
Abstract
Fabry disease (FD) is a rare X-linked lysosomal storage disorder caused by mutations in the α-galactosidase A (AGAL/GLA) gene. The lysosomal accumulation of the substrates globotriaosylceramide (Gb3) and globotriaosylsphingosine (lyso-Gb3) results in progressive renal failure, cardiomyopathy associated with cardiac arrhythmia, and recurrent strokes, significantly limiting life expectancy in affected patients. Current treatment options for FD include recombinant enzyme-replacement therapies (ERTs) with intravenous agalsidase-α (0.2 mg/kg body weight) or agalsidase-β (1 mg/kg body weight) every 2 weeks, facilitating cellular Gb3 clearance and an overall improvement of disease burden. However, ERT can lead to infusion-associated reactions, as well as the formation of neutralizing anti-drug antibodies (ADAs) in ERT-treated males, leading to an attenuation of therapy efficacy and thus disease progression. In this narrative review, we provide a brief overview of the clinical picture of FD and diagnostic confirmation. The focus is on the biochemical and clinical significance of neutralizing ADAs as a humoral response to ERT. In addition, we provide an overview of different methods for ADA measurement and characterization, as well as potential therapeutic approaches to prevent or eliminate ADAs in affected patients, which is representative for other ERT-treated lysosomal storage diseases.
Similar content being viewed by others


Classical male patients with Fabry disease (FD) have a high risk for a humoral response including anti-drug antibodies (ADAs) against enzyme-replacement therapy (ERT). |
The formation of ADAs is associated with worse clinical outcomes. |
ADAs already inhibit infused enzyme during infusions and impair cellular enzyme uptake. |
ADAs can be saturated by increased dosages of infused enzymes or suppressed by appropriate immune-modulatory approaches |
1 Introduction
Fabry disease (FD) is a rare X-linked inherited lysosomal storage disease (LSD) due to a deficiency in α-galactosidase A (AGAL) activity [1]. Loss of enzyme function causes a progressive, sometimes life-threatening, multisystem disease due to intracellular accumulation of glycosphingolipids such as globotriaosylceramide (Gb3) [1]. Treatment options for FD have included enzyme-replacement therapy (ERT) since 2001 [2, 3]. Although treatment with the two currently available recombinant enzymes (agalsidase-alfa and agalsidase-beta) generally improves disease load, many male patients are at high risk for the formation of neutralizing anti-drug antibodies (ADAs), which significantly reduce the efficacy of ERT [4,5,6,7]. Since ERT needs to be prescribed life-long with mean annual costs of ~ 250.000€ per patient, ADAs increase the economic burden in addition to individual disease load.
In this narrative review, we provide a brief overview of the clinical picture of FD and diagnostic confirmation. The focus is on the biochemical and clinical significance of neutralizing ADAs as a humoral response to ERT. In addition, we provide an overview of different methods for ADA measurement and characterization, as well as potential therapeutic approaches to prevent or eliminate ADAs in affected patients. Since a humoral response to ERT generally follows the same immunological mechanisms, this review can be applied to other ERT-treated LSDs such as Gaucher disease, Mucopolysaccharidosis, Pompe disease, etc.
1.1 Clinical Picture and Diagnostic Confirmation of Fabry Disease (FB)
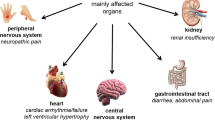
Affected patients with FD suffer from a multisystemic disease that includes progressive renal failure (requiring dialysis or transplantation), cardiomyopathy with sometimes life-threatening cardiac arrhythmias, recurrent transient ischemic attacks and strokes, gastrointestinal pain, and neuropathic pain of the extremities [1, 8]. Due to the random inactivation of one of the two X chromosomes, the clinical phenotype is more variable in affected females than in males [9].
FD is suspected when Fabry-typical symptoms or manifestations are detected and/or a positive family history is present. To confirm the diagnosis in men, determination of AGAL activity in leukocytes or from dried blood spots is the gold standard. A pathologically low AGAL activity confirms the presence of FD. In contrast, in women, a molecular genetic test detecting a disease-causing mutation in the GLA gene is required for confirmation, as women often have AGAL activities in blood that are within the reference range. Nowadays, men with pathologically decreased AGAL enzymatic activity should also undergo molecular genetic testing to determine the disease-causing mutation in order to select an appropriate FD-specific therapy, such as chaperone therapy [10]. As a biomarker for disease burden, pathologically elevated globotriaosylsphingosine (lyso-Gb3), the deacylated form of Gb3, can be measured in plasma or urine and contributes to diagnosis and therapy monitoring [11,12,13,14,15,16,17].
In case of unclear diagnostics (i.e., ambiguous AGAL activity, disputed controversial mutation, lyso-Gb3 levels and/or comorbidities), tissue biopsies can be performed to detect multilamellar myelin bodies (“zebra bodies”) by electron microscopy, which are pathognomonic for FD, but require special sample preparation.
2 Enzyme-Replacement Therapy (ERT)
2.1 Therapy Goals and Therapy Initiation
Once the FD diagnosis is confirmed, it is advisable to refer patients to an interdisciplinary Fabry center for initial evaluation and, if indicated, initiation of therapy. According to current expert opinion [18, 19], the following therapeutic goals should be aimed for: (1) a reduction of FD-specific complaints including pain reduction, (2) a prevention and/or delay of progressive organ manifestations with special focus on the kidney, heart, and central nervous system, (3) an improvement of quality of life, and (4) the normalization of life expectancy. Since male FD patients usually show a worse disease progression than females, the recommendations for the initiation of a FD-specific therapy in adult patients with a classic or milder late-onset clinical phenotype include a personalized treatment approach, taking into account the natural history of the specific disease phenotype [19]. In adult males with classic GLA mutations, FD-specific treatment (either ERT or migalastat) should be considered independent of the patient’s symptoms or manifestations. Treatment decisions may also be influenced by advanced age and additional comorbidities. In females with a classic mutation, treatment initiation is appropriate if patients are symptomatic with FD-typical organ involvement. In asymptomatic females with classic GLA mutations, treatment initiation needs to be considered in the presence of renal, cardiac, or central nervous system damage. In patients (females and males) with late-onset mutations (i.e., p.N215S), treatment should be considered in the presence of renal, cardiac, or central nervous system damage, even in the absence of other FD-typical symptoms (such as angiokeratoma, Cornea verticillata, etc.). However, symptoms should be caused by FD, requiring histologic evaluation (biopsies) or biochemical evidence of cellular Gb3 accumulation.
Patients with "benign," non-disease-causing GLA variants/polymorphisms should not be treated with any FD-specific therapy. Since the therapeutic goals are not always achievable, current consensus recommendations [18, 19] provide some guidance to establish appropriate goals between clinicians and FD patients. However, with respect to irreversible end-organ damage, early FD-specific therapy in combination with adjunctive therapy is advisable to delay or slow disease progression.
2.2 ERT—Two Approved Active Compounds
ERT as the first causal treatment for FD was approved in 2001 [2, 3]. Currently, two preparations are approved in Europe, which are administered intravenously every 2 weeks. Agalsidase-alfa (Replagal, Takeda/Shire) is produced in human fibrosarcoma cells HT-1080 with a recommended dosage of 0.2 mg/kg body weight. Agalsidase-beta (Fabrazyme, Sanofi Genzyme) is produced in Chinese hamster ovary (CHO) cells with a recommended dose of 1.0 mg/kg body weight. Over the past 20 years, ERT has shown good efficacy by stabilizing or even improving disease burden (reviewed by Lenders and Brand [20]), with beneficial effects especially regarding quality of life and reducing life-threatening events [21,22,23]. In short, with respect to the individual patients and their manifestations, the following therapeutic effects can be achieved by ERT: stabilization of kidney function (i.e., estimated glomerular filtration rate (eGFR), proteinuria) or delay of progression to terminal kidney failure, stabilization of cardiac mass and function and reduction of left ventricular hypertrophy, amelioration of FD-specific pain and gastrointestinal symptoms, and amelioration of hypohidrosis [20, 24,25,26,27].
In addition to ERT, concomitant nephroprotective and cardioprotective medication (angiotensin-converting enzyme or angiotensin II receptor blockers) is highly recommended to preserve renal [28] as well as cardiac [29] function. However, recent reviews and meta-analyses reported only a limited beneficial effect of ERT on patient outcome [30, 31]. An important mechanism responsible for the limited efficacy of therapy is the formation of neutralizing anti-drug antibodies (ADAs) to the infused enzymes, which are the focus of this review.
2.3 Humoral Response to ERT
ERT is based on the intravenous infusion of a (mostly) foreign recombinant protein [2, 3, 32]. Consequently, an infused enzyme might be recognized as foreign, and triggers an appropriate immune response, leading to the formation of specific antibodies against the enzyme [32]. Several studies in patients with different LSDs have shown that patients without any endogenous enzyme /protein, which is termed "cross-reactive immunologic material" (CRIM) negative, are at high risk of developing an immune response after ERT initiation [4, 5, 33, 34]. Due to the different effects, it is most important to distinguish between two different responses caused by continuous exposure to ERT.
2.4 Infusion-Associated Reactions
Infusion-associated reactions (IARs) most frequently occur in ERT-naïve FD patients directly after ERT initiation, and affected patients are mainly CRIM-negative males having zero endogenous AGAL activity [2, 3, 35, 36]. Symptoms are generally limited to fever and/or chills and require premedication with anti-histamines and steroids and a decreased infusion rate [36]. In addition to milder symptoms such as urticarial skin lesions, facial flushing or swelling, headache, and pain in the limbs, more severe IARs have been reported with chest and throat tightness, dyspnea, hypotension, and life-threatening anaphylactic reactions [37,38,39,40]. According to the manufacturer’s instructions, 24% of patients treated with agalsidase-alfa and 67% of patients treated with agalsidase-beta can suffer from these adverse effects. According to the literature, IARs are mainly the result of anaphylactoid (IgE-independent) rather than anaphylactic (IgE-mediated type 1 hypersensitivity) reactions, since only few affected patients were positive for IgEs against recombinant AGAL [21, 39, 41,42,43,44]. The risk for IARs seems to be increased in patients with anti-agalsidase IgG antibodies [5, 36]. The IgE-independent mechanisms resulting in hypersensitivity against the infused enzyme are not yet fully understood, but changes in osmotic pressure during infusion might lead to mast cell destabilization and histamine release. In addition, an abnormal activation of the complement system can result in a type II hypersensitivity. However, a recent study identified only 25% of patients with IARs with reduced C4 levels, suggesting that the primary cause for IARs is not complement-mediated cytotoxic hypersensitivity reactions [39]. In this study the authors further demonstrated that circulating dendritic cells were drastically reduced during IARs, suggesting their possible sequestration to the sites of inflammation [39]. An increase in natural killer cells and a decrease in T cells was also observed [39]. In addition, cytokines IL-4, IL-8, and TNF-α showed a significant increase, indicating nonspecific degranulation of mast cells, indicating a crosstalk between immune cells resulting in IgE-independent mast-cell-specific allergic inflammation [39].
In addition to the general recommendations for preventing IARs [36], more specialized protocols for managing severe reactions were established in the last decade. In patients with IgEs against agalsidase-beta, a graded dosing and individual infusion-rate regimen for a duration of 27 infusions resulted in successful reinstitution of agalsidase-beta in five of six patients [42]. A recent study demonstrated the tolerization of agalsidase-beta in eight patients (only one positive for IgEs and six patients with IgGs against AGAL) using a combination of premedications that included corticosteroids, mast cell stabilizers, H1 and H2 blockers, and intravenous (IV) fluids [39]. Aydin and colleagues used an adapted three-bag protocol [45], which was based on a consecutive 1:10 dilution of 14 mg agalsidase-alfa solved in 250 mL normal saline solution (solution 3: 14 mg agalsidase-alfa solved in 250 mL of saline solution; solution 2: 225 mL saline solution + 25 mL solution 3; solution 1: 225 mL saline solution + 25 mL solution 2) to successfully reinstitute agalsidase-alfa infusion in two brothers negative for IgEs [38]. This protocol starts with 1/40,000 of the therapeutic dose and increases dose over 12 steps, resulting in an infusion time of approximately 504 min [38]. However, a recent case study demonstrated that a three-bag protocol alone or in combination with pretreatment including glucocorticoids, H1/H2 antagonists, leukotriene receptor antagonists, and even B-cell depletion via rituximab was not able to reinstitute agalsidase-beta in a patient with IgEs against AGAL, but required a specific anti-IgE inhibition by omalizumab [40].
2.5 Clinical Impact of Neutralizing Anti-Drug Antibodies (ADAs)
Anti-drug AGAL IgG antibodies develop within 3–6 months after ERT initiation mainly in male CRIM-negative patients [4, 36, 41], which is the expected timeframe for an appropriate immune response with IgGs. In a recent prospective study, a significant increased risk for the formation of neutralizing ADA in male patients with FD was demonstrated if patients were treated with agalsidase-beta (1.0 mg/kg every other week) compared to agalsidase-alfa (0.2 mg/kg every other week) [46], underlining the importance of dosage for ADA formation. In detail, Arends and colleagues stated that risk calculations for ADAs were performed in classical males only and that antibody measurements were available for 92 classical patients, of whom 11 were excluded due to mixed antibody responses [46]. Of the remaining 81 classical patients, 11 of 39 agalsidase-alfa-treated patients (28%) and 22 of 42 agalsidase-beta-treated patients (52%) were positive for ADAs, resulting in an increased risk of 2.8 (95% confidence interval (CI) 1.02–7.88; p = 0.041) for agalsidase-beta [46]. However, although the authors only included classical male patients in their risk assessments, there is a lack of information on the type of mutations (nonsense, missense, etc.) and CRIM status in these cohorts, which represents a limitation of this study [46].
Furthermore, by using multivariate regression models, van der Veen and colleagues found that nonsense and frameshift mutations in the GLA gene, higher plasma lyso-Gb3 at baseline and agalsidase-beta as first treatment were significantly associated with ADA development [47]. However, the authors failed to reproduce the risk associated with the type of enzyme when they tried to validate the risk score in a second cohort and only the type of mutation remained significant [47]. To analyze if one of the two compounds is associated with a higher risk for ADA formation, more appropriate studies with comparable and larger patient cohorts in addition to a clearly defined mutation status (nonsense, missense, etc.) and CRIM status are warranted. In this respect, this highlights the general limitations of current studies in FD, analyzing an impact of different ERT dosages by comparing heterogeneous populations with different severities of GLA mutations, age at treatment initiation, comorbidities, etc.
Early studies demonstrated an inhibitory function of IgGs against infused AGAL, as well as a high cross-reactivity between agalsidase-alfa and agalsidase-beta [4]. The formation of inhibitory ADAs is generally assumed to be irreversible [48], although appropriate long-term studies are still required. The clinical impact of ADAs has been controversially discussed over the past years. High ADA titers (measured by ELISA) were associated with increased Gb3 accumulations in endothelial cells [49], but had an unclear effect on Gb3 levels in plasma [21, 50, 51]. However, on the basis of previously established serum-mediated inhibition assays [4], Rombach and colleagues demonstrated that patients with inhibitory antibodies suffered from higher (lyso)-Gb3 levels in plasma [48]. During follow-up, elevated lyso-Gb3 levels and increased disease burden (measured by patients' disease scores) were confirmed in patients with neutralizing ADAs [5]. Importantly, it was demonstrated that affected patients suffered from a more prominent loss of renal function and increasing septum thickness over time compared to those without ADAs [5]. The clinical outcome under therapy is influenced on the one hand by neutralizing ADAs and certainly by the severity of the disease burden before therapy initiation. To better differentiate the impact, the study populations being compared should have a comparable disease burden before therapy initiation and comparable types of mutations. However, recent case studies confirmed the deleterious effects of ADAs by showing massive Gb3 accumulation in various tissues in an affected male patient even after 6 years of ERT [52] and increasing lyso-Gb3 levels and lack of improvement in symptoms such as abdominal pain and acroparesthesias in two men after 3 years of ERT with high antibody titers [53].
One reason that may explain the different results of the various studies is the combined clinical relevance resulting from the level of ADA titers and the infused dose of the enzyme. A subgroup analysis of patients with neutralizing ADAs revealed a better biochemical response to aga lsidase-beta at 1.0 mg/kg in terms of decreasing lyso-Gb3 levels over time compared to agalsidase-alfa at 0.2 mg/kg [46], suggesting a link between antibody titer and dosages. These data were supported by a recent multicenter study showing a steeper decrease of lyso-Gb3 levels in plasma when patients were switched (back) from agalsidase-alfa (0.2 mg/kg) to agalsidase-beta (1.0 mg/kg) [54]. By measuring the amount of free (unbound) ADAs, it was demonstrated that titers can be saturated during infusions [55] and that a saturation was associated with better clinical outcomes [6]. A schematic model of the interaction between free antibodies and infused enzyme and the concept of ADA saturation is presented in Fig. 1.

Schematic model of anti-drug antibody (ADA) saturation with an interaction between free antibodies and infused enzyme. A Patients with saturated antibodies: Due to a low ADA titer or sufficient dosage of infused enzyme (α-galactosidase A excess), only a proportion of infused enzyme will be recognized and neutralized by free ADAs. B Patients with non-saturated antibodies: due to high ADA titers or insufficient dosage of infused enzyme (ADA excess), the majority of infused enzyme will be recognized and neutralized by free ADAs. AGAL α-galactosidase A, e.o.w. every other week
Early studies showed that IgGs against AGAL from FD patients have an inhibitory function [4], whereas some subsequent studies distinguished between inhibitory and non-inhibitory ADAs. Since individually formed ADAs are polyclonal, it seems unlikely that a patient’s ADAs recognize only non-important epitopes. Rather, it suggests that patients with positive IgGs without measurable inhibition are likely to have a generally low ADA titer.
2.6 Patients at Risk
In general, there are several patient- and product-specific factors that need to be considered in the assessment of the immunogenic risk for patients treated with a recombinant enzyme. Patient-specific factors include the route of therapeutic administration, with the highest risk associated with inhalation. The risk decreases in the following order: subcutaneous, intraperitoneal, intramuscular, and intravenous administration [33, 56]. In FD, the two currently approved products agalsidase-alfa and agalsidase-beta are intravenously infused, and are associated with the lowest risk of triggering an immune response [2, 3]. An additional factor is the dosage, since higher dosages more likely increase the risk [33, 56]. The frequency and duration, repeated dosages, and prolonged exposure may either break or lead to tolerance [33, 56]. Both compounds have relative short plasma half-lives and proteins are hardly detectable in plasma before the next infusion. One of the most important factors is the CRIM status, defined by the presence or absence of endogenous AGAL. CRIM-negative patients without endogenous enzyme have the highest risk of producing antibodies under therapy [33, 56]. Since FD is an X-chromosomal-linked disease, only hemizygous males or homozygous females (very rare) can be CRIM negative. Thus, classical male FD patients with nonsense or null mutations, which result in the absence of any detectable AGAL (functional or non-functional) are at a high risk for the formation of ADAs [4, 5, 46]. The absence of AGAL activity does not necessarily predict the absence of AGAL protein, since even missense mutations can result in a complete loss of function. Thus, quantitative methods are needed and are generally based on western blotting as suggested for Pompe disease [57], but require a polyclonal anti-AGAL reference antibody, recognizing a broad spectrum of epitopes in order to detect even small AGAL protein fragments (due to truncations). Product-specific factors can include host cell protein contaminants or non-human post-translational and artificial chemical modifications of the protein [33, 56]. In this respect, one of the first studies dealing with ADAs in FD suggested that there seems to be a comparable risk for ADAs if patients are treated with similar dosages of agalsidase-alfa or agalsidase-beta (both 0.2 mg/kg) [4]. This might also lead to the assumption that product-specific factors can likely be excluded. Although only five patients were included in each group, the severity of mutations was comparable [4]. Thus, it can be assumed that no differences in antibody production can be observed if males with comparable severity of mutations are analyzed.
2.7 Biochemical Impact of Neutralizing ADAs
Several studies have demonstrated that inhibitory ADAs formed against infused AGAL belong to the subclasses IgG1and IgG4[55, 58, 59]. Depending on the recognized epitopes, ADAs seem to have an impact on several different mechanisms (Fig. 2). In general, uptake of infused AGAL (agalsidase-alfa or agalsidase-beta) is cation-independent mannose-6-phosphate (M6P) receptor-mediated, resulting in increased lysosomal AGAL activity and subsequent Gb3 depletion (Fig. 2a). In addition, infused AGAL can deplete Gb3 and lyso-Gb3 directly within the plasma. In the presence of neutralizing ADAs, infused AGAL is recognized by ADAs, resulting in different effects. ADAs may recognize epitopes localized at uptake-relevant domains including amino acid positions N139, N192, and N215. Binding of ADAs may result in a masking of these positions, which are subsequently not accessible to the M6P receptor, resulting in a decreased enzymatic uptake followed by a reduced Gb3 depletion [7] (Fig. 2b). ADAs may also bind to a processing site and prevent maturation (Fig. 2c), an effect that has not been analyzed in FD so far (to the best of our knowledge). To date the most characterized is the enzymatic activity neutralizing function of ADAs during infusion [4,5,6, 48, 55], probably mediated by the recognition of epitopes within catalytic domains. First, activity-neutralized AGAL will not be able to reduce Gb3 and lyso-Gb3 in plasma. Second, even if these inactive ADA/AGAL complexes are internalized and translocated to the lysosomes, complexes seem to be unable to dissociate [7] (Fig. 2d). ADAs that do not have a direct effect of AGAL activity or uptake, can be termed “non-neutralizing” (Fig. 2e). However, these non-neutralizing ADAs may also affect pharmacokinetics, trafficking, and conformation, which is also currently yet insufficiently analyzed in FD. Finally, Fcγ-receptor-expressing cells, such as macrophages, recognize drug/ADA complexes via the Fc- fragment and lead to an increased drug clearance [4] (Fig. 2f). Although not directly demonstrated in FD, ADAs might also form large ADA-protein immune complexes, which are either insoluble or can mediate complement activation (planted antigens) and result in membranous nephritis as reported for Pompe disease [60,61,62] (Fig. 2g). Since ADAs in affected patients with FD are polyclonal and thus recognize different epitopes [7, 63], it can be assumed that in most individuals a combination of the mechanisms leads to decreased therapeutic efficacy.

Potential impact of anti-drug antibodies (ADAs) on infused α-galactosidase A (AGAL). a In the absence of ADAs, infused AGAL is internalized via the M6P-receptor, resulting in increased lysosomal AGAL activity and increased Gb3 clearance. b Binding of ADAs to the uptake domain (i.e., masking of M6P residues) can prevent AGAL from uptake via the M6P-receptor. c Binding of ADAs to processing domains may result in regular M6P-receptor-mediated uptake, but might lead to inefficiently processed AGAL and reduced intralysosomal enzymatic activity and reduced Gb3 clearance. d Binding of ADAs to catalytic domains can result in unimpaired enzyme uptake, but reduced lysosomal enzymatic activity and impaired Gb3 depletion, due to a missing dissociation. e ADA-binding to non-relevant domains might have no further impact on AGAL uptake and thus lysosomal activity. f Independent of recognized epitopes, ADA/AGAL complexes can be recognized by macrophages via the Fcγ receptor and thus eliminated during infusions. g Multivalent ADAs might cross-link AGAL molecules, resulting in large ADA/AGAL complexes with unknown effects in Fabry disease
3 Methods for Determination of ADAs
The controversial results of ADAs on treatment efficacy may be explained by the fact that no standardized assays and protocols have yet been developed to determine IgG levels in affected FD patients. Most studies analyzing IgG response are performed either by ERT manufacturers or different independent research laboratories using their own protocols and different methods [64]. Thus, titers from different studies are hardly comparable. Another pitfall in many studies is the lack of a corresponding methods section describing the protocols used. Some studies do not even state whether antibodies were measured by ELISA or not. In the following, we provide a brief overview of the most commonly used methods at present.
As early as 2001, ELISA-based measurements demonstrated the early formation of ADA s against infused agalsidase-beta within the first 6 months after therapy initiation [3]. Due to the lack of serial dilutions, ADA titers could be detected but not quantified in patients [3]. Using serial dilutions of sera from FD patients, Linthorst and colleagues provided relative ADA titers and demonstrated cross-reactivity of ADAs with respect to agalsidase-alfa and agalsidase-beta [4]. In general, relative titers can be expressed by using sera from healthy controls, from patients who ultimately did not develop ADAs, or from patients who are ERT-naïve [2, 4, 49, 65]. Some other studies used animal-derived anti-AGAL antibodies as positive controls [59]. However, due to the different species, different secondary antibodies are required for final detection, which do not allow for proper quantification. In addition, animal-derived polyclonal antibodies may differ in their individual binding affinity and epitope recognition compared to human antibodies, and are not recommended as proper controls for the quantification of human ADAs. Thus, the use of a human reference antibody with a known concentration and comparable biochemical characteristics for the quantification of ADA titers from human samples is strongly advised (Fig. 3A). To overcome the limitations, van der Veen and colleagues pooled sera from FD patients with ADAs to generate a positive control, allowing ADA measurements with the same secondary detection antibodies [58]. However, due to the unknown antibody concentration within the positive control, even this approach did not allow a quantification of measured ADA titers. Recently, Lenders and colleagues generated a polyclonal reference antibody by pooling purified and characterized anti-AGAL antibodies from 22 patients, allowing an ELISA-based measurement and ADA titer quantification expressed as ng/mL serum [66]. The use of ELISA-based assays also allows determination of and distinguishing between appropriate IgG isotypes.

Methods for the detection and characterization of α-galactosidase A (AGAL)-specific anti-drug antibodies (ADAs). A ELISA-based quantification of ADAs: AGAL-specific IgGs of serial dilutions from blood samples bind to immobilized AGAL and are detected with enzyme-labeled secondary antibodies. A serial dilution of a proper reference antibody allows a subsequent quantification. B Inhibition assay: AGAL hydrolyzes the artificial substrate 4-MU-α-Gal to fluorescent 4-MU if not blocked by inhibitory ADAs. A titration of individual free IgGs determines the amount of enzyme-replacement therapy (ERT) required to saturate ADAs during infusion. C Cellular uptake assays: Fluorescence-labeled AGAL is pre-incubated with the patient’s IgGs and added to appropriate cells. Subsequent assays can analyze whether ADA/AGA L complexes are formed (1), inhibit intracellular enzymatic activity (2) and/ or uptake (3). (i) Intracellular fluorescence signal from labeled AGAL can be quantified by appropriate techniques. Also, indirect AGAL or ADA detection can be performed using appropriate primary and secondary antibodies. (ii) Cells can be lysed to determine intracellular AGAL activity after uptake. (iii) Cell lysates can also be used for ELISA-based detection of AGAL uptake (free or as ADA/AGAL complexes). The use of appropriate primary detection antibodies directly allows the determination of AGAL/ADA complex-forming IgG subclasses
Linthorst and colleagues also reported an easily measurable neutralizing activity of ADAs in vitro [4]. This assay is based on the hydrolysis of the artificial AGAL substrate 4-methylumbelliferyl-α-D-galactopyranoside (4-MU-α-Gal) to 4-methyl-umbelliferone (4-MU) (Fig. 3B). In short, a fixed amount of AGAL will be pre-incubated with sera of interest and subsequently the residual enzymatic activity will be measured and compared to a negative and a positive control. Thus, an inhibition assay is a functional assay, detects activity-neutralizing ADAs, and can classify patients as “neutralization-negative” or “-positive” [4]. However, probably due to the proteolytic effects of the serum in combination with a basic to neutral pH, the standard serum-mediated inhibition assays have a relatively high background, resulting in decreased sensitivity [55]. To improve sensitivity, Lenders and colleagues separate whole IgG fractions from raw sera, allowing the authors to improve sensitivity and to estimate the required amount of infused AGAL to overcome and saturate existing free and unbound ADAs during infusion [6, 55] (Fig. 3B).
Cell-based antibody assays are mainly important to further analyze the functionality of ADAs (Fig. 3C). In their initial study, Linthorst and colleagues demonstrated the uptake of AGAL/ADA complexes by leukocytes, suggesting the IgG-mediated uptake of AGAL/ADA complexes via Fcγ receptors [4]. Later on, Mauhin and colleagues showed that ADAs from FD patients can be internalized by blood mononuclear cells and inhibit intracellular AGAL activity [59]. However, since FD is a small-vessel disease and the endothelium should internalize ERT rather than peripheral blood mononuclear cells (PBMCs), the use of endothelial cells for uptake analyses is recommended more than using PBMCs. The use of fluorescence-labeled AGAL enabled Stappers and colleagues to demonstrate a patient- and thus antibody/epitope-dependent inhibitory effect of ADAs on cellular uptake [7] (Fig. 3C(i)). Furthermore, by combining the fluorescence-based in vivo measures with subsequent cell lysis and AGAL activity measures (Fig. 3C(ii)) and ELISAs (Fig. 3C(iii)), the authors demonstrated that ADA/AGAL complexes are internalized, but lack enzymatic activity within cells due to lack of dissociation [7].
For completeness, a rapid immunochromatographic bedside test has also been developed, allowing fast detection of ADAs in sera [67]. However, since this assay requires an optical estimation of signal strength, its use for titer estimations is limited.
Other, more sophisticated methods for quantifying and characterizing ADAs may include epitope mappings and affinity measurements. Recently, Kukacka and colleagues used a proteolytic epitope excision with subsequent mass spectrometry to identify the single epitope and binding affinity of a monoclonal rabbit antibody against human AGAL [68]. Due to the complexity, such an approach would not be feasible for measuring comparable characteristics for human polyclonal antibodies. Instead, epitope mappings with fragmented AGAL successfully revealed individual epitopes for patients, suggesting polyclonal antibodies [63].
However, the exclusive use of single peptides covering the AGAL sequence is a limitation, since conformational epitopes cannot be detected with this approach. Antibody affinities as a further important characteristic can be measured for example by ELISA, surface plasmon resonance immunoassays, or quartz crystal microbalance with dissipation monitoring (QCM-D). Recently, Lenders and colleagues demonstrated no significant differences for QCM-D-based ADA affinity measures between agalsidase-alfa, agalsidase-beta, and mossAGAL [66]. However, these data were based on the reference antibody pooled from 22 patients and do not necessarily reflect individual patient-specific affinities. Since high ADA affinities seem to be associated with increasing inhibitory capacities [69], while decreasing affinities might indicate the onset of tolerization [70], appropriate analyses will be of interest in future studies to identify FD patients at risk.
4 Methods to Prevent or Reduce ADAs
Specific protocols to prevent or reduce pre-existing neutralizing ADAs in patients with FD have not been reported in detail so far. However, Lenders and colleagues demonstrated that an immunosuppressive therapy in transplanted male FD patients might prevent ADA formation in ERT-naïve patients and reduce ADA titers in patients already under ERT [71].
In addition to FD, the most frequent LSDs currently treatable with ERT are Gaucher disease, MPS I, MPS II, MPS IVa, MPS VI, and Pompe disease (Table 1). For all diseases, a humoral response has been reported [72]. However, risk for ADA formation differs between these diseases [72], which is due to the underlying heredity (autosomal recessive or X-chromosomal) as well as disease-causing mutations. FD is an X-chromosomal linked disease due to ~ 1.000 different variants within the GLA gene. On the other hand, Pompe disease is an autosomal recessive disease, with only a few known specific mutations within the GAA gene. Whereas only male patients and homozygous females with FD and nonsense mutations can be CRIM negative, nearly all affected Pompe disease patients are CRIM negative with an increased risk for ADA formation [72]. In addition, awareness among treating physicians of the impact of ADAs on treatment efficacy varies across diseases. In a disease with rapid disease progression that eventually leads to early death, such as infantile Pompe disease, the deleterious effects of neutralizing ADAs are much more apparent than in FD. In conclusion, research concerning ADA formation, impact, prevention, and suppression is advanced in Pompe and Gaucher disease, which is reflected by the various reported immune-modulating protocols [72, 73]. Since the main underlying mechanisms (for an immune response) are the same in all diseases treated with ERT, existing knowledge and appropriate treatment protocols can be transferred to other diseases treated with ERT.
Several immune adsorption protocols based on non-specific IgG depletion showed high therapeutic efficiencies [74,75,76]. However, nonspecific adsorption systems remove complete Ig fractions and thus may further affect and weaken the humoral immune response [76]. In contrast, an antigen-specific immune adsorption results in depletion of pathogenic antibodies, while other antibodies and the humoral immune response remain unaffected [77,78,79,80]. In this respect, Lenders and colleagues demonstrated that AGAL-specific ADAs can be selectively re moved from the sera of FD patients in vitro [7, 66].
However, titers appear to recover very rapidly, which would require a high frequency of immunoadsorptions. Immunomodulation protocols appear to be clinically easier to implement. Most successful approaches included the inhibition of folic acid metabolism, DNA alkylation, and antibody-mediated B-cell depletion [81,82,83,84,85,86,87,88,89,90,91].
However, since memory B cells are not depleted by rituximab, the additional administration of bortezomib has been reported to effectively reduce ADA titers in infantile Pompe disease [83] and of omalizumab to reduce IgEs in FD [40]. Other options would be to use specific blocking peptides identified by epitope mapping or simply to increase the infused enzyme dosage to overcome existing ADA titers [6]. However, the maximum approved dosage of agalsidase-beta is currently restricted to 1 mg/kg body weight, making a dosage increase only implementable when switching from the regular approved agalsidase-alfa dosage (0.2 mg/kg) or lower agalsidase-beta dosage (< 1 mg/kg) to the regular approved agalsidase-beta dosage (1 mg/kg) [6]. Furthermore, this approach might not be suitable for every patient, since a dosage increase can result in increasing ADA titers [6, 40].
5 Knowledge Gaps and Outlook
Although major progress has been made in recent years in the characterization of neutralizing ADAs in FD, there are still important gaps in our knowledge that need to be addressed in future studies. Key tasks include developing strategies to prevent or reduce ADAs in affected patients. As reported above, appropriate already existing protocols for immunomodulation could be adapted for FD patients at risk. In addition, new approaches may also be conceivable for patients with FD.
For example, oral antigen administration resulted in significantly lower ADA titers in mice with Pompe disease or hemophilia A [92, 93]. To date, it has not been investigated whether oral administration of low-dose AGAL might support immune tolerance, resulting in lower ADA titers in an AGAL-deficient background. Of note, patients treated orally with human recombinant AGAL might benefit twice in that the risk for ADAs could be reduced, in addition to a reduction in gastrointestinal symptoms [94].
Finally, novel treatment strategies will also present new questions and obstacles. A prolonged half-life in plasma either by more stable compounds such as pegunigalsidase-alfa or by constant gene therapy-mediated expression will probably require modified assays to detect antibodies. The currently used assays aim to detect free antibodies in plasma and fail to detect ADA/AGAL complexes.
Samples from patients treated with next-generation ERTs or gene therapy might therefore require special sample preparation to dissociate existing ADA/AGAL complexes, or additional assays may need to be developed for the detection of these complexes.
6 Conclusion
CRIM-negative patients such as classical male FD patients with nonsense GLA mutations suffer from a high risk of a humoral response including ADAs against ERT.
The formation of ADAs is often associated with worse clinical outcome and disease progression. ADAs can already inhibit infused enzyme during infusions and reduce cellular enzyme uptake. Already existing protocols for immune tolerance induction to avoid or reduce ADAs can be transferred from other ERT-treated diseases to improve therapeutic efficacy in FD and other LSDs.
References
Zarate YA, Hopkin RJ. Fabry’s disease. Lancet. 2008;372:1427–35.
Schiffmann R, Kopp JB, Austin HA 3rd, Sabnis S, Moore DF, Weibel T, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285:2743–9.
Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, et al. Safety and efficacy of recombinant human a-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med. 2001;345:9–16.
Linthorst GE, Hollak CEM, Donker-Koopman WE, Strijland A, Aerts JMFG. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004;66:1589–95.
Lenders M, Stypmann J, Duning T, Schmitz B, Brand SM, Brand E. Serum-mediated inhibition of enzyme replacement therapy in Fabry disease. J Am Soc Nephrol. 2016;27:256–64.
Lenders M, Neußer LP, Rudnicki M, Nordbeck P, Canaan-Kühl S, Nowak A, et al. Dose-dependent effect of enzyme replacement therapy on neutralizing antidrug antibody titers and clinical outcome in patients with Fabry disease. J Am Soc Nephrol. 2018;29:2879–89.
Stappers F, Scharnetzki D, Schmitz B, Manikowski D, Brand SM, Grobe K, et al. Neutralising anti-drug antibodies in Fabry disease can inhibit endothelial enzyme uptake and activity. J Inherit Metab Dis. 2020;43:334–47.
Eng CM, Fletcher J, Wilcox WR, Waldek S, Scott CR, Sillence DO, et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007;30:184–92.
Echevarria L, Benistan K, Toussaint A, Dubourg O, Hagege AA, Eladari D, et al. X-chromosome inactivation in female patients with Fabry disease. Clin Genet. 2016;89:44–54.
Germain DP, Hughes DA, Nicholls K, Bichet DG, Giugliani R, Wilcox WR, et al. Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N Engl J Med. 2016;375:545–55.
Auray-Blais C, Ntwari A, Clarke JT, Warnock DG, Oliveira JP, Young SP, et al. How well does urinary lyso-Gb3 function as a biomarker in Fabry disease? Clin Chim Acta. 2010;411:1906–14.
Niemann M, Rolfs A, Störk S, Bijnens B, Breunig F, Beer M, et al. Gene mutations versus clinically relevant phenotypes: lyso-Gb3 defines Fabry disease. Circ Cardiovasc Genet. 2014;7:8–16.
Auray-Blais C, Blais CM, Ramaswami U, Boutin M, Germain DP, Dyack S, et al. Urinary biomarker investigation in children with Fabry disease using tandem mass spectrometry. Clin Chim Acta. 2015;438:195–204.
Boutin M, Menkovic I, Martineau T, Vaillancourt-Lavigueur V, Toupin A, Auray-Blais C. Separation and analysis of lactosylceramide, galabiosylceramide, and globotriaosylceramide by LC-MS/MS in urine of Fabry disease patients. Anal Chem. 2017;89:13382–90.
Nowak A, Mechtler TP, Desnick RJ, Kasper DC. Plasma LysoGb3: A useful biomarker for the diagnosis and treatment of Fabry disease heterozygotes. Mol Genet Metab. 2017;120:57–61.
Auray-Blais C, Lavoie P, Boutin M, Ntwari A, Hsu TR, Huang CK, et al. Biomarkers associated with clinical manifestations in Fabry disease patients with a late-onset cardiac variant mutation. Clin Chim Acta. 2017;466:185–93.
Arends M, Wijburg FA, Wanner C, Vaz FM, van Kuilenburg ABP, Hughes DA, et al. Favourable effect of early versus late start of enzyme replacement therapy on plasma globotriaosylsphingosine levels in men with classical Fabry disease. Mol Genet Metab. 2017;121:157–61.
Wanner C, Arad M, Baron R, Burlina A, Elliott PM, Feldt-Rasmussen U, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018;124:189–203.
Ortiz A, Germain DP, Desnick RJ, Politei J, Mauer M, Burlina A, et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123:416–27.
Lenders M, Brand E. Effects of enzyme replacement therapy and antidrug antibodies in patients with Fabry disease. J Am Soc Nephrol. 2018;29:2265–78.
Banikazemi M, Bultas J, Waldek S, Wilcox WR, Whitley CB, McDonald M, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007;146:77–86.
Watt T, Burlina AP, Cazzorla C, Schönfeld D, Banikazemi M, Hopkin RJ, et al. Agalsidase beta treatment is associated with improved quality of life in patients with Fabry disease: findings from the Fabry Registry. Genet Med. 2010;12:703–12.
Hughes DA, Barba Romero MÁ, Hollak CE, Giugliani R, Deegan PB. Response of women with Fabry disease to enzyme replacement therapy: comparison with men, using data from FOS–the Fabry Outcome Survey. Mol Genet Metab. 2011;103:207–14.
Germain DP, Charrow J, Desnick RJ, Guffon N, Kempf J, Lachmann RH, et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet. 2015;52:353–8.
Kampmann C, Perrin A, Beck M. Effectiveness of agalsidase alfa enzyme replacement in Fabry disease: cardiac outcomes after 10 years’ treatment. Orphanet J Rare Dis. 2015;10:125.
Schiffmann R, Swift C, Wang X, Blankenship D, Ries M. A prospective 10-year study of individualized, intensified enzyme replacement therapy in advanced Fabry disease. J Inherit Metab Dis. 2015;38:1129–36.
Lenders M, Brand E. Fabry Disease: The Current Treatment Landscape. Drugs. 2021;81:635–45.
Warnock DG, Thomas CP, Vujkovac B, Campbell RC, DA CharrowJ L, et al. Antiproteinuric therapy and Fabry nephropathy: factors associated with preserved kidney function during agalsidase-beta therapy. J Med Genet. 2015;52:860–6.
Seydelmann N, Wanner C, Störk S, Ertl G, Weidemann F. Fabry disease and the heart. Best Pract Res Clin Endocrinol Metab. 2015;29:195–204.
Rombach SM, Smid BE, Linthorst GE, Dijkgraaf MG, Hollak CE. Natural course of Fabry disease and the effectiveness of enzyme replacement therapy: a systematic review and meta-analysis: effectiveness of ERT in different disease stages. J Inherit Metab Dis. 2014;37:341–52.
El Dib R, Gomaa H, Ortiz A, Politei J, Kapoor A, Barreto F. Enzyme replacement therapy for Anderson-Fabry disease: a complementary overview of a Cochrane publication through a linear regression and a pooled analysis of proportions from cohort studies. PLoS ONE. 2017;12:e173358.
Lusher JM, Arkin S, Abildgaard CF, Schwartz RS; Kogenate Previously Untreated Patient Study Group. Recombinant factor VIII for the treatment of previously untreated patients with hemophilia A. Safety, efficacy, and development of inhibitors. N Engl J Med. 1993;328:453–9.
Wang J, Lozier J, Johnson G, Kirshner S, Verthelyi D, Pariser A, et al. Neutralizing antibodies to therapeutic enzymes: considerations for testing, prevention and treatment. Nat Biotechnol. 2008;26:901–8.
de Vries JM, van der Beek NAME, Kroos MA, Ozkan L, van Doorn PA, Richards SM, et al. High antibody titer in an adult with Pompe disease affects treatment with alglucosidase alfa. Mol Genet Metab. 2010;101:338–45.
Wilcox WR, Linthorst GE, Germain DP, Feldt-Rasmussen U, Waldek S, Richards SM, et al. Anti-a-galactosidase A antibody response to agalsidase beta treatment: data from the Fabry Registry. Mol Genet Metab. 2012;105:443–9.
Smid BE, Hoogendijk SL, Wijburg FA, Hollak CE, Linthorst GE. A revised home treatment algorithm for Fabry disease: influence of antibody formation. Mol Genet Metab. 2013;108:132–7.
Nicholls K, Bleasel K, Becker G. Severe infusion reactions to Fabry enzyme replacement therapy: rechallenge after tracheostomy. JIMD Rep. 2012;5:109–12.
Aydin O, Kasapkara CS, Celik GE. Successful desensitization with agalsidase alfa in 2 brothers with Fabry disease. J Investig Allergol Clin Immunol. 2013;23:367–8.
Limgala RP, Fikry J, Veligatla V, Goker-Alpan O. The interaction of innate and adaptive ammunity and stabilization of mast cell activation in management of infusion related reactions in patients with Fabry disease. Int J Mol Sci. 2020;21:7213.
DuBuske I, Schmidlin K, Bernstein JA. Successful desensitization of a patient with Fabry disease with agalsidase beta (Fabrazyme) anaphylaxis after omalizumab pretreatment. Ann Allergy Asthma Immunol. 2021;126:96.
Wilcox WR, Banikazemi M, Guffon N, Waldek S, Lee P, Linthorst GE, et al. International Fabry Disease Study Group: long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet. 2004;75:65–74.
Bodensteiner D, Scott CR, Sims KB, Shepherd GM, Cintron RD, Germain DP. Successful reinstitution of agalsidase beta therapy in Fabry disease patients with previous IgE-antibody or skin-test reactivity to the recombinant enzyme. Genet Med. 2008;10:353–8.
Wraith JE, Tylki-Szymanska A, Guffon N, Lien YH, Tsimaratos M, Vellodi A, et al. Safety and efficacy of enzyme replacement therapy with agalsidase beta: an international, open-label study in pediatric patients with Fabry disease. J Pediatr. 2008;152:563–70.
Tanaka A, Takeda T, Hoshina T, Fukai K, Yamano T. Enzyme replacement therapy in a patient with Fabry disease and the development of IgE antibodies against agalsidase beta but not agalsidase alpha. J Inherit Metab Dis. 2010;33(Suppl 3):S249–52.
Castells M. Rapid desensitization for hypersensitivity reactions to medications. Immunol Allergy Clin North Am. 2009;29:585–606.
Arends M, Biegstraaten M, Wanner C, Sirrs S, Mehta A, Elliott PM, et al. Agalsidase alfa versus agalsidase beta for the treatment of Fabry disease: an international cohort study. J Med Genet. 2018;55:351–8.
van der Veen SJ, Vlietstra WJ, van Dussen L, van Kuilenburg ABP, Dijkgraaf MGW, et al. Predicting the development of anti-drug antibodies against recombinant alpha-galactosidase A in male patients with classical Fabry disease. Int J Mol Sci. 2020;21:5784.
Rombach SM, Aerts JM, Poorthuis BJ, Groener JE, Donker-Koopman W, Hendriks E, et al. Long-term effect of antibodies against infused alpha-galactosidase A in Fabry disease on plasma and urinary (lyso)Gb3 reduction and treatment outcome. PLoS ONE. 2012;7:e47805.
Bénichou B, Goyal S, Sung C, Norfleet AM, O’Brien F. A retrospective analysis of the potential impact of IgG antibodies to agalsidase beta on efficacy during enzyme replacement therapy for Fabry disease. Mol Genet Metab. 2009;96:4–12.
Schiffmann R, Warnock DG, Banikazemi M, Bultas J, Linthorst GE, Packman S, et al. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009;24:2102–11.
Vedder AC, Linthorst GE, Houge G, Groener JE, Ormel EE, Bouma BJ, et al. Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2 mg/kg. PLoS ONE. 2007;2:e598.
Hongo K, Harada T, Fukuro E, Kobayashi M, Ohashi T, Eto Y. Massive accumulation of globotriaosylceramide in various tissues from a Fabry patient with a high antibody titer against alpha-galactosidase A after 6 years of enzyme replacement therapy. Mol Genet Metab Rep. 2020;24:100623.
Mhanni AA, Auray-Blais C, Boutin M, Johnston A, LeMoine K, Patterson J, et al. Therapeutic challenges in two adolescent male patients with Fabry disease and high antibody titres. Mol Genet Metab Rep. 2020;24:100618.
Lenders M, Nordbeck P, Canaan-Kühl S, Kreul L, Duning T, Lorenz L, et al. Treatment switch in Fabry disease—a matter of dose? J Med Genet. 2021;58:342–50.
Lenders M, Schmitz B, Brand SM, Foell D, Brand E. Characterization of drug-neutralizing antibodies in patients with Fabry disease during infusion. J Allergy Clin Immunol. 2018;141:2289-92.e7.
Krishna M, Nadler SG. Immunogenicity to biotherapeutics—the role of anti-drug immune complexes. Front Immunol. 2016;7:21.
Wang Z, Okamoto P, Keutzer J. A new assay for fast, reliable CRIM status determination in infantile-onset Pompe disease. Mol Genet Metab. 2014;111:92–100.
van der Veen SJ, van Kuilenburg ABP, Hollak CEM, Kaijen PHP, Voorberg J, Langeveld M. Antibodies against recombinant alpha-galactosidase A in Fabry disease: subclass analysis and impact on response to treatment. Mol Genet Metab. 2019;126:162–8.
Mauhin W, Lidove O, Amelin D, Lamari F, Caillaud C, Mingozzi F, et al. Deep characterization of the anti-drug antibodies developed in Fabry disease patients, a prospective analysis from the French multicenter cohort FFABRY. Orphanet J Rare Dis. 2018;13:127.
Hunley TE, Corzo D, Dudek M, Kishnani P, Amalfitano A, Chen YT, et al. Nephrotic syndrome complicating alpha-glucosidase replacement therapy for Pompe disease. Pediatrics. 2004;114:e532–5.
Debiec H, Valayannopoulos V, Boyer O, Nöel LH, Callard P, Sarda H, et al. Allo-immune membranous nephropathy and recombinant aryl sulfatase replacement therapy: a need for tolerance induction therapy. J Am Soc Nephrol. 2014;25:675–80.
Ronco P, Debiec H. Pathophysiological advances in membranous nephropathy: time for a shift in patient’s care. Lancet. 2015;385:1983–92.
Scharnetzki D, Stappers F, Lenders M, Brand E. Detailed epitope mapping of neutralizing anti-drug antibodies against recombinant α-galactosidase A in patients with Fabry disease. Mol Genet Metab. 2020;131:229–34.
Mauhin W, Lidove O, Masat E, Mingozzi F, Mariampillai K, Ziza JM, et al. Innate and adaptive immune response in Fabry disease. JIMD Rep. 2015;22:1–10.
Tsukimura T, Tayama Y, Shiga T, Hirai K, Togawa T, Sakuraba H. Anti-drug antibody formation in Japanese Fabry patients following enzyme replacement therapy. Mol Genet Metab Rep. 2020;25:100650.
Lenders M, Scharnetzki D, Heidari A, Di Iorio D, Wegner SV, Brand E. Generation and characterization of a polyclonal human reference antibody to measure anti-drug antibody titers in patients with Fabry disease. Int J Mol Sci. 2021;22:2680.
Nakano S, Tsukimura T, Togawa T, Ohashi T, Kobayashi M, Takayama K, Kobayashi Y, Abiko H, Satou M, Nakahata T, Warnock DG, Sakuraba H, Shibasaki F. Rapid immunochromatographic detection of serum anti-α-galactosidase A antibodies in Fabry patients after enzyme replacement therapy. PLoS ONE. 2015;10:e0128351.
Kukacka Z, Iurascu M, Lupu L, Rusche H, Murphy M, Altamore L, et al. Antibody epitope of human α-galactosidase A revealed by affinity mass spectrometry: a basis for reversing immunoreactivity in enzyme replacement therapy of Fabry disease. ChemMedChem. 2018;13:909–15.
Hofbauer CJ, Whelan SF, Hirschler M, Allacher P, Horling FM, Lawo JP, Oldenburg J, Tiede A, Male C, Windyga J, Greinacher A, Knöbl PN, Schrenk G, Koehn J, Scheiflinger F, Reipert BM. Affinity of FVIII-specific antibodies reveals major differences between neutralizing and nonneutralizing antibodies in humans. Blood. 2015;125:1180–8.
Pratt KP. Anti-Drug antibodies: emerging approaches to predict, reduce or reverse biotherapeutic immunogenicity. Antibodies. 2018;7:19.
Lenders M, Oder D, Nowak A, Canaan-Kühl S, Arash-Kaps L, Drechsler C, et al. Impact of immunosuppressive therapy on therapy-neutralizing antibodies in transplanted patients with Fabry disease. J Intern Med. 2017;282:241–53.
Kishnani PS, Dickson PI, Muldowney L, Lee JJ, Rosenberg A, Abichandani R, et al. Immune response to enzyme replacement therapies in lysosomal storage diseases and the role of immune tolerance induction. Mol Genet Metab. 2016;117:66–83.
Doerfler PA, Nayak S, Corti M, Morel L, Herzog RW, Byrne BJ. Targeted approaches to induce immune tolerance for Pompe disease therapy. Mol Ther Methods Clin Dev. 2016;3:15053.
Biesenbach P, Kain R, Derfler K, Perkmann T, Soleiman A, Benharkou A, et al. Long-term outcome of anti-glomerular basement membrane antibody disease treated with immunoadsorption. PLoS ONE. 2014;9:e103568.
Ohlow MA, Brunelli M, Schreiber M, Lauer B. Therapeutic effect of immunoadsorption and subsequent immunoglobulin substitution in patients with dilated cardiomyopathy: results from the observational prospective Bad Berka Registry. J Cardiol. 2017;69:409–16.
Hamilton P, Harris R, Mitra S. Chapter four—immunoadsorption techniques and its current role in the intensive care unit, aspects in continuous renal replacement therapy. In: A. Karkar (Ed.), IntechOpen. 2019.
Sun C, Meng F, Li Y, Jin Q, Li H, Li F. Antigen-specific immunoadsorption of anti-acetylcholine receptor antibodies from sera of patients with myastenia gravis. Artif Cells Blood Substit Immobil Biotechnol. 2010;38:99–102.
Lazaridis K, Baltatzidou V, Tektonidis N, Tzartos SJ. Antigen-specific immunoadsorption of MuSK autoantibodies as a treatment of MuSK-induced experimental autoimmune myasthenia gravis. J Neuroimmunol. 2020;339:577136.
Handisurya A, Worel N, Rabitsch W, Bojic M, Pajenda S, Reindl-Schwaighofer R, et al. Antigen-specific immunoadsorption with the Glycosorb® ABO immunoadsorption system as a novel treatment modality in pure red cell aplasia following major and bidirectional ABO-incompatible allogeneic hematopoietic stem cell transplantation. Front Med. 2020;7:585628.
Pavenski K, Bucholz M, Cheatley PL, Krok E, Anderson M, Prasad GR, et al. The first north American experience using Glycosorb immunoadsorption columns for blood group-incompatible kidney transplantation. Can J Kidney Health Dis. 2020;7:2054358120962586.
Markic J, Polic B, Kuzmanic-Samija R, Marusic E, Stricevic L, Metlicic V, et al. Immune modulation therapy in a CRIM-positive and IgG antibody positive infant with Pompe disease treated with alglucosidase alpha: a case report. JIMD Rep. 2012;2:11–5.
Elder ME, Nayak S, Collins SW, Lawson LA, Kelley JS, Herzog RW, et al. B-Cell depletion and immunomodulation before initiation of enzyme replacement therapy blocks the immune response to acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2013;163:847-54.e1.
Banugaria SG, Prater SN, McGann JK, Feldman JD, Tannenbaum JA, Bailey C, et al. Bortezomib in the rapid reduction of high sustained antibody titers in disorders treated with therapeutic protein: Lessons learned from Pompe disease. Genet Med. 2013;15:123–31.
Kazi ZB, Desai AK, Berrier KL, Troxler RB, Wang RY, Abdul-Rahman OA, et al. Sustained immune tolerance induction in enzyme replacement therapy-treated CRIM-negative patients with infantile Pompe disease. JCI Insight. 2017;2:e94328.
Banugaria SG, Prater SN, Patel TT, Dearmey SM, Milleson C, Sheets KB, et al. Algorithm for the early diagnosis and treatment of patients with cross reactive immunologic material-negative classic infantile Pompe disease: a step towards improving the efficacy of ERT. PLoS ONE. 2013;8:e67052.
Banugaria SG, Patel TT, Mackey J, Das S, Amalfitano A, Rosenberg AS, et al. Persistence of high sustained antibodies to enzyme replacement therapy despite extensive immunomodulatory therapy in an infant with Pompe disease: need for agents to target antibody-secreting plasma cells. Mol Genet Metab. 2012;105:677–80.
Markic J, Polic B, Stricevic L, Metlicic V, Kuzmanic-Samija R, Kovacevic T, et al. Effects of immune modulation therapy in the first Croatian infant diagnosed with Pompe disease: a 3-year follow-up study. Wien Klin Wochenschr. 2014;126:133–7.
Mendelsohn NJ, Messinger YH, Rosenberg AS, Kishnani PS. Elimination of antibodies to recombinant enzyme in Pompe’s disease. N Engl J Med. 2009;360:194–5.
Kim KH, Messinger YH, Burton BK. Successful reduction of high-sustained anti-idursulfase antibody titers by immune modulation therapy in a patient with severe mucopolysaccharidosis type II. Mol Genet Metab Rep. 2014;2:20–4.
Julien DC, Woolgar K, Pollard L, Miller H, Desai A, Lindstrom K, Kishnani PS. Immune modulation for enzyme replacement therapy in a female patient with Hunter Syndrome. Front Immunol. 2020;11:1000.
Sun A, Alshuaibi W, Petroni D, Skoda-Smith S, Goldberg MJ, Hale S. Immune modulation in a patient with Morquio syndrome treated with enzyme replacement therapy. J Allergy Clin Immunol Pract. 2018;6:1749–51.
Ohashi T, Iizuka S, Shimada Y, Eto Y, Ida H, Hachimura S, Kobayashi H. Oral administration of recombinant human acid α-glucosidase reduces specific antibody formation against enzyme in mouse. Mol Genet Metab. 2011;103:98–100.
Sherman A, Su J, Lin S, Wang X, Herzog RW, Daniell H. Suppression of inhibitor formation against FVIII in a murine model of hemophilia A by oral delivery of antigens bioencapsulated in plant cells. Blood. 2014;124:1659–68.
Lenders M, Boutin M, Auray-Blais C, Brand E. Effects of orally delivered alpha-galactosidase A on gastrointestinal symptoms in patients with Fabry disease. Gastroenterology. 2020;159:1602–4.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL.
Conflicts of interest
M.L. received speaker honoraria, travel funding, and research grants from Amicus Therapeutics, Sanofi Genzyme, and Takeda. E.B. received research grants and speaker honoraria from Amicus Therapeutics, Sanofi Genzyme, and Takeda.
Availability of data and material
Not applicable.
Author’ contributions
Malte Lenders and Eva Brand both wrote the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Lenders, M., Brand, E. Mechanisms of Neutralizing Anti-drug Antibody Formation and Clinical Relevance on Therapeutic Efficacy of Enzyme Replacement Therapies in Fabry Disease. Drugs 81, 1969–1981 (2021). https://doi.org/10.1007/s40265-021-01621-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-021-01621-y