathy. Exp Neurol. 2011;232(2):154–61.
Ta LE, Espeset L, Podratz J, Windebank AJ. Neurotoxicity of oxaliplatin and cisplatin for dorsal root ganglion neurons correlates with platinum-DNA binding. Neurotoxicology. 2006;27:992.
Di Cesare ML, Zanardelli M, Failli P, Ghelardini C. Oxaliplatin-induced oxidative stress in nervous system-derived cellular models: Could it correlate with in vivo neuropathy? Free Radical Biol Med. 2013;61:143–50.
Sharawy N, Rashed L, Youakim MF. Evaluation of multi-neuroprotective effects of erythropoietin using cisplatin induced peripheral neurotoxicity model. Exp Toxicol Pathol. 2015;67(4):315–22.
Joseph EK, Chen X, Bogen O, Levine JD. Oxaliplatin acts on IB4-positive nociceptors to induce an oxidative stress-dependent acute painful peripheral neuropathy. J Pain. 2008;9(5):463–72.
Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12(10):1161–208.
Di Cesare ML, Zanardelli M, Failli P, Ghelardini C. Oxaliplatin-induced neuropathy: oxidative stress as pathological mechanism. Protective effect of silibinin. J Pain. 2012;13(3):276–84.
Shim HS, Bae C, Wang J, Lee KH, Hankerd KM, Kim HK, et al. Peripheral and central oxidative stress in chemotherapy-induced neuropathic pain. Mol Pain. 2019;15:1744806919840098.
Di Cesare ML, Zanardelli M, Landini I, Pacini A, Ghelardini C, Mini E, et al. Effect of the SOD mimetic MnL4 on in vitro and in vivo oxaliplatin toxicity: possible aid in chemotherapy induced neuropathy. Free Radical Biol Med. 2016;93:67–76.
Viatchenko-Karpinski V, Ling J, Gu JG. Down-regulation of Kv43 channels and a-type K+ currents in V2 trigeminal ganglion neurons of rats following oxaliplatin treatment. Mol Pain. 2018;14:1.
Makker PGS, White D, Lees JG, Parmar J, Goldstein D, Park SB, et al. Acute changes in nerve excitability following oxaliplatin treatment in mice. J Neurophysiol. 2020;124(1):232–44.
Alberti P, Canta A, Chiorazzi A, Fumagalli G, Meregalli C, Monza L, et al. Topiramate prevents oxaliplatin-related axonal hyperexcitability and oxaliplatin induced peripheral neurotoxicity. Neuropharmacology. 2020;164:107905.
Kono T, Satomi M, Suno M, Kimura N, Yamazaki H, Furukawa H, et al. Oxaliplatin-induced neurotoxicity involves TRPM8 in the mechanism of acute hypersensitivity to cold sensation. Brain Behav. 2012;2(1):68–73.
Descoeur J, Pereira V, Pizzoccaro A, Francois A, Ling B, Maffre V, et al. Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol Med. 2011;2:266–78.
Benoit E, Brienza S, Dubois JM. Oxaliplatin, an anticancer agent that affects both Na^+ and K^+ channels in frog peripheral myelinated axons. Gen Physiol Biophys. 2006;25:263–76.
Wahlman C, Doyle TM, Little JW, Luongo L, Janes K, Chen Z, et al. Chemotherapy-induced pain is promoted by enhanced spinal adenosine kinase levels through astrocyte-dependent mechanisms. Pain. 2018;159(6):1025–34.
Robinson CR, Zhang H, Dougherty PM. Astrocytes, but not microglia, are activated in oxaliplatin and bortezomib-induced peripheral neuropathy in the rat. Neuroscience. 2014;274:308–17.
Hu LY, Zhou Y, Cui WQ, Hu XM, Du LX, Mi WL, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) dependent microglial activation promotes cisplatin-induced peripheral neuropathy in mice. Brain Behav Immun. 2018;68:132–45.
Mekhail TM, Markman M. Paclitaxel in cancer therapy. Expert Opin Pharmacother. 2002;3(6):755–66.
Yared JA, Tkaczuk KH. Update on taxane development: new analogs and new formulations. Drug Des Devel Ther. 2012;6:371–84.
Brewer JR, Morrison G, Dolan ME, Fleming GF. Chemotherapy-induced peripheral neuropathy: current status and progress. Gynecol Oncol. 2016;140(1):176–83.
Swain SM, Arezzo JC. Neuropathy associated with microtubule inhibitors: diagnosis, incidence, and management. Clin Adv Hematol Oncol. 2008;6(6):455–67.
Dougherty PM, Cata JP, Cordella JV, Burton A, Weng HR. Taxol-induced sensory disturbance is characterized by preferential impairment of myelinated fiber function in cancer patients. Pain. 2004;109(1–2):132–42.
Hilkens PH, Verweij J, Vecht CJ, Stoter G, van den Bent MJ. Clinical characteristics of severe peripheral neuropathy induced by docetaxel (Taxotere). Ann Oncol. 1997;8(2):187–90.
Mustafa Ali M, Moeller M, Rybicki L, Moore HCF. Long-term peripheral neuropathy symptoms in breast cancer survivors. Breast Cancer Res Treat. 2017;166(2):519–26.
Bandos H, Melnikow J, Rivera DR, Swain SM, Sturtz K, Fehrenbacher L, et al. Long-term Peripheral Neuropathy in Breast Cancer Patients Treated With Adjuvant Chemotherapy: NRG Oncology/NSABP B-30. J Natl Cancer Inst. 2018. https://doi.org/10.1093/jnci/djx162.
de la Morena BP, Conesa M, González-Billalabeitia E, Urrego E, García-Garre E, García-Martínez E, et al. Delayed recovery and increased severity of Paclitaxel-induced peripheral neuropathy in patients with diabetes. J Natl Compr Canc Netw. 2015;13(4):417–23.
Bridges CM, Smith EM. What about Alice? Peripheral neuropathy from taxane-containing treatment for advanced nonsmall cell lung cancer. Support Care Cancer. 2014;22(9):2581–92.
Bonomi P, Kim K, Fairclough D, Cella D, Kugler J, Rowinsky E, et al. Comparison of survival and quality of life in advanced non-small-cell lung cancer patients treated with two dose levels of paclitaxel combined with cisplatin versus etoposide with cisplatin: results of an Eastern Cooperative Oncology Group trial. J Clin Oncol. 2000;18(3):623–31.
Shimozuma K, Ohashi Y, Takeuchi A, Aranishi T, Morita S, Kuroi K, et al. Taxane-induced peripheral neuropathy and health-related quality of life in postoperative breast cancer patients undergoing adjuvant chemotherapy: N-SAS BC 02, a randomized clinical trial. Support Care Cancer. 2012;20(12):3355–64.
Lindå H, Sköld MK, Ochsmann T. Activating transcription factor 3, a useful marker for regenerative response after nerve root injury. Front Neurol. 2011;2:30.
Kidd JF, Pilkington MF, Schell MJ, Fogarty KE, Skepper JN, Taylor CW, et al. Paclitaxel affects cytosolic calcium signals by opening the mitochondrial permeability transition pore. J Biol Chem. 2002;277(8):6504–10.
Krukowski K, Ma J, Laumet GO, Gutti T, Heijnen CJ, Kavelaars A, et al. HDAC6 inhibition effectively reverses chemotherapy-induced peripheral neuropathy. Pain. 2017;158(6):1126–37.
Windebank AJ, Grisold W. Chemotherapy-induced neuropathy. J Peripher Nerv Syst. 2008;13(1):27–46.
McCormick B, Lowes DA, Colvin L, Torsney C, Galley HF. MitoVitE, a mitochondria-targeted antioxidant, limits paclitaxel-induced oxidative stress and mitochondrial damage in vitro, and paclitaxel-induced mechanical hypersensitivity in a rat pain model. Br J Anaesth. 2016;117(5):659–66.
Li Y, Tatsui CE, Rhines LD, North RY, Harrison DS, Cassidy RM, et al. Dorsal root ganglion neurons become hyperexcitable and increase expression of voltage-gated T-type calcium channels (Cav3.2) in paclitaxel-induced peripheral neuropathy. Pain. 2017;158(3):417–29.
Bennett GJ, Liu GK, Xiao WH, Jin HW, Siau C. Terminal arbor degeneration—a novel lesion produced by the antineoplastic agent paclitaxel. Eur J Neurosci. 2011;33(9):1667–76.
Li Y, North RY, Rhines LD, Tatsui CE, Rao G, Edwards DD, et al. DRG voltage-gated sodium channel 1.7 is upregulated in paclitaxel-induced neuropathy in rats and in humans with neuropathic pain. J Neurosci. 2018;38(5):1124–36.
Chang W, Berta T, Kim YH, Ji RR, Lee S, Lee SY. Expression and role of voltage-gated sodium channels in human dorsal root ganglion neurons with special focus on Nav1.7, species differences, and regulation by paclitaxel. Neurosci Bull. 2018;34(1):4–12.
Li Y, Zhang H, Zhang H, Kosturakis AK, Jawad AB, Dougherty PM. Toll-like receptor 4 signaling contributes to Paclitaxel-induced peripheral neuropathy. J Pain. 2014;15(7):712–25.
Li Y, Zhang H, Kosturakis AK, Cassidy RM, Zhang H, Kennamer-Chapman RM, et al. MAPK signaling downstream to TLR4 contributes to paclitaxel-induced peripheral neuropathy. Brain Behav Immun. 2015;49:255–66.
Li Y, Adamek P, Zhang H, Tatsui CE, Rhines LD, Mrozkova P, et al. The cancer chemotherapeutic paclitaxel increases human and rodent sensory neuron responses to TRPV1 by activation of TLR4. J Neurosci. 2015;35(39):13487–500.
Penson RT, Kronish K, Duan Z, Feller A, Stark P, Cook SE, et al. Cytokines IL-1b, IL-2, IL-6, IL-8, MCP-1, GM-CSF and TNFa in patients with epithelial ovarian cancer and their relationship to treatment with paclitaxel. Eur J Cancer. 1999;35:S239.
Siau C, Xiao W, Bennett GJ. Paclitaxel- and vincristine-evoked painful peripheral neuropathies: loss of epidermal innervation and activation of Langerhans cells. Exp Neurol. 2006;201(2):507–14.
Krukowski K, Eijkelkamp N, Laumet G, Hack CE, Yan L, Dougherty PM, et al. CD8+ T cells and endogenous IL-10 are required for resolution of chemotherapy-induced neuropathic pain. J Neurosci. 2016;36(43):11074–83.
Ledeboer A, Jekich BM, Sloane EM, Mahoney JH, Langer SJ, Milligan ED, et al. Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain Behav Immun. 2007;21(5):686–98.
Peters CM, Jimenez-Andrade JM, Kuskowski MA, Ghilardi JR, Mantyh PW. An evolving cellular pathology occurs in dorsal root ganglia, peripheral nerve and spinal cord following intravenous administration of paclitaxel in the rat. Brain Res. 2007;1168:46.
Islam B, Lustberg M, Staff NP, Kolb N, Alberti P, Argyriou AA. Vinca alkaloids, thalidomide and eribulin-induced peripheral neurotoxicity: from pathogenesis to treatment. J Peripher Nerv Syst. 2019;24(Suppl 2):S63–73.
Verstappen CCP, Koeppen S, Heimans JJ, Huijgens PC, Scheulen ME, Strumberg D, et al. Dose-related vincristine-induced peripheral neuropathy with unexpected off-therapy worsening. Neurology. 2005;64(6):1076–7.
Madsen ML, Due H, Ejskjær N, Jensen P, Madsen J, Dybkær K. Aspects of vincristine-induced neuropathy in hematologic malignancies: a systematic review. Cancer Chemother Pharmacol. 2019;84(3):471–85.
Kanbayashi Y, Hosokawa T, Okamoto K, Konishi H, Otsuji E, Yoshikawa T, et al. Statistical identification of predictors for peripheral neuropathy associated with administration of bortezomib, taxanes, oxaliplatin or vincristine using ordered logistic regression analysis. Anticancer Drugs. 2010;21(9):877–81.
Chauvenet AR, Shashi V, Selsky C, Morgan E, Kurtzberg J, Bell B. Vincristine-induced neuropathy as the initial presentation of charcot-marie-tooth disease in acute lymphoblastic leukemia: a Pediatric Oncology Group study. J Pediatr Hematol Oncol. 2003;25(4):316–20.
Ness KK, Jones KE, Smith WA, Spunt SL, Wilson CL, Armstrong GT, et al. Chemotherapy-related neuropathic symptoms and functional impairment in adult survivors of extracranial solid tumors of childhood: results from the St Jude Lifetime Cohort Study. Arch Phys Med Rehabil. 2013;94(8):1451–7.
Andersson M, López-Vega JM, Petit T, Zamagni C, Easton V, Kamber J, et al. Efficacy and safety of pertuzumab and trastuzumab administered in a single infusion bag, followed by vinorelbine: VELVET Cohort 2 final results. Oncologist. 2017;22(10):1160–8.
Haim N, Epelbaum R, Ben-Shahar M, Yarnitsky D, Simri W, Robinson E. Full dose vincristine (without 2-mg dose limit) in the treatment of lymphomas. Cancer. 1994;73(10):2515–9.
Kandula T, Farrar MA, Cohn RJ, Mizrahi D, Carey K, Johnston K, et al. Chemotherapy-induced peripheral neuropathy in long-term survivors of childhood cancer: clinical, neurophysiological, functional, and patient-reported outcomes. JAMA Neurol. 2018;75(8):980–8.
Topp KS, Tanner KD, Levine JD. Damage to the cytoskeleton of large diameter sensory neurons and myelinated axons in vincristine-induced painful peripheral neuropathy in the rat. J Comp Neurol. 2000;424(4):563–76.
Stanton RA, Gernert KM, Nettles JH, Aneja R. Drugs that target dynamic microtubules: a new molecular perspective. Med Res Rev. 2011;31(3):443–81.
Cioroiu C, Weimer LH. Update on chemotherapy-induced peripheral neuropathy. Curr Neurol Neurosci Rep. 2017;17(6):47.
Joseph EK, Levine JD. Mitochondrial electron transport in models of neuropathic and inflammatory pain. Pain. 2006;121(1–2):105–14.
Gerdts J, Summers DW, Milbrandt J, DiAntonio A. Axon self-destruction: new links among SARM1, MAPKs, and NAD+ metabolism. Neuron. 2016;89(3):449–60.
Zhou L, Ao L, Yan Y, Li C, Li W, Ye A, et al. Levo-corydalmine attenuates vincristine-induced neuropathic pain in mice by upregulating the Nrf2/HO-1/CO pathway to inhibit connexin 43 expression. Neurotherapeutics. 2020;17(1):340–55.
Amirkhanloo F, Karimi G, Yousefi-Manesh H, Abdollahi A, Roohbakhsh A, Dehpour AR. The protective effect of modafinil on vincristine-induced peripheral neuropathy in rats: a possible role for TRPA1 receptors. Basic Clin Pharmacol Toxicol. 2020. https://doi.org/10.1111/bcpt.13454.
Liu W, Ye J, Yan H. Investigation of key genes and pathways in inhibition of oxycodone on vincristine-induced microglia activation by using bioinformatics analysis. Dis Markers. 2019. https://doi.org/10.1155/2019/3521746.
Food and Drug Administration (2014) THALOMID® (thalidomide)
Bramuzzo M, Stocco G, Montico M, Arrigo S, Calvi A, Lanteri P, et al. Risk factors and outcomes of thalidomide-induced peripheral neuropathy in a pediatric inflammatory bowel disease cohort. Inflamm Bowel Dis. 2017;23(10):1810–6.
Briani C, Zara G, Rondinone R, Iaccarino L, Ruggero S, Toffanin E, et al. Positive and negative effects of thalidomide on refractory cutaneous lupus erythematosus. Autoimmunity. 2005;38(7):549–55.
Bastuji-Garin S, Ochonisky S, Bouche P, Gherardi RK, Duguet C, Djerradine Z, et al. Incidence and risk factors for thalidomide neuropathy: a prospective study of 135 dermatologic patients. J Invest Dermatol. 2002;119(5):1020–6.
Zara G, Ermani M, Rondinone R, Arienti S, Doria A. Thalidomide and sensory neurotoxicity: a neurophysiological study. J Neurol Neurosurg Psychiatry. 2008;79(11):1258–61.
Morawska M, Grzasko N, Kostyra M, Wojciechowicz J, Hus M. Therapy-related peripheral neuropathy in multiple myeloma patients. Hematol Oncol. 2015;33(4):113–9.
Mileshkin L, Stark R, Day B, Seymour JF, Zeldis JB, Prince HM. Development of neuropathy in patients with myeloma treated with thalidomide: patterns of occurrence and the role of electrophysiologic monitoring. J Clin Oncol. 2006;24(27):4507–14.
Chaudhry V, Cornblath DR, Corse A, Freimer M, Simmons-O’Brien E, Vogelsang G. Thalidomide-induced neuropathy. Neurology. 2002;59(12):1872–5.
Chaudhry V, Cornblath DR, Polydefkis M, Ferguson A, Borrello I. Characteristics of bortezomib- and thalidomide-induced peripheral neuropathy. J Peripher Nerv Syst. 2008;13(4):275–82.
Isoardo G, Bergui M, Durelli L, Barbero P, Boccadoro M, Bertola A, et al. Thalidomide neuropathy: clinical, electrophysiological and neuroradiological features. Acta Neurol Scand. 2004;109(3):188–93.
Richardson PG, Briemberg H, Jagannath S, Wen PY, Barlogie B, Berenson J, et al. Frequency, characteristics, and reversibility of peripheral neuropathy during treatment of advanced multiple myeloma with bortezomib. J Clin Oncol. 2006;24(19):3113–20.
Tosi P, Zamagni E, Cellini C, Plasmati R, Cangini D, Tacchetti P, et al. Neurological toxicity of long-term (>1 yr) thalidomide therapy in patients with multiple myeloma. Eur J Haematol. 2005;74(3):212–6.
Cavaletti G, Beronio A, Reni L, Ghiglione E, Schenone A, Briani C, et al. Thalidomide sensory neurotoxicity: a clinical and neurophysiologic study. Neurology. 2004;62(12):2291–3.
Katodritou E, Vadikolia C, Lalagianni C, Kotsopoulou M, Papageorgiou G, Kyrtsonis MC, et al. “Real-world” data on the efficacy and safety of lenalidomide and dexamethasone in patients with relapsed/refractory multiple myeloma who were treated according to the standard clinical practice: a study of the Greek Myeloma Study Group. Ann Hematol. 2014;93(1):129–39.
Glasmacher A, Hahn C, Hoffmann F, Naumann R, Goldschmidt H, von Lilienfeld-Toal M, et al. A systematic review of phase-II trials of thalidomide monotherapy in patients with relapsed or refractory multiple myeloma. Br J Haematol. 2006;132(5):584–93.
Plasmati R, Pastorelli F, Cavo M, Petracci E, Zamagni E, Tosi P, et al. Neuropathy in multiple myeloma treated with thalidomide: a prospective study. Neurology. 2007;69(6):573–81.
Dimopoulos M, Spencer A, Attal M, Prince HM, Harousseau J-L, Dmoszynska A, et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;357(21):2123–32.
Briani C, Torre CD, Campagnolo M, Lucchetta M, Berno T, Candiotto L, et al. Lenalidomide in patients with chemotherapy-induced polyneuropathy and relapsed or refractory multiple myeloma: results from a single-centre prospective study. J Peripher Nerv Syst. 2013;18(1):19–24.
Miguel JS, Weisel K, Moreau P, Lacy M, Song K, Delforge M, et al. Pomalidomide plus low-dose dexamethasone versus high-dose dexamethasone alone for patients with relapsed and refractory multiple myeloma (MM-003): a randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14(11):1055–66.
Latif T, Chauhan N, Khan R, Moran A, Usmani SZ. Thalidomide and its analogues in the treatment of multiple myeloma. Exp Hematol Oncol. 2012;1(1):27.
Fonseca R, Jena AB, Peneva D, Clancy Z. Survival gains in multiple myeloma from 2003 to 2014. J Clin Oncol. 2018;36(30 suppl):98.
Barlogie B, Tricot G, Anaissie E, Shaughnessy J, Rasmussen E, van Rhee F, et al. Thalidomide and hematopoietic-cell transplantation for multiple myeloma. N Engl J Med. 2006;354(10):1021–30.
Fullerton PM, O’Sullivan DJ. Thalidomide neuropathy: a clinical electrophysiological, and histological follow-up study. J Neurol Neurosurg Psychiatry. 1968;31(6):543–51.
Hafström T. Polyneuropathy after neurosedyn (thalidomide) and its prognosis. Acta Neurol Scand. 1967;43(S32):5–41.
Banach M, Jurczyszyn A, Skotnicki A. Thalidomide induced peripheral neuropathy in multiple myeloma patients. Przegl Lek. 2015;72(11):629–35.
Luczkowska K, Litwinska Z, Paczkowska E, Machalinski B. Pathophysiology of drug-induce peripheral neuropathy in patients with multiple myeloma. J Physiol Pharmacol. 2018. https://doi.org/10.26402/jpp.2018.2.02.
Tamilarasan KP, Kolluru GK, Rajaram M, Indhumathy M, Saranya R, Chatterjee S. Thalidomide attenuates nitric oxide mediated angiogenesis by blocking migration of endothelial cells. BMC Cell Biol. 2006;7(1):17.
Keifer JA, Guttridge DC, Ashburner BP, Baldwin AS Jr. Inhibition of NF-kappa B activity by thalidomide through suppression of IkappaB kinase activity. J Biol Chem. 2001;276(25):22382–7.
Kirchmair R, Tietz AB, Panagiotou E, Walter DH, Silver M, Yoon YS, et al. Therapeutic angiogenesis inhibits or rescues chemotherapy-induced peripheral neuropathy: taxol- and thalidomide-induced injury of vasa nervorum is ameliorated by VEGF. Mol Ther. 2007;15(1):69–75.
Herndon TM, Deisseroth A, Kaminskas E, Kane RC, Koti KM, Rothmann MD, et al. US Food and Drug Administration approval: carfilzomib for the treatment of multiple myeloma. Clin Cancer Res. 2013;19(17):4559–63.
Kane RC, Bross PF, Farrell AT, Pazdur R. Velcade: US FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist. 2003;8(6):508–13.
Pantani L, Zamagni E, Zannetti BA, Pezzi A, Tacchetti P, Brioli A, et al. Bortezomib and dexamethasone as salvage therapy in patients with relapsed/refractory multiple myeloma: analysis of long-term clinical outcomes. Ann Hematol. 2014;93(1):123–8.
Electronic Medicines Compendium. Bortezomib 2.5mg powder for solution for injection 2019. https://www.medicines.org.uk/emc/product/10568/smpc
Argyriou AA, Iconomou G, Kalofonos HP. Bortezomib-induced peripheral neuropathy in multiple myeloma: a comprehensive review of the literature. Blood. 2008;112(5):1593–9.
Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348(26):2609–17.
Kropff M, Bisping G, Schuck E, Liebisch P, Lang N, Hentrich M, et al. Bortezomib in combination with intermediate-dose dexamethasone and continuous low-dose oral cyclophosphamide for relapsed multiple myeloma. Br J Haematol. 2007;138(3):330–7.
Kane RC, Dagher R, Farrell A, Ko CW, Sridhara R, Justice R, et al. Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res. 2007;13(18 Pt 1):5291–4.
Martin TG. Peripheral neuropathy experience in patients with relapsed and/or refractory multiple myeloma treated with carfilzomib. Oncology (Williston Park). 2013;27(Suppl 3):4–10.
Kumar SK, Berdeja JG, Niesvizky R, Lonial S, Laubach JP, Hamadani M, et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: an open-label phase 1/2 study. Lancet Oncol. 2014;15(13):1503–12.
Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Špička I, Oriol A, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015;372(2):142–52.
Sonneveld P. Management of multiple myeloma in the relapsed/refractory patient. Hematology Am Soc Hematol Educ Program. 2017;2017(1):508–17.
Mohan M, Matin A, Davies FE. Update on the optimal use of bortezomib in the treatment of multiple myeloma. Cancer Manag Res. 2017;9:51–63.
Landowski TH, Megli CJ, Nullmeyer KD, Lynch RM, Dorr RT. Mitochondrial-mediated disregulation of Ca2+ is a critical determinant of Velcade (PS-341/bortezomib) cytotoxicity in myeloma cell lines. Cancer Res. 2005;65(9):3828–36.
Cavaletti G, Gilardini A, Canta A, Rigamonti L, Rodriguez-Menendez V, Ceresa C, et al. Bortezomib-induced peripheral neurotoxicity: a neurophysiological and pathological study in the rat. Exp Neurol. 2007;204(1):317–25.
Meregalli C, Canta A, Carozzi VA, Chiorazzi A, Oggioni N, Gilardini A, et al. Bortezomib-induced painful neuropathy in rats: a behavioral, neurophysiological and pathological study in rats. Eur J Pain. 2010;14(4):343–50.
Zheng H, Xiao WH, Bennett GJ. Mitotoxicity and bortezomib-induced chronic painful peripheral neuropathy. Exp Neurol. 2012;238(2):225–34.
Iijima Y, Bandow K, Amano S, Sano M, Hino S, Kaneko T, et al. Protection of bortezomib-induced neurotoxicity by antioxidants. Anticancer Res. 2020;40(7):3685–96.
Alé A, Bruna J, Calls A, Karamita M, Haralambous S, Probert L, et al. Inhibition of the neuronal NFκB pathway attenuates bortezomib-induced neuropathy in a mouse model. Neurotoxicology. 2016;55:58–64.
Zhao W, Wang W, Li X, Liu Y, Gao H, Jiang Y, et al. Peripheral neuropathy following bortezomib therapy in multiple myeloma patients: association with cumulative dose, heparanase, and TNF-α. Ann Hematol. 2019;98(12):2793–803.
Zheng Y, Sun Y, Yang Y, Zhang S, Xu T, Xin W, et al. GATA3-dependent epigenetic upregulation of CCL21 is involved in the development of neuropathic pain induced by bortezomib. Mol Pain. 2019;15:1744806919863292.
Park SB, Alberti P, Kolb NA, Gewandter JS, Schenone A, Argyriou AA. Overview and critical revision of clinical assessment tools in chemotherapy-induced peripheral neurotoxicity. J Peripher Nerv Syst. 2019;24(Suppl 2):S13-s25.
Cavaletti G, Frigeni B, Lanzani F, Mattavelli L, Susani E, Alberti P, et al. Chemotherapy-induced peripheral neurotoxicity assessment: a critical revision of the currently available tools. Eur J Cancer. 2010;46(3):479–94.
Alberti P, Bernasconi DP, Cornblath DR, Jose Merkies IS, Park SB, Velasco R, et al. Prospective evaluation of health care provider and patient assessments in chemotherapy induced peripheral neurotoxicity. Neurology. 2021. https://doi.org/10.1212/WNL.0000000000012300.
Kandula T, Farrar MA, Kiernan MC, Krishnan AV, Goldstein D, Horvath L, et al. Neurophysiological and clinical outcomes in chemotherapy-induced neuropathy in cancer. Clin Neurophysiol. 2017;128(7):1166–75.
Park SB, Goldstein D, Lin CS, Krishnan AV, Friedlander ML, Kiernan MC. Acute abnormalities of sensory nerve function associated with oxaliplatin-induced neurotoxicity. J Clin Oncol. 2009;27(8):1243–9.
Krishnan AV, Goldstein D, Friedlander M, Kiernan MC. Oxaliplatin and axonal Na+ channel function in vivo. Clin Cancer Res. 2006;12(15):4481–4.
Kiernan MC, Krishnan AV. The pathophysiology of oxaliplatin-induced neurotoxicity. Curr Med Chem. 2006;13(24):2901–7.
Argyriou AA, Polychronopoulos P, Iconomou G, Chroni E, Kalofonos HP. A review on oxaliplatin-induced peripheral nerve damage. Cancer Treat Rev. 2008;34(4):368–77.
Argyriou AA, Koltzenburg M, Polychronopoulos P, Papapetropoulos S, Kalofonos HP. Peripheral nerve damage associated with administration of taxanes in patients with cancer. Crit Rev Oncol Hematol. 2008;66(3):218–28.
Park SB, Lin CSY, Krishnan AV, Goldstein D, Friedlander ML, Kiernan MC. Oxaliplatin-induced neurotoxicity: changes in axonal excitability precede development of neuropathy. Brain. 2009;132(Pt 10):2712–23.
Park SB, Lin CSY, Krishnan AV, Goldstein D, Friedlander ML, Kiernan MC. Dose effects of oxaliplatin on persistent and transient Na+ conductances and the development of neurotoxicity. PLoS ONE. 2011;6(4):e18469.
Krøigård T, Svendsen TK, Wirenfeldt M, Schrøder HD, Qvortrup C, Pfeiffer P, et al. Early changes in tests of peripheral nerve function during oxaliplatin treatment and their correlation with chemotherapy-induced polyneuropathy symptoms and signs. Eur J Neurol. 2020;27(1):68–76.
Murray JE, Pickering HR, Lin CSY, Goldstein D, Friedlander ML, Kiernan MC, et al. 6 Functional impact of neuropathy in patients receiving oxaliplatin chemotherapy. Clin Neurophysiol. 2014;125:e3.
Osmani K, Vignes S, Aissi M, Wade F, Milani P, Lévy BI, et al. Taxane-induced peripheral neuropathy has good long-term prognosis: a 1- to 13-year evaluation. J Neurol. 2012;259(9):1936–43.
Pal PK. Clinical and electrophysiological studies in vincristine induced neuropathy. Electromyogr Clin Neurophysiol. 1999;39(6):323–30.
Timmins HC, Li T, Kiernan MC, Baron-Hay S, Marx G, Boyle F, et al. Taxane-induced peripheral neuropathy: differences in patient report and objective assessment. Support Care Cancer. 2020;28:459–4466.
Timmins HC, Li T, Huynh W, Kiernan MC, Baron-Hay S, Boyle F, et al. Electrophysiological and phenotypic profiles of taxane-induced neuropathy. Clin Neurophysiol. 2020;131:1979–85.
Matsuoka A, Mitsuma A, Maeda O, Kajiyama H, Kiyoi H, Kodera Y, et al. Quantitative assessment of chemotherapy-induced peripheral neurotoxicity using a point-of-care nerve conduction device. Cancer Sci. 2016;107(10):1453–7.
Kocer B, Sucak G, Kuruoglu R, Aki Z, Haznedar R, Erdogmus NI. Clinical and electrophysiological evaluation of patients with thalidomide-induced neuropathy. Acta Neurol Belg. 2009;109(2):120–6.
Fruhstorfer H. Thermal sensibility changes during ischemie nerve block. Pain. 1984;20(4):355–61.
Magerl W, Fuchs PN, Meyer RA, Treede R-D. Roles of capsaicin-insensitive nociceptors in cutaneous pain and secondary hyperalgesia. Brain. 2001;124(9):1754–64.
Ziegler EA, Magerl W, Meyer RA, Treede R-D. Secondary hyperalgesia to punctate mechanical stimuli: central sensitization to A-fibre nociceptor input. Brain. 1999;122(12):2245–57.
Thomas S, Ajroud-Driss S, Dimachkie MM, Gibbons C, Freeman R, Simpson DM, et al. Peripheral neuropathy research registry: a prospective cohort. J Peripher Nerv Syst. 2019;24(1):39–47.
Krøigård T, Schrøder HD, Qvortrup C, Eckhoff L, Pfeiffer P, Gaist D, et al. Characterization and diagnostic evaluation of chronic polyneuropathies induced by oxaliplatin and docetaxel comparing skin biopsy to quantitative sensory testing and nerve conduction studies. Eur J Neurol. 2014;21(4):623–9.
Velasco R, Videla S, Villoria J, Ortiz E, Navarro X, Bruna J. Reliability and accuracy of quantitative sensory testing for oxaliplatin-induced neurotoxicity. Acta Neurol Scand. 2015;131(5):282–9.
Zhi WI, Chen P, Kwon A, Chen C, Harte SE, Piulson L, et al. Chemotherapy-induced peripheral neuropathy (CIPN) in breast cancer survivors: a comparison of patient-reported outcomes and quantitative sensory testing. Breast Cancer Res Treat. 2019;178(3):587–95.
Dougherty PM, Cata JP, Burton AW, Vu K, Weng HR. Dysfunction in multiple primary afferent fiber subtypes revealed by quantitative sensory testing in patients with chronic vincristine-induced pain. J Pain Symptom Manag. 2007;33(2):166–79.
Cata JP, Weng HR, Burton AW, Villareal H, Giralt S, Dougherty PM. Quantitative sensory findings in patients with bortezomib-induced pain. J Pain. 2007;8(4):296–306.
Lauria G, Cornblath DR, Johansson O, McArthur JC, Mellgren SI, Nolano M, et al. EFNS guidelines on the use of skin biopsy in the diagnosis of peripheral neuropathy. Eur J Neurol. 2005;12(10):747–58.
Lauria G, Lombardi R. Skin biopsy: a new tool for diagnosing peripheral neuropathy. BMJ. 2007;334(7604):1159–62.
Collongues N, Samama B, Schmidt-Mutter C, Chamard-Witkowski L, Debouverie M, Chanson J-B, et al. Quantitative and qualitative normative dataset for intraepidermal nerve fibers using skin biopsy. PLoS ONE. 2018;13(1):e0191614.
Burakgazi AZ, Messersmith W, Vaidya D, Hauer P, Hoke A, Polydefkis M. Longitudinal assessment of oxaliplatininduced neuropathy. Neurology. 2011;77(10):980–6.
Bechakra M, Nieuwenhoff MD, van Rosmalen J, Groeneveld GJ, Scheltens-de Boer M, Sonneveld P, et al. Clinical, electrophysiological, and cutaneous innervation changes in patients with bortezomib-induced peripheral neuropathy reveal insight into mechanisms of neuropathic pain. Mol Pain. 2018. https://doi.org/10.1177/1744806918797042.
Alam U, Jeziorska M, Petropoulos IN, Asghar O, Fadavi H, Ponirakis G, et al. Diagnostic utility of corneal confocal microscopy and intra-epidermal nerve fibre density in diabetic neuropathy. PLoS ONE. 2017;12(7):e0180175.
Chen X, Graham J, Dabbah MA, Petropoulos IN, Ponirakis G, Asghar O, et al. Small nerve fiber quantification in the diagnosis of diabetic sensorimotor polyneuropathy: comparing corneal confocal microscopy with intraepidermal nerve fiber density. Diabetes Care. 2015;38(6):1138–44.
Kemp HI, Petropoulos IN, Rice ASC, Vollert J, Maier C, Strum D, et al. Use of corneal confocal microscopy to evaluate small nerve fibers in patients with human immunodeficiency virus. JAMA Ophthalmol. 2017;135(7):795–800.
Ferdousi M, Azmi S, Petropoulos IN, Fadavi H, Ponirakis G, Marshall A, et al. Corneal confocal microscopy detects small fibre neuropathy in patients with upper gastrointestinal cancer and nerve regeneration in chemotherapy induced peripheral neuropathy. PLoS ONE. 2015;10(10):e0139394.
Quattrini C, Tavakoli M, Jeziorska M, Kallinikos P, Tesfaye S, Finnigan J, et al. Surrogate markers of small fiber damage in human diabetic neuropathy. Diabetes. 2007;56:2148.
Stettner M, Hinrichs L, Guthoff R, Bairov S, Petropoulos IN, Warnke C, et al. Corneal confocal microscopy in chronic inflammatory demyelinating polyneuropathy. Ann Clin Transl Neurol. 2015;3(2):88–100.
Kautio A-L, Haanpaa M, Kautiainen H, Kalso E, Saarto T. Burden of chemotherapy-induced neuropathy—a cross-sectional study. Support Care Cancer. 2011;12:1991.
Azmi S, Ferdousi M, Alam U, Petropoulos IN, Ponirakis G, Marshall A, et al. Small-fibre neuropathy in men with type 1 diabetes and erectile dysfunction: a cross-sectional study. Diabetologia. 2017;60(6):1094–101.
Evdokimov D, Frank J, Klitsch A, Unterecker S, Warrings B, Serra J, et al. Reduction of skin innervation is associated with a severe fibromyalgia phenotype. Ann Neurol. 2019;86(4):504–16.
Perkins BA, Lovblom LE, Bril V, Scarr D, Ostrovski I, Orszag A, et al. Corneal confocal microscopy for identification of diabetic sensorimotor polyneuropathy: a pooled multinational consortium study. Diabetologia. 2018;61(8):1856–61.
Lewis EJH, Lovblom LE, Ferdousi M, Halpern EM, Jeziorska M, Pacaud D, et al. Rapid corneal nerve fiber loss: a marker of diabetic neuropathy onset and progression. Diabetes Care. 2020;48:1829–35.
Argyriou AA, Park SB, Islam B, Tamburin S, Velasco R, Alberti P, et al. Neurophysiological, nerve imaging and other techniques to assess chemotherapy-induced peripheral neurotoxicity in the clinical and research settings. J Neurol Neurosurg Psychiatry. 2019;90(12):1361–9.
Campagnolo M, Lazzarini D, Cacciavillani M, Fregona I, Bergamo F, Lonardi S, et al. Corneal confocal microscopy in patients with chemotherapy-induced neuropathy. PLoS ONE. 2012;10:S11–2.
Cocito F, Ricciardelli G, Mangiacavalli S, Pompa A, Pochintesta L, Ferretti V, et al. Corneal sub-basal neural damage pattern in multiple myeloma patients treated with bortezomib: an in vivo confocal study. Leuk Lymphoma. 2015;56(12):3440–1.
Bennedsgaard K, Ventzel L, Andersen NT, Themistocleous AC, Bennett DL, Jensen TS, et al. Oxaliplatin- and docetaxel-induced polyneuropathy: clinical and neurophysiological characteristics. J Peripher Nerv Syst. 2020;25(4):377–87.
Petropoulos IN, Al-Mohammedi A, Chen X, Ferdousi M, Ponirakis G, Kemp H, et al. The utility of corneal nerve fractal dimension analysis in peripheral neuropathies of different etiology. Transl Vis Sci Technol. 2020;9(9):43.
Chiang JCB, Goldstein D, Trinh T, Au K, Mizrahi D, Muhlmann M, et al. A cross-sectional study of sub-basal corneal nerve reduction following neurotoxic chemotherapy. Transl Vis Sci Technol. 2021;10(1):24.
Bennett MI, Rayment C, Hjermstad M, Aass N, Caraceni A, Kaasa S. Prevalence and aetiology of neuropathic pain in cancer patients: a systematic review. Pain. 2012;153(2):359–65.
Oh SY, Shin SW, Koh S-J, Bae SB, Chang H, Kim JH, et al. Multicenter, cross-sectional observational study of the impact of neuropathic pain on quality of life in cancer patients. Support Care Cancer. 2017;25(12):3759–67.
Kautio AL, Haanpää M, Kautiainen H, Kalso E, Saarto T. Burden of chemotherapy-induced neuropathy—a cross-sectional study. Support Care Cancer. 2011;19(12):1991–6.
Ezendam NP, Pijlman B, Bhugwandass C, Pruijt JF, Mols F, Vos MC, et al. Chemotherapy-induced peripheral neuropathy and its impact on health-related quality of life among ovarian cancer survivors: results from the population-based PROFILES registry. Gynecol Oncol. 2014;135(3):510–7.
Gewandter JS, Fan L, Magnuson A, Mustian K, Peppone L, Heckler C, et al. Falls and functional impairments in cancer survivors with chemotherapy-induced peripheral neuropathy (CIPN): a University of Rochester CCOP study. Support Care Cancer. 2013;21(7):2059–66.
Kuroi K, Shimozuma K. Neurotoxicity of taxanes: symptoms and quality of life assessment. Breast Cancer. 2004;11(1):92.
Miaskowski C, Mastick J, Paul SM, Topp K, Smoot B, Abrams G, et al. Chemotherapy-Induced Neuropathy in Cancer Survivors. J Pain Symptom Manag. 2017;54(2):204-18.e2.
Mols F, Beijers T, Lemmens V, van den Hurk CJ, Vreugdenhil G, van de Poll-Franse LV. Chemotherapy-induced neuropathy and its association with quality of life among 2- to 11-year colorectal cancer survivors: results from the population-based PROFILES registry. J Clin Oncol. 2013;31(21):2699–707.
Mols F, Beijers T, Vreugdenhil G, van de Poll-Franse L. Chemotherapy-induced peripheral neuropathy and its association with quality of life: a systematic review. Support Care Cancer. 2014;22(8):2261–9.
Zanville NR, Nudelman KN, Smith DJ, Von Ah D, McDonald BC, Champion VL, et al. Evaluating the impact of chemotherapy-induced peripheral neuropathy symptoms (CIPN-sx) on perceived ability to work in breast cancer survivors during the first year post-treatment. Support Care Cancer. 2016;24(11):4779–89.
Song X, Wilson KL, Kagan J, Panjabi S. Cost of peripheral neuropathy in patients receiving treatment for multiple myeloma: a US administrative claims analysis. Ther Adv Hematol. 2019. https://doi.org/10.1177/2040620719839025.
Pike CT, Birnbaum HG, Muehlenbein CE, Pohl GM, Natale RB. Healthcare costs and workloss burden of patients with chemotherapy-associated peripheral neuropathy in breast, ovarian, head and neck, and nonsmall cell lung cancer. Chemother Res Pract. 2012. https://doi.org/10.1155/2012/913848.
Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. The Lancet Neurol. 2015;14(2):162–73.
Loprinzi CL, Lacchetti C, Bleeker J, Cavaletti G, Chauhan C, Hertz DL, et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: ASCO guideline update. J Clin Oncol. 2020;38:3325–48.
Albers JW, Chaudhry V, Cavaletti G, Donehower RC. Interventions for preventing neuropathy caused by cisplatin and related compounds. Cochrane Database Syst Rev. 2014. https://doi.org/10.1002/14651858.CD005228.
Hershman DL, Unger JM, Crew KD, Minasian LM, Awad D, Moinpour CM, et al. Randomized double-blind placebo-controlled trial of acetyl-L-carnitine for the prevention of taxane-induced neuropathy in women undergoing adjuvant breast cancer therapy. J Clin Oncol. 2013;31(20):2627–33.
Gewandter JS, Brell J, Cavaletti G, Dougherty PM, Evans S, Howie L, et al. Trial designs for chemotherapy-induced peripheral neuropathy prevention: ACTTION recommendations. Neurology. 2018;91(9):403–13.
Guo Y, Jones D, Palmer JL, Forman A, Dakhil SR, Velasco MR, et al. Oral alpha-lipoic acid to prevent chemotherapy-induced peripheral neuropathy: a randomized, double-blind, placebo-controlled trial. Support Care Cancer. 2014;22(5):1223–31.
Desideri I, Francolini G, Becherini C, Terziani F, Delli Paoli C, Olmetto E, et al. Use of an alpha lipoic, methylsulfonylmethane and bromelain dietary supplement (Opera®) for chemotherapy-induced peripheral neuropathy management, a prospective study. Med Oncol. 2017;34(3):1–5.
Howells LM, Iwuji COO, Irving GRB, Barber S, Walter H, Sidat Z, et al. Curcumin combined with FOLFOX chemotherapy is safe and tolerable in patients with metastatic colorectal cancer in a randomized phase IIa trial. J Nutr. 2019;149(7):1133–9.
Maschio M, Zarabla A, Maialetti A, Marchesi F, Giannarelli D, Gumenyuk S, et al. Prevention of bortezomib-related peripheral neuropathy with docosahexaenoic acid and α-lipoic acid in patients with multiple myeloma: preliminary data. Integr Cancer Ther. 2018;17(4):1115.
Pace A, Savarese A, Picardo M, Maresca V, Pacetti U, Del Monte G, et al. Neuroprotective effect of vitamin E supplementation in patients treated with cisplatin chemotherapy. J Clin Oncol. 2003;21(5):927–31.
Pace A, Nisticò C, Cuppone F, Bria E, Galiè E, Graziano G, et al. Peripheral neurotoxicity of weekly paclitaxel chemotherapy: a schedule or a dose issue? Clin Breast Cancer. 2007;7(7):550–4.
Kottschade LA, Sloan JA, Mazurczak MA, Johnson DB, Murphy BP, Rowland KM, et al. The use of vitamin E for the prevention of chemotherapy-induced peripheral neuropathy: results of a randomized phase III clinical trial. Support Care Cancer. 2011;19(11):1769–77.
Argyriou AA, Chroni E, Koutras A, Ellul J, Papapetropoulos S, Katsoulas G, et al. Vitamin E for prophylaxis against chemotherapy-induced neuropathy: a randomized controlled trial. Neurology. 2005;64(1):26–31.
Cascinu S, Catalano V, Cordella L, Labianca R, Giordani P, Baldelli AM, et al. Neuroprotective effect of reduced glutathione on oxaliplatin-based chemotherapy in advanced colorectal cancer: a randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2002;20(16):3478–83.
Cascinu S, Cordella L, Del Ferro E, Fronzoni M, Catalano G. Neuroprotective effect of reduced glutathione on cisplatin-based chemotherapy in advanced gastric cancer: a randomized double-blind placebo-controlled trial. J Clin Oncol. 1995;13(1):26–32.
Leal AD, Qin R, Atherton PJ, Haluska P, Behrens RJ, Tiber CH, et al. North Central Cancer Treatment Group/Alliance trial N08CA-the use of glutathione for prevention of paclitaxel/carboplatin-induced peripheral neuropathy: a phase 3 randomized, double-blind, placebo-controlled study. Cancer. 2014;120(12):1890–7.
Wang WS, Lin JK, Lin TC, Chen WS, Jiang JK, Wang HS, et al. Oral glutamine is effective for preventing oxaliplatin-induced neuropathy in colorectal cancer patients. Oncologist. 2007;12(3):312–9.
Vahdat L, Papadopoulos K, Lange D, Leuin S, Kaufman E, Donovan D, et al. Reduction of paclitaxel-induced peripheral neuropathy with glutamine. Clin Cancer Res. 2001;7(5):1192–7.
Stubblefield MD, Vahdat LT, Balmaceda CM, Troxel AB, Hesdorffer CS, Gooch CL. Glutamine as a neuroprotective agent in high-dose paclitaxel-induced peripheral neuropathy: a clinical and electrophysiologic study. Clin Oncol (R Coll Radiol). 2005;17(4):271–6.
Leong SS, Tan EH, Fong KW, Wilder-Smith E, Ong YK, Tai BC, et al. Randomized double-blind trial of combined modality treatment with or without amifostine in unresectable stage III non-small-cell lung cancer. J Clin Oncol. 2003;21(9):1767–74.
Hilpert F, Stähle A, Tomé O, Burges A, Rossner D, Späthe K, et al. Neuroprotection with amifostine in the first-line treatment of advanced ovarian cancer with carboplatin/paclitaxel-based chemotherapy—a double-blind, placebo-controlled, randomized phase II study from the Arbeitsgemeinschaft Gynäkologische Onkologoie (AGO) Ovarian Cancer Study Group. Support Care Cancer. 2005;13(10):797–805.
Gurney JG, Bass JK, Onar-Thomas A, Huang J, Chintagumpala M, Bouffet E, et al. Evaluation of amifostine for protection against cisplatin-induced serious hearing loss in children treated for average-risk or high-risk medulloblastoma. Neuro Oncol. 2014;16(6):848–55.
Gandara DR, Nahhas WA, Adelson MD, Lichtman SM, Podczaski ES, Yanovich S, et al. Randomized placebo-controlled multicenter evaluation of diethyldithiocarbamate for chemoprotection against cisplatin-induced toxicities. J Clin Oncol. 1995;13(2):490–6.
Knijn N, Tol J, Koopman M, Werter MJ, Imholz AL, Valster FA, et al. The effect of prophylactic calcium and magnesium infusions on the incidence of neurotoxicity and clinical outcome of oxaliplatin-based systemic treatment in advanced colorectal cancer patients. Eur J Cancer. 2011;47(3):369–74.
Gamelin L, Boisdron-Celle M, Delva R, Guérin-Meyer V, Ifrah N, Morel A, et al. Prevention of oxaliplatin-related neurotoxicity by calcium and magnesium infusions: a retrospective study of 161 patients receiving oxaliplatin combined with 5-Fluorouracil and leucovorin for advanced colorectal cancer. Clin Cancer Res. 2004;10(12 Pt 1):4055–61.
Ao R, Wang YH, Li RW, Wang ZR. Effects of calcium and magnesium on acute and chronic neurotoxicity caused by oxaliplatin: a meta-analysis. Exp Ther Med. 2012;4(5):933–7.
Loprinzi CL, Qin R, Dakhil SR, Fehrenbacher L, Stella PJ, Atherton PJ, et al. Phase III randomized, placebo (PL)-controlled, double-blind study of intravenous calcium/magnesium (CaMg) to prevent oxaliplatin-induced sensory neurotoxicity (sNT), N08CB: An alliance for clinical trials in oncology study. J Clin Oncol. 2013;31(15 suppl):3501.
Loprinzi CL, Qin R, Dakhil SR, Fehrenbacher L, Flynn KA, Atherton P, et al. Phase III randomized, placebo-controlled, double-blind study of intravenous calcium and magnesium to prevent oxaliplatin-induced sensory neurotoxicity (N08CB/Alliance). J Clin Oncol. 2014;32(10):997–1005.
Han CH, Khwaounjoo P, Kilfoyle DH, Hill A, McKeage MJ. Phase I drug-interaction study of effects of calcium and magnesium infusions on oxaliplatin pharmacokinetics and acute neurotoxicity in colorectal cancer patients. BMC Cancer. 2013;13(1):495.
Flatters SJL, Dougherty PM, Colvin LA. Clinical and preclinical perspectives on chemotherapy-induced peripheral neuropathy (CIPN): a narrative review. Br J Anaesth. 2017;119(4):737–49.
Gewandter JS, Freeman R, Kitt RA, Cavaletti G, Gauthier LR, McDermott MP, et al. Chemotherapy-induced peripheral neuropathy clinical trials: review and recommendations. Neurology. 2017;89(8):859–69.
Lu Z, Moody J, Marx BL, Hammerstrom T. Treatment of chemotherapy-induced peripheral neuropathy in integrative oncology: a survey of acupuncture and oriental medicine practitioners. J Altern Complement Med. 2017;23(12):964–70.
Li K, Giustini D, Seely D. A systematic review of acupuncture for chemotherapy-induced peripheral neuropathy. Curr Oncol (Toronto, Ont). 2019;26(2):e147–54.
Han X, Wang L, Shi H, Zheng G, He J, Wu W, et al. Acupuncture combined with methylcobalamin for the treatment of chemotherapy-induced peripheral neuropathy in patients with multiple myeloma. BMC Cancer. 2017;17(1):40.
Rostock M, Jaroslawski K, Guethlin C, Ludtke R, Schröder S, Bartsch HH. Chemotherapy-induced peripheral neuropathy in cancer patients: a four-arm randomized trial on the effectiveness of electroacupuncture. Evid Based Complement Altern Med. 2013. https://doi.org/10.1155/2013/349653.
Jin Y, Wang Y, Zhang J, Xiao X, Zhang Q. Efficacy and safety of acupuncture against chemotherapy-induced peripheral neuropathy: a systematic review and meta-analysis. Evid Based Complement Altern Med. 2020;2020:8875433.
Hsieh YL, Chou LW, Hong SF, Chang FC, Tseng SW, Huang CC, et al. Laser acupuncture attenuates oxaliplatin-induced peripheral neuropathy in patients with gastrointestinal cancer: a pilot prospective cohort study. Acupunct Med. 2016;34(5):398–405.
Garcia MK, Cohen L, Guo Y, Zhou Y, You B, Chiang J, et al. Electroacupuncture for thalidomide/bortezomib-induced peripheral neuropathy in multiple myeloma: a feasibility study. J Hematol Oncol. 2014;7(1):41.
Kleckner IR, Kamen C, Gewandter JS, Mohile NA, Heckler CE, Culakova E, et al. Effects of exercise during chemotherapy on chemotherapy-induced peripheral neuropathy: a multicenter, randomized controlled trial. Support Care Cancer. 2018;26(4):1019–28.
Lin WL, Wang RH, Chou FH, Feng IJ, Fang CJ, Wang HH. The effects of exercise on chemotherapy-induced peripheral neuropathy symptoms in cancer patients: a systematic review and meta-analysis. Support Care Cancer. 2021;29:5303–11.
Gewandter JS, Mohile SG, Heckler CE, Ryan JL, Kirshner JJ, Flynn PJ, et al. A phase III randomized, placebo-controlled study of topical amitriptyline and ketamine for chemotherapy-induced peripheral neuropathy (CIPN): a University of Rochester CCOP Study of 462 cancer survivors. Support Care Cancer. 2014;22:1807–14.
Rossignol J, Cozzi B, Liebaert F, Hatton S, Viallard ML, Hermine O, et al. High concentration of topical amitriptyline for treating chemotherapy-induced neuropathies. Support Care Cancer. 2019;27(8):3053–9.
Anand U, Otto WR, Anand P. Sensitization of capsaicin and icilin responses in oxaliplatin treated adult rat DRG neurons. Mol Pain. 2010;6:82.
Filipczak-Bryniarska I, Krzyzewski RM, Kucharz J, Michalowska-Kaczmarczyk A, Kleja J, Woron J, et al. High-dose 8% capsaicin patch in treatment of chemotherapy-induced peripheral neuropathy: single-center experience. Med Oncol. 2017;9:1.
Anand P, Elsafa E, Privitera R, Naidoo K, Yiangou Y, Donatien P, et al. Rational treatment of chemotherapy-induced peripheral neuropathy with capsaicin 8% patch: from pain relief towards disease modification. J Pain Res. 2019;12:2039.
Kim B-S, Jin J-Y, Kwon JH, Woo IS, Ko YH, Park S-Y, et al. Efficacy and safety of oxycodone/naloxone as add-on therapy to gabapentin or pregabalin for the management of chemotherapy-induced peripheral neuropathy in Korea. Asia Pac J Clin Oncol. 2018;14:e448–54.
Cartoni C, Brunetti GA, Federico V, Efficace F, Grammatico S, Tendas A, et al. Controlled-release oxycodone for the treatment of bortezomib-induced neuropathic pain in patients with multiple myeloma. Support Care Cancer. 2012;10:2621.
Bykov K, Bateman BT, Franklin JM, Vine SM, Patorno E. Association of gabapentinoids with the risk of opioid-related adverse events in surgical patients in the United States. JAMA Netw Open. 2020;3(12):e2031647.
Mishra S, Bhatnagar S, Goyal GN, Rana SP, Upadhya SP. A comparative efficacy of amitriptyline, gabapentin, and pregabalin in neuropathic cancer pain: a prospective randomized double-blind placebo-controlled study. Am J Hosp Palliat Care. 2012;29(3):177–82.
Tsavaris N, Kopterides P, Kosmas C, Efthymiou A, Skopelitis H, Dimitrakopoulos A, et al. Gabapentin monotherapy for the treatment of chemotherapy-induced neuropathic pain: a pilot study. Pain Med. 2008;9:1209.
Magnowska M, Izycka N, Kapola-Czyz J, Romala A, Lorek J, Spaczynski M, et al. Effectiveness of gabapentin pharmacotherapy in chemotherapy-induced peripheral neuropathy. Ginekol Pol. 2018;89:200–4.
Rao RD, Loprinzi CL, Michalak JC, Sloan JA, Novotny P, Soori GS, et al. Efficacy of gabapentin in the management of chemotherapy-induced peripheral neuropathy: a phase 3 randomized, double-blind, placebo-controlled, crossover trial (N00C3). Cancer. 2007;110(9):2110–8.
de Andrade DC, Jacobsen Teixeira M, Galhardoni R, Ferreira KSL, Braz Mileno P, Scisci N, et al. Pregabalin for the prevention of oxaliplatin-induced painful neuropathy: a randomized Double-Blind Trial. Oncologist. 2017;22(10):1154-e105.
Hammack JE, Michalak JC, Loprinzi CL, Sloan JA, Novotny PJ, Soori GS, et al. Phase III evaluation of nortriptyline for alleviation of symptoms of cis-platinum-induced peripheral neuropathy. Pain. 2002;98(1–2):195–203.
Kautio A-L, Haanpää M, Saarto T, Kalso E. Amitriptyline in the treatment of chemotherapy-induced neuropathic symptoms. J Pain Symptom Manag. 2008;35(1):31–9.
Kautio AL, Haanpää M, Leminen A, Kalso E, Kautiainen H, Saarto T. Amitriptyline in the prevention of chemotherapy-induced neuropathic symptoms. Anticancer Res. 2009;29(7):2601–6.
Hall FS, Schwarzbaum JM, Perona MTG, Templin JS, Caron MG, Lesch KP, et al. A greater role for the norepinephrine transporter than the serotonin transporter in murine nociception. Neuroscience. 2011;175:315–27.
Bellingham GA, Peng PW. Duloxetine: a review of its pharmacology and use in chronic pain management. Reg Anesth Pain Med. 2010;35(3):294–303.
Smith EM, Pang H, Cirrincione C, Fleishman S, Paskett ED, Ahles T, et al. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. JAMA. 2013;309(13):1359–67.
Smith EML, Pang H, Cirrincione C, Fleishman S, Paskett ED, Ahles T, et al. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy. A randomized clinical trial. JAMA. 2013;309:1359–67.
Yang YH, Lin JK, Chen WS, Lin TC, Yang SH, Jiang JK, et al. Duloxetine improves oxaliplatin-induced neuropathy in patients with colorectal cancer: an open-label pilot study. Support Care Cancer. 2012;20(7):1491–7.
Hirayama Y, Ishitani K, Sato Y, Iyama S, Takada K, Murase K, et al. Effect of duloxetine in Japanese patients with chemotherapy-induced peripheral neuropathy: a pilot randomized trial. Int J Clin Oncol. 2015;20(5):866–71.
Smith EM, Pang H, Ye C, Cirrincione C, Fleishman S, Paskett ED, et al. Predictors of duloxetine response in patients with oxaliplatin-induced painful chemotherapy-induced peripheral neuropathy (CIPN): a secondary analysis of randomised controlled trial—CALGB/alliance 170601. Eur J Cancer Care (Engl). 2017. https://doi.org/10.1111/ecc.12421.
Farshchian N, Alavi A, Heydarheydari S, Moradian N. Comparative study of the effects of venlafaxine and duloxetine on chemotherapy-induced peripheral neuropathy. Cancer Chemother Pharmacol. 2018;82(5):787–93.
Durand JP, Deplanque G, Montheil V, Gornet JM, Scotte F, Mir O, et al. Efficacy of venlafaxine for the prevention and relief of oxaliplatin-induced acute neurotoxicity: results of EFFOX, a randomized, double-blind, placebo-controlled phase III trial. Ann Oncol. 2012;23(1):200–5.
Kus T, Aktas G, Alpak G, Kalender ME, Sevinc A, Kul S, et al. Efficacy of venlafaxine for the relief of taxane and oxaliplatin-induced acute neurotoxicity: a single-center retrospective case–control study. Support Care Cancer. 2016;24:2085.
Zimmerman C, Atherton PJ, Pachman D, Seisler D, Wagner-Johnston N, Dakhil S, et al. MC11C4: a pilot randomized, placebo-controlled, double-blind study of venlafaxine to prevent oxaliplatin-induced neuropathy. Support Care Cancer. 2016;24(3):1071–8.
Leonard GD, Wright MA, Quinn MG, Fioravanti S, Harold N, Schuler B, et al. Survey of oxaliplatin-associated neurotoxicity using an interview-based questionnaire in patients with metastatic colorectal cancer. BMC Cancer. 2005;5:116.
Alejandro LM, Behrendt CE, Chen K, Openshaw H, Shibata S. Predicting acute and persistent neuropathy associated with oxaliplatin. Am J Clin Oncol. 2013;36(4):331–7.
Rothenberg ML, Oza AM, Bigelow RH, Berlin JD, Marshall JL, Ramanathan RK, et al. Superiority of oxaliplatin and fluorouracil-leucovorin compared with either therapy alone in patients with progressive colorectal cancer after irinotecan and fluorouracil-leucovorin: interim results of a phase III trial. J Clin Oncol. 2003;21(11):2059–69.
Yamada Y, Higuchi K, Nishikawa K, Gotoh M, Fuse N, Sugimoto N, et al. Phase III study comparing oxaliplatin plus S-1 with cisplatin plus S-1 in chemotherapy-naïve patients with advanced gastric cancer. Ann Oncol. 2015;26(1):141–8.
Bando H, Yamada Y, Tanabe S, Nishikawa K, Gotoh M, Sugimoto N, et al. Efficacy and safety of S-1 and oxaliplatin combination therapy in elderly patients with advanced gastric cancer. Gastric Cancer. 2016;19(3):919–26.
Lonardi S, Sobrero A, Rosati G, Di Bartolomeo M, Ronzoni M, Aprile G, et al. Phase III trial comparing 3–6 months of adjuvant FOLFOX4/XELOX in stage II-III colon cancer: safety and compliance in the TOSCA trial. Ann Oncol. 2016;27(11):2074–81.
Al-Batran SE, Hartmann JT, Probst S, Schmalenberg H, Hollerbach S, Hofheinz R, et al. Phase III trial in metastatic gastroesophageal adenocarcinoma with fluorouracil, leucovorin plus either oxaliplatin or cisplatin: a study of the Arbeitsgemeinschaft Internistische Onkologie. J Clin Oncol. 2008;26(9):1435–42.
Cassidy J, Clarke S, Díaz-Rubio E, Scheithauer W, Figer A, Wong R, et al. Randomized phase III study of capecitabine plus oxaliplatin compared with fluorouracil/folinic acid plus oxaliplatin as first-line therapy for metastatic colorectal cancer. J Clin Oncol. 2008;26(12):2006–12.
Tournigand C, Cervantes A, Figer A, Lledo G, Flesch M, Buyse M, et al. OPTIMOX1: a randomized study of FOLFOX4 or FOLFOX7 with oxaliplatin in a stop-and-Go fashion in advanced colorectal cancer–a GERCOR study. J Clin Oncol. 2006;24(3):394–400.
André T, Boni C, Mounedji-Boudiaf L, Navarro M, Tabernero J, Hickish T, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004;350(23):2343–51.
Beijers AJ, Mols F, Vreugdenhil G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Support Care Cancer. 2014;22(7):1999–2007.
Land SR, Kopec JA, Cecchini RS, Ganz PA, Wieand HS, Colangelo LH, et al. Neurotoxicity from oxaliplatin combined with weekly bolus fluorouracil and leucovorin as surgical adjuvant chemotherapy for Stage II and III colon cancer: NSABP C-07. J Clin Oncol. 2007;25:2205.
Ibrahim A, Hirschfeld S, Cohen MH, Griebel DJ, Williams GA, Pazdur R. FDA drug approval summaries: oxaliplatin. Oncologist. 2004;9(1):8–12.
Argyriou AA, Polychronopoulos P, Iconomou G, Koutras A, Kalofonos HP, Chroni E. Paclitaxel plus carboplatin-induced peripheral neuropathy. A prospective clinical and electrophysiological study in patients suffering from solid malignancies. J Neurol. 2005;252(12):1459–64.
Peng L, Bu Z, Ye X, Zhou Y, Zhao Q. Incidence and risk of peripheral neuropathy with nab-paclitaxel in patients with cancer: a meta-analysis. Eur J Cancer Care (Engl). 2017. https://doi.org/10.1111/ecc.12407.
Socinski MA, Bondarenko I, Karaseva NA, Makhson AM, Vynnychenko I, Okamoto I, et al. Weekly nab-paclitaxel in combination with carboplatin versus solvent-based paclitaxel plus carboplatin as first-line therapy in patients with advanced non-small-cell lung cancer: final results of a phase III trial. J Clin Oncol. 2012;30(17):2055–62.
Baldwin RM, Owzar K, Zembutsu H, Chhibber A, Kubo M, Jiang C, et al. A genome-wide association study identifies novel loci for paclitaxel-induced sensory peripheral neuropathy in CALGB 40101. Clin Cancer Res. 2012;18(18):5099–109.
Dorling L, Kar S, Michailidou K, Hiller L, Vallier AL, Ingle S, et al. The relationship between common genetic markers of breast cancer risk and chemotherapy-induced toxicity: a case–control study. PLoS ONE. 2016;11(7):e0158984.
Scagliotti G, Novello S, von Pawel J, Reck M, Pereira JR, Thomas M, et al. Phase III study of carboplatin and paclitaxel alone or with sorafenib in advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(11):1835–42.
Scagliotti GV, De Marinis F, Rinaldi M, Crinò L, Gridelli C, Ricci S, et al. Phase III randomized trial comparing three platinum-based doublets in advanced non-small-cell lung cancer. J Clin Oncol. 2002;20(21):4285–91.
Gao G, Chu H, Zhao L, Gui T, Xu Q, Shi J. A meta-analysis of paclitaxel-based chemotherapies administered once every week compared with once every 3 weeks first-line treatment of advanced non-small-cell lung cancer. Lung Cancer. 2012;76(3):380–6.
Ramchandren S, Leonard M, Mody RJ, Donohue JE, Moyer J, Hutchinson R, et al. Peripheral neuropathy in survivors of childhood acute lymphoblastic leukemia. J Peripher Nerv Syst. 2009;14(3):184–9.
Lavoie Smith EM, Li L, Chiang C, Thomas K, Hutchinson RJ, Wells EM, et al. Patterns and severity of vincristine-induced peripheral neuropathy in children with acute lymphoblastic leukemia. J Peripher Nerv Syst. 2015;20(1):37–46.
Dimopoulos MA, Zervas K, Kouvatseas G, Galani E, Grigoraki V, Kiamouris C, et al. Thalidomide and dexamethasone combination for refractory multiple myeloma. Ann Oncol. 2001;12(7):991–5.
Prince HM, Mileshkin L, Roberts A, Ganju V, Underhill C, Catalano J, et al. A multicenter phase II trial of thalidomide and celecoxib for patients with relapsed and refractory multiple myeloma. Clin Cancer Res. 2005;11(15):5504–14.
von Lilienfeld-Toal M, Hahn-Ast C, Furkert K, Hoffmann F, Naumann R, Bargou R, et al. A systematic review of phase II trials of thalidomide/dexamethasone combination therapy in patients with relapsed or refractory multiple myeloma. Eur J Haematol. 2008;81(4):247–52.
Prince HM, Schenkel B, Mileshkin L. An analysis of clinical trials assessing the efficacy and safety of single-agent thalidomide in patients with relapsed or refractory multiple myeloma. Leuk Lymphoma. 2007;48(1):46–55.
Grover JK, Uppal G, Raina V. The adverse effects of thalidomide in relapsed and refractory patients of multiple myeloma. Ann Oncol. 2002;13(10):1636–40.
Facon T, Mary JY, Hulin C, Benboubker L, Attal M, Pegourie B, et al. Melphalan and prednisone plus thalidomide versus melphalan and prednisone alone or reduced-intensity autologous stem cell transplantation in elderly patients with multiple myeloma (IFM 99-06): a randomised trial. Lancet. 2007;370(9594):1209–18.
Richardson PG, Blood E, Mitsiades CS, Jagannath S, Zeldenrust SR, Alsina M, et al. A randomized phase 2 study of lenalidomide therapy for patients with relapsed or relapsed and refractory multiple myeloma. Blood. 2006;108(10):3458–64.
Peng L, Ye X, Zhou Y, Zhang J, Zhao Q. Meta-analysis of incidence and risk of peripheral neuropathy associated with intravenous bortezomib. Support Care Cancer. 2015;23(9):2813–24.
Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352(24):2487–98.
Richardson PG, Sonneveld P, Schuster MW, Stadtmauer EA, Facon T, Harousseau J-L, et al. Reversibility of symptomatic peripheral neuropathy with bortezomib in the phase III APEX trial in relapsed multiple myeloma: impact of a dose-modification guideline. Br J Haematol. 2009;144(6):895–903.
Aguiar PM, de Mendonça LT, Colleoni GWB, Storpirtis S. Efficacy and safety of bortezomib, thalidomide, and lenalidomide in multiple myeloma: an overview of systematic reviews with meta-analyses. Crit Rev Oncol Hematol. 2017;113:195–212.
van der Hoop RG, van der Burg ME, ten Bokkel Huinink WW, van Houwelingen C, Neijt JP. Incidence of neuropathy in 395 patients with ovarian cancer treated with or without cisplatin. Cancer. 1990;66(8):1697–702.
Tofthagen CS, Cheville AL, Loprinzi CL. The physical consequences of chemotherapy-induced peripheral neuropathy. Curr Oncol Rep. 2020;22(5):50.
Ruddy KJ, Le-Rademacher J, Lacouture ME, Wilkinson M, Onitilo AA, Vander Woude AC, et al. Randomized controlled trial of cryotherapy to prevent paclitaxel-induced peripheral neuropathy (RU221511I); an ACCRU trial. Breast. 2019;48:89–97.
Reeves BN, Dakhil SR, Sloan JA, Wolf SL, Burger KN, Kamal A, et al. Further data supporting that paclitaxel-associated acute pain syndrome is associated with development of peripheral neuropathy: North Central Cancer Treatment Group trial N08C1. Cancer. 2012;118(20):5171–8.
Molassiotis A, Cheng HL, Leung KT, Li YC, Wong KH, Au JSK, et al. Risk factors for chemotherapy-induced peripheral neuropathy in patients receiving taxane- and platinum-based chemotherapy. Brain Behav. 2019;9(6):1312.
Miltenburg NC, Boogerd W. Chemotherapy-induced neuropathy:a comprehensive survey. Cancer Treat Rev. 2014;40(7):872–82.
Loprinzi CL, Reeves BN, Dakhil SR, Sloan JA, Wolf SL, Burger KN, et al. Natural history of paclitaxel-associated acute pain syndrome: prospective cohort study NCCTG N08C1. J Clin Oncol. 2011;29(11):1472–8.
Liu H, Xu R, Huang H. Peripheral neuropathy outcomes and efficacy of subcutaneous bortezomib when combined with thalidomide and dexamethasone in the treatment of multiple myeloma. Exp Ther Med. 2016;12(5):3041–6.
Kang L, Tian Y, Xu S, Chen H. Oxaliplatin-induced peripheral neuropathy: clinical features, mechanisms, prevention and treatment. J Neurol. 2020;268:3269–82.
Hershman DL, Lacchetti C, Dworkin RH, Lavoie Smith EM, Bleeker J, Cavaletti G, et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2014;32(18):1941–67.
Glendenning JL, Barbachano Y, Norman AR, Dearnaley DP, Horwich A, Huddart RA. Long-term neurologic and peripheral vascular toxicity after chemotherapy treatment of testicular cancer. Cancer. 2010;116(10):2322–31.
Eckhoff L, Feddersen S, Knoop AS, Ewertz M, Bergmann TK. Docetaxel-induced neuropathy: a pharmacogenetic case-control study of 150 women with early-stage breast cancer. Acta Oncol. 2015;54(4):530–7.
Cavaletti G, Marmiroli P. Management of oxaliplatin-induced peripheral sensory neuropathy. Cancers (Basel). 2020;12(6):1370.
Cavaletti G, Marmiroli P. Chemotherapy-induced peripheral neurotoxicity. Curr Opin Neurol. 2015;28(5):500–7.
Brydøy M, Oldenburg J, Klepp O, Bremnes RM, Wist EA, Wentzel-Larsen T, et al. Observational study of prevalence of long-term raynaud-like phenomena and neurological side effects in testicular cancer survivors. JNCI. 2009;101(24):1682–95.
Argyriou AA, Polychronopoulos P, Koutras A, Iconomou G, Gourzis P, Assimakopoulos K, et al. Is advanced age associated with increased incidence and severity of chemotherapy-induced peripheral neuropathy? Support Care Cancer. 2006;14(3):223–9.
Argyriou AA, Cavaletti G, Bruna J, Kyritsis AP, Kalofonos HP. Bortezomib-induced peripheral neurotoxicity: an update. Arch Toxicol. 2014;88(9):1669–79.
Anyanwu CO, Stewart CL, Werth VP. Thalidomide-induced orofacial neuropathy. J Clin Rheumatol. 2014;20(7):399–400.
Albany C, Dockter T, Wolfe E, Le-Rademacher J, Wagner-Johnston N, Einhorn L, et al. Cisplatin-associated neuropathy characteristics compared with those associated with other neurotoxic chemotherapy agents (Alliance A151724). Support Care Cancer. 2020;29:833–40.
Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, et al. DrugBank 50: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–82.
U.S. Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0 National Institute of Health National Cancer Institute 2017 (updated 27/11/2017). https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_5x7.pdf.
Gedlicka C, Scheithauer W, Schüll B, Kornek GV. Effective treatment of oxaliplatin-induced cumulative polyneuropathy with alpha-lipoic acid. J Clin Oncol. 2002;20(15):3359–61.
Gedlicka C, Kornek GV, Schmid K, Scheithauer W. Amelioration of docetaxel/cisplatin induced polyneuropathy by {alpha}-lipoic acid. Ann Oncol. 2003;14:339–40.
Maschio M, Zarabla A, Maialetti A, Marchesi F, Giannarelli D, Gumenyuk S, et al. The effect of docosahexaenoic acid and α-lipoic acid as prevention of bortezomib-related neurotoxicity in patients with multiple myeloma. Integr Cancer Ther. 2019. https://doi.org/10.1177/1534735419888584.
Roberts JA, Jenison EL, Kim K, Clarke-Pearson D, Langleben A. A randomized, multicenter, double-blind, placebo-controlled, dose-finding study of ORG 2766 in the prevention or delay of cisplatin-induced neuropathies in women with ovarian cancer. Gynecol Oncol. 1997;67(2):172–7.
Koeppen S, Verstappen CC, Körte R, Scheulen ME, Strumberg D, Postma TJ, et al. Lack of neuroprotection by an ACTH (4–9) analogue. a randomized trial in patients treated with vincristine for Hodgkin’s or non-Hodgkin’s lymphoma. J Cancer Res Clin Oncol. 2004;130(3):153–60.
Milla P, Airoldi M, Weber G, Drescher A, Jaehde U, Cattel L. Administration of reduced glutathione in FOLFOX4 adjuvant treatment for colorectal cancer: effect on oxaliplatin pharmacokinetics, Pt-DNA adduct formation, and neurotoxicity. Anticancer Drugs. 2009;20(5):396–402.
Izgu N, Metin ZG, Karadas C, Ozdemir L, Cetin N, Demirci U. Prevention of chemotherapy-induced peripheral neuropathy with classical massage in breast cancer patients receiving paclitaxel: an assessor-blinded randomized controlled trial. Eur J Oncol Nurs. 2019;40:36–43.
Greenlee H, Crew KD, Capodice J, Awad D, Buono D, Shi Z, et al. Randomized sham-controlled pilot trial of weekly electro-acupuncture for the prevention of taxane-induced peripheral neuropathy in women with early stage breast cancer. Breast Cancer Res Treat. 2016;156(3):453–64.
Glimelius B, Manojlovic N, Pfeiffer P, Mosidze B, Kurteva G, Karlberg M, et al. Persistent prevention of oxaliplatin-induced peripheral neuropathy using calmangafodipir (PledOx(®)): a placebo-controlled randomised phase II study (PLIANT). Acta Oncol. 2018;57(3):393–402.
Argyriou AA, Chroni E, Polychronopoulos P, Iconomou G, Koutras A, Makatsoris T, et al. Efficacy of oxcarbazepine for prophylaxis against cumulative oxaliplatin-induced neuropathy. Neurology. 2006;67(12):2253.
Wilson RH, Lehky T, Thomas RR, Quinn MG, Floeter MK, Grem JL. Acute oxaliplatin-induced peripheral nerve hyperexcitability. J Clin Oncol. 2002;20(7):1767–74.
Izgu N, Ozdemir L, Bugdayci BF. Effect of aromatherapy massage on chemotherapy-induced peripheral neuropathic pain and fatigue in patients receiving oxaliplatin: an open label quasi-randomized controlled pilot study. Cancer Nurs. 2019;2:139.
Molassiotis A, Suen LKP, Cheng HL, Mok TSK, Lee SCY, Wang CH, et al. A randomized assessor-blinded wait-list-controlled trial to assess the effectiveness of acupuncture in the management of chemotherapy-induced peripheral neuropathy. Integr Cancer Ther. 2019;18:1534735419836501.
van den Heuvel SAS, van der Wal SEI, Smedes LA, Radema SA, van Alfen N, Vissers KCP, et al. Intravenous lidocaine old-school drug, new purpose—reduction of intractable pain in patients with chemotherapy induced peripheral neuropathy. Pain Res Manag. 2017. https://doi.org/10.1155/2017/8053474.
Rao RD, Loprinzi CL, Flynn PJ, Nashawaty M, Sloan JA, Novotny P, et al. Efficacy of lamotrigine in the management of chemotherapy-induced peripheral neuropathy: a phase 3 randomized, double-blind, placebo-controlled trial, N01C3. Cancer. 2008;112(12):2802–8.
Lynch ME, Cesar-Rittenberg P, Hohmann AG. A double-blind, placebo-controlled, crossover pilot trial with extension using an oral mucosal cannabinoid extract for treatment of chemotherapy-induced neuropathic pain. J Pain Symptom Manag. 2014;1:166.
Barton DL, Wos EJ, Qin R, Mattar BI, Green NB, Lanier KS, et al. A double-blind, placebo-controlled trial of a topical treatment for chemotherapy-induced peripheral neuropathy: NCCTG trial N06CA. Support Care Cancer. 2011;19(6):833–41.
Fallon MT, Storey DJ, Krishan A, Weir CJ, Mitchell R, Fleetwood-Walker SM, et al. Cancer treatment-related neuropathic pain: proof of concept study with menthol—a TRPM8 agonist. Support Care Cancer. 2015;23(9):2769–77.
Ripamonti CI, Santini D, Maranzano E, Berti M, Roila F. Management of cancer pain: ESMO Clinical Practice Guidelines. Ann Oncol. 2012;23(Suppl 7):139–54.
Massey RL, Kim HK, Abdi S. Brief review: chemotherapy-induced painful peripheral neuropathy (CIPPN): current status and future directions. Can J Anesth. 2014;61(8):754–62.
Fradkin M, Batash R, Elmaleh S, Debi R, Schaffer P, Schaffer M, et al. Management of peripheral neuropathy induced by chemotherapy. Curr Med Chem. 2019;26(25):4698–708.
Dorsey SG, Kleckner IR, Barton D, Mustian K, O’Mara A, St Germain D, et al. The National Cancer Institute Clinical Trials planning meeting for prevention and treatment of chemotherapy-induced peripheral neuropathy. J Natl Cancer Inst. 2019;111(6):531–7.
Attal N, Cruccu G, Baron R, Haanpaa M, Hansson P, Jensen TS, et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol. 2010;17(9):1113-e88.
Otake A, Yoshino K, Ueda Y, Sawada K, Mabuchi S, Kimura T, et al. Usefulness of duloxetine for paclitaxel-induced peripheral neuropathy treatment in gynecological cancer patients. Anticancer Res. 2015;35:359–63.
Özdoǧan M, Samur M, Bozcuk HŞ, Çoban E, Savaş B, Aydin H. Venlafaxine for treatment of chemotherapy-induced neuropathic pain. Turkish J Cancer. 2004;34(3):110–3.
Durand JP, Alexandre J, Guillevin L, Goldwasser F. Clinical activity of venlafaxine and topiramate against oxaliplatin-induced disabling permanent neuropathy. Anticancer Drugs. 2005;16:587–91.
Saif MW, Syrigos K, Kaley K, Isufi I. Role of pregabalin in treatment of oxaliplatin-induced sensory neuropathy. Anticancer Res. 2010;30:2927–33.
Acknowledgements
Funding
Jamie Burgess gratefully acknowledges the support given by the Pain Relief Foundation in the form of a PhD studentship whilst writing this manuscript. No funding or sponsorship was received for the publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship contributions
All persons who meet authorship criteria are listed as authors. All authors certify that they have participated sufficiently in the work to take public responsibility for the content, including participation in the concept, design, analysis, writing, or revision of the manuscript. Conceptualisation, Uazman Alam and Jamie Burgess; writing—original draft preparation, Jamie Burgess, Maryam Ferdousi, Anne Marshall and Uazman Alam; writing—review and editing, Jamie Burgess, Kohei Matsumoto, David Gosal, C.B., Andrew Marshall, Tony Mak, Anne Marshall, Bernhard Frank, Rayaz A Malik, Uazman Alam; visualisation, Jamie Burgess and Uazman Alam; supervision, Uazman Alam. All authors have read and agreed to the published version of the manuscript. All included figures were created with Biorender.com.
Disclosures
Jamie Burgess, Uazman Alam, Maryam Ferdousi, David Gosal, Anne Marshall, Tony Mak, Andrew Marshall, Bernhard Frank, Rayaz A Malik and Uazman Alam have nothing to disclose. Cheng Boon’s affiliation changed during the manuscript’s production from the Department of Clinical Oncology, The Clatterbridge Cancer Centre, Wirral, UK to the Department of Clinical Oncology, The Royal Wolverhampton NHS Trust, Wolverhampton, UK.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Data availability
Data sharing is not applicable for this article, as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Burgess, J., Ferdousi, M., Gosal, D. et al. Chemotherapy-Induced Peripheral Neuropathy: Epidemiology, Pathomechanisms and Treatment. Oncol Ther 9, 385–450 (2021). https://doi.org/10.1007/s40487-021-00168-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40487-021-00168-y