
Abstract
Ketoconazole (Ketoconazole HRA™), an imidazole derivative, is a useful treatment option in the management of endogenous Cushing’s syndrome in adults and adolescents > 12 years of age, based on evidence from more than three decades of use of the drug in clinical practice. Originally developed as an antifungal agent, ketoconazole is a potent steroidogenesis inhibitor. Approximately 60% (range, 45–88% across key studies) of patients with Cushing’s syndrome who are treated with ketoconazole achieve control of hypercortisolism, with efficacy demonstrated in all aetiologies of the disease. Furthermore, reductions in cortisol levels in patients treated with ketoconazole are associated with improvements in clinical and biochemical features of Cushing’s syndrome and common comorbidities. Hepatotoxicity, the main safety concern with ketoconazole, can be managed effectively with careful monitoring of hepatic enzymes, with hepatic enzyme abnormalities generally being mild to moderate, asymptomatic and reversible upon dose reduction or drug withdrawal.
Similar content being viewed by others


Digital Features for this Adis Drug Q&A can be found at https://doi.org/10.6084/m9.figshare.13198988. |
Indicated in the EU for the treatment of endogenous Cushing’s syndrome in adults and adolescents > 12 years of age |
Normalisation of urinary free cortisol levels achieved in ~ 60% (range, 45–88% across key studies) of treated patients, with further patients achieving partial control |
Risk of hepatotoxicity can be managed effectively with careful monitoring and dose reduction or drug withdrawal if required |
Other potential adverse events include gastrointestinal events, adrenal insufficiency and rash |
Has a high potential for drug-drug interactions |
What is the rationale for using ketoconazole in Cushing’s syndrome?
Ketoconazole, an imidazole derivative, was originally developed and approved for use as an antifungal agent [1]. In the early 1980s, following observations that ketoconazole was a potent inhibitor of steroidogenesis through its broad inhibition of cytochrome P450 (CYP) enzymes in the adrenal glands, it was hypothesised that ketoconazole may have a role in clinical situations where steroidogenesis inhibition is a therapeutic goal [2, 3]. One such situation is in the treatment of endogenous Cushing’s syndrome, a rare but life-threatening condition resulting from cortisol hypersecretion [4, 5]. In Cushing’s syndrome, chronic hypercortisolism can result in a range of complications or co-morbidities, including hypertension, hypokalaemia, growth retardation in children and diabetes mellitus. Endogenous Cushing’s syndrome can have a range of aetiologies, broadly divided into adrenocorticotropic hormone (ACTH)-dependent disease [including pituitary corticotroph adenoma (Cushing’s disease; ~ 60–70%) and extrapituitary or ectopic ACTH syndrome (EAS; ~ 5–10%)] and ACTH-independent disease (~ 20–30%). Although surgical resection of the underlying tumour is the recommended first-line treatment for Cushing’s syndrome, medical therapies (including steroidogenesis inhibitors) also play a role in treatment, including when surgery is not possible, unsuccessful or contraindicated, or while awaiting the effect of radiation therapy of the pituitary gland [6, 7].
The EMA approval of ketoconazole for use as an antifungal agent was suspended in 2013 after the benefit-risk assessment was deemed to no longer be favourable [8]; however, in 2014, ketoconazole (Ketoconazole HRA™) was approved in the EU for use in the treatment of endogenous Cushing’s syndrome in adults and adolescents above the age of 12 years (Table 1) [9], based on an assessment of the safety and efficacy of the drug from more than three decades of off-label use.
How does ketoconazole work in Cushing’s syndrome?
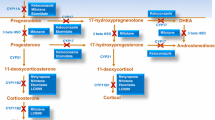
Ketoconazole inhibits several CYP enzymes in the adrenal glands resulting in a rapid and marked reduction in adrenal steroid production (Fig. 1) [1, 2, 9]. Most notably, ketoconazole reduces cortisol synthesis through inhibition of CYP17A1 17α-hydroxylase activity [1, 9,10,11,12]. Ketoconazole also inhibits cortisol and aldosterone synthesis through inhibitory effects on 11β-hydroxylase (CYP11B1), 18-hydroxylase (CYP11B2) and (at higher concentrations) the cholesterol side chain cleavage enzyme (P450scc) [1, 9,10,11,12].

adapted from the EMA Public Assessment Report: Ketoconazole HRA [1]. A red cross indicates ketoconazole inhibition of the enzymatic step. HSD hydroxysteroid dehydrogenase, OH hydroxy-, P450scc cholesterol side chain cleavage enzyme
Ketoconazole inhibition of adrenal steroid biosynthesis. Figure
Ketoconazole also inhibits androgen synthesis through its inhibitory effects on CYP17A1 17,20-desmolase activity in the adrenals and also in Leydig cells (Fig. 1) [1, 9, 11, 12]. Ketoconazole’s inhibitory effect on androgen production may be beneficial in the treatment of hirsutism (present in many women with Cushing's syndrome) [13] but also has the potential to lead to hypogonadism in males [13, 14].
Ketoconazole also inhibits cholesterol synthesis, resulting in decreases in total and LDL cholesterol of 10–25%, suggesting a potential beneficial effect in the management of dyslipidaemia [1, 15, 16].
Ketoconazole requires acidity for dissolution and absorption [9]. Following oral administration, peak plasma concentrations are reached within 1–2 h. Drug exposure increases more than dose proportionally over the therapeutic dose range. Ketoconazole undergoes extensive metabolism to a large number of inactive metabolites. Plasma elimination occurs in a biphasic manner, with the drug having a half-life of 2 h during the first 10 h after administration and 8 h thereafter, although the half-life increases with dose and duration of treatment. Most excretion occurs through the faeces via the bile [9]. In conclusion, these pharmacokinetic data explain the rapid onset of action of ketoconazole and the need to administer the daily dosage in two ot three divided doses.
How should ketoconazole be used in Cushing’s syndrome?
Ketoconazole tablets are indicated in the EU for the treatment of endogenous Cushing’s syndrome in adults and adolescents > 12 years of age (Table 1) [9]. The ketoconazole daily dosage should be periodically adjusted on an individual basis with the aim to normalise UFC and/or plasma cortisol levels. Thus, treatment with ketoconazole should be supervised by an endocrinologist or internist having the appropriate facilities for the monitoring of biochemical responses [9].
The recommended ketoconazole dosage at initiation is 400–600 mg/day taken orally in two or three divided doses [9]. During the initial treatment period, cortisol levels should be monitored every few days or weeks. If UFC and/or plasma cortisol levels remain above normal, a dosage increase of 200 mg/day every 7–28 days (up-titration) can be considered if tolerated, up to a maximum of 1200 mg/day in two or three divided doses [9]. Most typically, a maintenance dosage of 600–800 mg/day may be required to restore normal cortisol levels [1, 9].
Once the effective maintenance dosage is established, monitoring of cortisol levels should be performed every 3–6 months [9]. The maintenance dosage can be continued as described above (block-only regimen); alternatively, the ketoconazole dosage can be further increased by 200 mg/day with the addition of concomitant corticosteroid replacement therapy (block-and-replace regimen) [9]. With a block-and-replace regimen, the aim is to completely suppress endogenous cortisol production, with compensation through replacement with exogenous glucocorticoids to avoid hypoadrenalism [17]. The choice between a block-only regimen or a block-and-replace regimen can be made based on clinician experience and preference. A block-and-replace regimen may be preferable if there is evidence of cyclical disease [6, 17]. Furthermore, in cases of severe hypercortisolism which are unlikely to respond to low dosages of the steroidogenesis inhibitor, a block-and-replace regimen can be used to safely control hypercortisolism more quickly through more rapid up-titration without the need for multiple interim biochemical checks [17]. Although a block-and-replace regimen can be used safely, care is required to ensure that incompletely treated and persistent hypercortisolism is not worsened by the unnecessary addition of exogenous glucocorticoids [17].
Treatment with ketoconazole can be stopped abruptly without a need for progressive dosage reduction, for example when a change in the therapeutic strategy (e.g. surgery) is desired [9]. Before initiating treatment with ketoconazole, liver function must be measured, with regular monitoring continuing during treatment (Table 1). Also, given the drug’s high potential for drug-drug interactions, including some that can result in potentially life-threatening adverse reactions (Table 2), before initiating ketoconazole treatment, local prescribing information should be consulted for details and recommendations, including further information on concomitant therapies that are contraindicated.
What is the efficacy of ketoconazole in Cushing’s syndrome?
Based on available data, oral ketoconazole is effective in the treatment of Cushing’s syndrome, having demonstrated good efficacy in controlling hypercortisolism as well as in improving common clinical and biochemical features of the disease [1, 7, 17]. Evidence for the efficacy of ketoconazole in the treatment of Cushing’s syndrome is primarily drawn from (non-controlled) retrospective studies of real-life data, with supplementary evidence from small single-arm prospective trials and individual case reports (see reference [1] for a full list of citations for individual studies and case reports). Published data are available for ~ 800 patients (from paediatric to elderly patients; ~ 75% female) who received treatment with ketoconazole, either as monotherapy or in combination with other drugs, for Cushing’s syndrome [1, 9]. Of these, ~ 79% had Cushing’s disease, ~ 16% had EAS and ~ 5% had ACTH-independent Cushing’s syndrome, based on patients for whom the subgroup could be identified from the publications. Approximately 200 of these patients were treated with ketoconazole for ≥ 6 months [1, 9].
Across the published data, ketoconazole was used in a range of clinical situations, including as a presurgery treatment, as a primary treatment because surgery was contraindicated, or as a secondary treatment after unsuccessful surgery or while waiting for radiotherapy to be effective [1, 7, 17]. Ketoconazole was generally used at 200–1200 mg/day, with the dosage individualised based on patient response and tolerability. The average maintenance dosage used was 600–800 mg/day. The duration of ketoconazole treatment varied considerably (from days, to 13 years), largely reflecting the different clinical situations in which the drug was used [1]. In most studies, the key efficacy outcome measure was the change in urinary free cortisol (UFC) levels.
Summaries of the key findings from the main studies demonstrating the efficacy of ketoconazole in the treatment of Cushing’s syndrome are presented in Tables 3 and 4. Overall, ~ 60% of patients with Cushing’s syndrome treated with ketoconazole monotherapy achieve normalisation of UFC levels [17], with the percentages of patients who achieve control ranging from 45% to 88% across the key studies (Tables 3 and 4). Among the remaining patients, a good proportion have a marked reduction in UFC levels without achieving normalisation. Reductions in cortisol levels in patients treated with ketoconazole are also commonly associated with improvements in clinical and biochemical features of Cushing’s syndrome and common comorbidities [13, 18,19,20,21,22,23]. For example, in one key study [22], improvements were reported in hypertension [in 33/41 (80%) of affected patients], diabetes [8/11 (73%)], myopathy [20/27 (74%)], menstrual disturbances [16/21 (76%)], overweight/obesity [23/45 (51%)], lower limb oedema [10/20 (50%)], psychiatric symptoms and hirsutism (patient numbers not reported). Ketoconazole treatment is effective across the different forms of Cushing’s syndrome (i.e. Cushing’s disease, EAS and ACTH-independent Cushing’s syndrome) [Tables 3 and 4]. The proportions of patients with reductions in UFC levels appears to be somewhat lower among patients with EAS (~ 40–50%) than in the overall population [1, 6]; however, this is not surprising given that patients with EAS commonly have very severe hypercortisolism (and thus may be candidates for combination therapy - see below) [24]. In patients with adrenal carcinomas (where ketoconazole is most typically used in preparation for adrenalectomy), the response to ketoconazole treatment in terms of UFC levels also appears to be more variable (albeit based on lower patient numbers); however, improvement in clinical symptoms is usually achieved [1].
In ~ 16% of the 800 patients who were treated with ketoconazole for Cushing’s syndrome described in the literature ketoconazole was used in combination with (one or two) other drugs (e.g. metyrapone, cabergoline, pasireotide, mitotane) [1]. In particular, combination therapy was used to treat patients with persistent or severe hypercortisolism [24,25,26,27], and ketoconazole demonstrated efficacy in this setting (Table 3). The average ketoconazole dosage when used in combination with other drugs (~ 400 mg/day) was lower than when the drug was used as monotherapy, although, again, ketoconazole posology was individualised based on response and tolerability [1].
Although based on a small number of patients (< 20 for whom the age could be identified in publications), ketoconazole has demonstrated efficacy in the treatment of adolescent patients (aged 12–17 years) [1, 9]. In addition to controlling UFC levels, treatment with ketoconazole in these patients can produce clinical improvements, including the restoration of normal growth and gonadal function. Dosages of ketoconazole used in adolescent patients were generally similar to those used in the overall population. The number of patients aged < 12 years treated with ketoconazole for Cushing’s syndrome is too small for any determination of efficacy, and the drug is not approved for use in this age group [1, 9].
The largest individual study on ketoconazole in Cushing’s syndrome involved 200 patients (44 males, 136 females; mean age 42 years, range 8–87 years) who were treated with ketoconazole as a single agent for active Cushing’s disease across 14 centres in France between 1995 and 2012 (FReSKO study) [20]. In the study, 40 patients received ketoconazole as a presurgery treatment, 32 received ketoconazole as a primary treatment because surgery was contraindicated or refused, and 128 received ketoconazole as a secondary treatment, either after unsuccessful surgery (n = 93) or while waiting for radiotherapy to be effective (n = 35). Among all patients, macro- and micro-adenomas were identified by pituitary MRI in 18 and 53% of patients, with a lack of obvious adenoma in 29% of patients [20].
In the study, approximately three quarters of all patients had control (49.2%) or partial control (25.9%) of hypercortisolism at last follow-up (Table 4) [20]. Furthermore, the effect of ketoconazole treatment on UFC levels was generally similar between the groups of patients who received treatment either presurgery or as a primary or secondary treatment (Table 4). Patients who received ketoconazole presurgery were treated for a mean of 4.05 months (range, 0.03–15 months), with mean UFC decreasing from 6.89 × the upper limit of normal (ULN) at the initial visit to 2.5 × ULN at the final visit (p < 0.05). Furthermore, clinical signs, hypertension, hypokalaemia and diabetes improved during ketoconazole treatment in 38.4–50.0% of affected patients (Table 4). Patients who received ketoconazole as a primary or secondary treatment were treated for a mean of 24.8 months (range, 0.2–135 months). Among these patients, mean UFC decreased during treatment from 3.3 × ULN at the initial visit to 1.8 × ULN at the final visit (p < 0.0001). Again, clinical signs, hypertension, hypokalaemia and diabetes improved during ketoconazole treatment in a good proportion of patients (Table 4). The antisecretory effect of ketoconazole used as a primary or secondary treatment was generally maintained in the long term [20]. Among 51 patients who were treated with ketoconazole for > 24 months (mean, 108.5 months; maximum, 135 months), UFC levels were normal in 33 patients (64.7%) and were decreased ≥ 50% without normalisation in 12 patients (23.5%); six patients (11.8%), treated for a mean of 64.3 months, had initial control but a final increase in UFC levels, indicating cortisol escape [20].
What is the safety and tolerability profile of ketoconazole tablets?
Oral ketoconazole is generally well tolerated when used in the treatment of patients with Cushing’s syndrome, based on available evidence [1]. Safety and tolerability data are available in published literature on ~ 800 patients with Cushing’s syndrome treated with ketoconazole, as well as from a long history of use of the drug as an antifungal agent (although use in this latter indication was generally at lower dosages and for a shorter duration than its use in Cushing’s syndrome) [1]. Overall, the most commonly reported adverse reactions in patients treated with ketoconazole (occurring in ≥ 1% of patients) are hepatic enzymes increased, nausea, vomiting, abdominal pain, diarrhoea, adrenal insufficiency, pruritus and rash [9]. In the FReSKO study, 41 (20.5%) of 200 patients with Cushing’s disease treated with ketoconazole had ketoconazole treatment withdrawn because of adverse effects [20].
The main safety concern associated with ketoconazole is a risk of hepatotoxicity [1, 9], with hepatotoxicity reported in ~15% of patients with Cushing’s syndrome treated with the drug [7]. However, serious hepatotoxicity is rare (incidence of ~ 1/2000 to 1/15,000) [29, 30]. The occurrence of hepatotoxicity in patients treated with ketoconazole is not fully predictable, and no clear dose relationship has been established [1, 31]. In the FReSKO study, increases in liver enzymes ≤ 5 × ULN were reported in ~ 13.5% of patients, with increases > 5 × ULN reported in ~ 2.5% of patients [20]. In all cases, hepatic enzyme levels returned to normal after ketoconazole dosage reduction or withdrawal (typically within 2–4 weeks). No cases of fatal hepatotoxicity were observed in the study [20]. The occurrence of hepatic enzyme elevations is generally observed within 6 months of ketoconazole initiation (most typically within 4 weeks), or around dosage increases, based on the findings of an observational, prospective French cohort study involving 108 patients with Cushing’s syndrome treated with ketoconazole [31]. Although later emergence has been reported, long term treatment with ketoconazole does not appear to increase the risk of hepatotoxicity [1, 31]. Increases in hepatic enzymes during ketoconazole treatment are generally mild to moderate and asymptomatic [31]; however, fatal hepatotoxicity related to ketoconazole use has been reported, particularly when ketoconazole is continued despite hepatic enzyme elevations [1, 9]. There is also some evidence that ketoconazole hepatotoxicity may occur more commonly in adolescents than in adults [1, 9].
As has been observed with other Cushing’s syndrome therapies, adrenal insufficiency can occur in patients treated with ketoconazole (reported in ~12% of patients [7]), related to the pharmacodynamic effects of the drug [1, 9]. In the FReSKO study, clinical and biological adrenal insufficiency was reported in 5.4% of patients (or 3.7% of patients when excluding those on a block-and-replace regimen) [20]. Adrenal insufficiency may occur through overtreatment with ketoconazole (and/or other steroidogenesis inhibitors), through insufficient glucocorticoid replacement therapy under a block-and-replace regimen, or due to a relative cortisol deficiency (as might occur in situations of stress, e.g. infection) [1, 9]. Also likely related to the pharmacodynamics effects of the drug, gynecomastia has also been observed in patients treated with ketoconazole [32, 33].
Gastrointestinal adverse events (e.g. nausea, vomiting, abdominal pain, diarrhoea) are also common amongst patients treated with ketoconazole [1, 9], occurring in ~13% of Cushing’s syndrome patients treated with the drug [7]. Most gastrointestinal events are mild and transient [1, 9].
Ketoconazole can also prolong the QT interval and increase the risk of torsade de pointes [1, 9]. Ketoconazole is contraindicated in patients with congenital or documented acquired QTc prolongation; in other patients, monitoring by ECG is required (Table 1). Furthermore, other medications that have QT-prolonging potential may be contraindicated during ketoconazole treatment (Table 2), with a key mechanism of the arrhythmogenic effects of ketoconazole being through drug-drug interactions (given its broad inhibition of CYP enzymes) that lead to an increase in the plasma concentrations of QT interval prolonging drugs [9]. There is also evidence that ketoconazole may prolong the QT interval through a mechanism involving a direct inhibition of current through the hERG potassium channel [34].
What is the current clinical position of ketoconazole in the management of Cushing’s syndrome?
Oral ketoconazole is a useful treatment option in the management of Cushing’s syndrome, based on available evidence. Although data from randomised controlled trials are lacking, evidence from more than three decades of use in clinical practice suggests that ketoconazole is safe and effective in the treatment of Cushing’s syndrome [1]. Approximately 60% of patients with Cushing’s syndrome who are treated with ketoconazole achieve control of hypercortisolism, with further patients achieving partial control. In addition to the effects on cortisol levels, ketoconazole treatment can produce improvements in clinical and biochemical features of Cushing’s syndrome. Interestingly, there is evidence that inter-individual variation in the responsiveness to steroidogenesis inhibition may (at least in part) be due to genetic differences in CYP enzymes, with a polymorphism in the CYP17A1 gene found to influence the therapeutic response in patients with Cushing’s syndrome treated with ketoconazole and/or metyrapone [35]. Control of hypercortisolism through ketoconazole treatment is usually maintained long term [1, 9]. Approximately 10–15% of patients treated with ketoconazole experience an increase in UFC levels after initially achieving control [1, 9, 20]; however, there is no consensus definition of “escape” in the current guidelines [6]. Hepatotoxicity, the main safety concern associated with ketoconazole, can be managed effectively with careful monitoring of hepatic enzymes, particularly around treatment initiation and any dosage increases. With adherence to recommended management practices, hepatic adverse events associated with ketoconazole use are generally mild and reversible; serious hepatotoxicity is rare (incidence of ~ 1/2000 to 1/15,000). One notable limitation of ketoconazole is the high potential for drug-drug interactions [1, 9].
Ketoconazole has been used effectively in a range of clinical situations in the treatment of Cushing’s syndrome, including as a presurgery treatment, as a primary treatment because surgery was contraindicated, or as a secondary treatment after unsuccessful surgery or while waiting for radiotherapy to be effective [1]. Although generally effective as monotherapy, ketoconazole can also be used as part of combination therapy, particularly for the control of severe hypercortisolism [1, 6, 24,25,26,27]. Aside from potentially benefiting from synergistic effects, use of different drugs in combination may permit lower dosages of individual drugs, which can be used to manage adverse events [1]. Current Endocrine Society and European Society for Endocrinology guidelines acknowledge that steroidogenesis inhibitors (including ketoconazole) play a role in the treatment of Cushing’s syndrome, and may be useful to control hypercortisolism in Cushing’s syndrome of all aetiologies [6]. In particular, the guidelines recommend steroidogenesis inhibitors for use as second-line treatment after pituitary surgery in Cushing’s disease (with or without radiotherapy/radiosurgery); as primary treatment in patients with occult or metastatic EAS; and as adjunctive treatment to reduce cortisol levels in adrenocortical carcinoma [6]. Given their rapid onset of action [6], another clinical situation where steroidogenesis inhibitors can be useful options is in the management of acute complications of hypercortisolism (e.g. psychosis) [4].
Change history
02 February 2021
A Correction to this paper has been published: https://doi.org/10.1007/s40267-021-00812-7
References
European Medicines Agency. Ketoconazole HRA: public accessment report. 2015. http://www.ema.europa.eu/en/documents/assessment-report/ketoconazole-hra-epar-public-assessment-report_en.pdf. Accessed 08 Oct 2020.
Loose DS, Kan PB, Hirst MA, et al. Ketoconazole blocks adrenal steroidogenesis by inhibiting cytochrome P450-dependent enzymes. J Clin Invest. 1983;71(5):1495–9.
Pont A, Williams PL, Loose DS, et al. Ketoconazole blocks adrenal steroid synthesis. Ann Intern Med. 1982;97(3):370–2.
Lacroix A, Feelders RA, Stratakis CA, et al. Cushing’s syndrome. Lancet. 2015;386(9996):913–27.
Chabre O, Cristante J. Treatment of Cushing’s syndrome: what place for medical treatment? Acta Endocrinol (Buchar). 2019;15(2):237–43.
Nieman LK, Biller BM, Findling JW, et al. Treatment of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2015;100(8):2807–31.
Pivonello R, De Leo M, Cozzolino A, et al. The treatment of Cushing’s disease. Endocr Rev. 2015;36(4):385–486.
European Medicines Agency. European Medicines Agency recommends suspension of marketing authorisations for oral ketoconazole [media release]. 2013. http://www.ema.europa.eu/en/news/european-medicines-agency-recommends-suspension-marketing-authorisations-oral-ketoconazole. Accessed 08 Oct 2020.
European Medicines Agency. Ketoconazole HRA: summary of product characteristics. 2020. http://www.ema.europa.eu/documents/product-information/ketoconazole-hra-epar-product-information_en.pdf. Accessed 08 Oct 2020.
Engelhardt D, Jacob K, Doerr HG. Different therapeutic efficacy of ketoconazole in patients with Cushing’s syndrome. Klin Wochenschr. 1989;67(4):241–7.
Engelhardt D, Weber MM, Miksch T, et al. The influence of ketoconazole on human adrenal steroidogenesis: incubation studies with tissue slices. Clin Endocrinol (Oxf). 1991;35(2):163–8.
Weber MM, Lang J, Abedinpour F, et al. Different inhibitory effect of etomidate and ketoconazole on the human adrenal steroid biosynthesis. Clin Investig. 1993;71(11):933–8.
Sonino N, Boscaro M, Paoletta A, et al. Ketoconazole treatment in Cushing’s syndrome: experience in 34 patients. Clin Endocrinol (Oxf). 1991;35(4):347–52.
Pont A, Williams PL, Azhar S, et al. Ketoconazole blocks testosterone synthesis. Arch Intern Med. 1982;142(12):2137–40.
Gylling H, Vanhanen H, Miettinen TA. Hypolipidemic effect and mechanism of ketoconazole without and with cholestyramine in familial hypercholesterolemia. Metabolism. 1991;40(1):35–41.
Gylling H, Vanhanen H, Miettinen TA. Effects of ketoconazole on cholesterol precursors and low density lipoprotein kinetics in hypercholesterolemia. J Lipid Res. 1993;34(1):59–67.
Daniel E, Newell-Price JD. Therapy of endocrine disease: steroidogenesis enzyme inhibitors in Cushing’s syndrome. Eur J Endocrinol. 2015;172(6):R263–80.
Gómez RM, Albiger NM, Diaz AG, et al. Effect of hypercortisolism control on high blood pressure in Cushing’s syndrome. Medicina (B Aires). 2007;67(5):439–44.
Fallo F, Paoletta A, Tona F, et al. Response of hypertension to conventional antihypertensive treatment and/or steroidogenesis inhibitors in Cushing’s syndrome. J Intern Med. 1993;234(6):595–8.
Castinetti F, Guignat L, Giraud P, et al. Ketoconazole in Cushing’s disease: is it worth a try? J Clin Endocrinol Metab. 2014;99(5):1623–30.
Castinetti F, Morange I, Jaquet P, et al. Ketoconazole revisited: a preoperative or postoperative treatment in Cushing’s disease. Eur J Endocrinol. 2008;158(1):91–9.
Moncet D, Morando DJ, Pitoia F, et al. Ketoconazole therapy: an efficacious alternative to achieve eucortisolism in patients with Cushing’s syndrome. Medicina (B Aires). 2007;67(1):26–31.
Valassi E, Crespo I, Gich I, et al. A reappraisal of the medical therapy with steroidogenesis inhibitors in Cushing’s syndrome. Clin Endocrinol (Oxf). 2012;77(5):735–42.
Corcuff J-B, Young J, Masquefa-Giraud P, et al. Rapid control of severe neoplastic hypercortisolism with metyrapone and ketoconazole. Eur J Endocrinol. 2015;172(4):473–81.
Feelders RA, de Bruin C, Pereira AM, et al. Pasireotide alone or with cabergoline and ketoconazole in Cushing’s disease. N Engl J Med. 2010;362(19):1846–8.
Kamenický P, Droumaguet C, Salenave S, et al. Mitotane, metyrapone, and ketoconazole combination therapy as an alternative to rescue adrenalectomy for severe ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 2011;96(9):2796–804.
Vilar L, Naves LA, Azevedo MF, et al. Effectiveness of cabergoline in monotherapy and combined with ketoconazole in the management of Cushing’s disease. Pituitary. 2010;13(2):123–9.
van den Bosch OF, Stades AM, Zelissen PM. Increased long-term remission after adequate medical cortisol suppression therapy as presurgical treatment in Cushing’s disease. Clin Endocrinol (Oxf). 2014;80(2):184–90.
Lewis JH, Zimmerman HJ, Benson GD, et al. Hepatic injury associated with ketoconazole therapy: analysis of 33 cases. Gastroenterology. 1984;86(3):503–13.
Lo Re V, 3rd, Carbonari DM, Lewis JD, et al. Oral azole antifungal medications and risk of acute liver injury, overall and by chronic liver disease status. Am J Med. 2016;129(3):283-91 e5.
Young J, Bertherat J, Vantyghem MC, et al. Hepatic safety of ketoconazole in Cushing’s syndrome: results of a Compassionate Use Programme in France. Eur J Endocrinol. 2018;178(5):447–58.
Chou SC, Lin JD. Long-term effects of ketoconazole in the treatment of residual or recurrent Cushing’s disease. Endocr J. 2000;47(4):401–6.
DeFelice R, Johnson DG, Galgiani JN. Gynecomastia with ketoconazole. Antimicrob Agents Chemother. 1981;19(6):1073–4.
Takemasa H, Nagatomo T, Abe H, et al. Coexistence of hERG current block and disruption of protein trafficking in ketoconazole-induced long QT syndrome. Br J Pharmacol. 2008;153(3):439–47.
Valassi E, Aulinas A, Glad CA, et al. A polymorphism in the CYP17A1 gene influences the therapeutic response to steroidogenesis inhibitors in Cushing’s syndrome. Clin Endocrinol (Oxf). 2017;87(5):433–9.
Acknowledgements
The manuscript was reviewed by: M. O. Savage, Centre for Endocrinology, William Harvey Research Institute, Queen Mary University of London, London, UK; S. M. Webb, Department of Endocrinology/Medicine, Hospital Sant Pau, Universitat Autònoma de Barcelona, CIBERER group747, Barcelona, Spain. During the peer review process, HRA Pharma, the marketing-authorisation holder of ketoconazole HRA™, was also offered an opportunity to provide a scientific accuracy review of their data. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of interest
M. Shirley is an employee of Adis International Ltd./Springer Nature and declares no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent for publication, Availability of data and material, Code availability
Not applicable.
Additional information
The original version of this article was revised due to a retrospective Open Access order.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Comm ons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Shirley, M. Ketoconazole in Cushing’s syndrome: a profile of its use. Drugs Ther Perspect 37, 55–64 (2021). https://doi.org/10.1007/s40267-020-00799-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40267-020-00799-7