
Abstract
Background
Data collected for medical, filing and administrative purposes in electronic patient records (EPRs) represent a rich source of individualised clinical data, which has great potential for improved detection of patients experiencing adverse drug reactions (ADRs), across all approved drugs and across all indication areas.
Objectives
The aim of this study was to take advantage of techniques for temporal data mining of EPRs in order to detect ADRs in a patient- and dose-specific manner.
Methods
We used a psychiatric hospital’s EPR system to investigate undesired drug effects. Within one workflow the method identified patient-specific adverse events (AEs) and links these to specific drugs and dosages in a temporal manner, based on integration of text mining results and structured data. The structured data contained precise information on drug identity, dosage and strength.
Results
When applying the method to the 3,394 patients in the cohort, we identified AEs linked with a drug in 2,402 patients (70.8 %). Of the 43,528 patient-specific drug substances prescribed, 14,736 (33.9 %) were linked with AEs. From these links we identified multiple ADRs (p < 0.05) and found them to occur at similar frequencies, as stated by the manufacturer and in the literature. We showed that drugs displaying similar ADR profiles share targets, and we compared submitted spontaneous AE reports with our findings. For nine of the ten most prescribed antipsychotics in the patient population, larger doses were prescribed to sedated patients than non-sedated patients; five patients exhibited a significant difference (p < 0.05). Finally, we present two cases (p < 0.05) identified by the workflow. The method identified the potentially fatal AE QT prolongation caused by methadone, and a non-described likely ADR between levomepromazine and nightmares found among the hundreds of identified novel links between drugs and AEs (p < 0.05).
Conclusions
The developed method can be used to extract dose-dependent ADR information from already collected EPR data. Large-scale AE extraction from EPRs may complement or even replace current drug safety monitoring methods in the future, reducing or eliminating manual reporting and enabling much faster ADR detection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Temporal data mining of patient-specific notes stored in electronic patient record (EPR) systems can be used for pharmacovigilance in a clinical, multi-diseased and polypharmacy population. |
Structured and unstructured data accumulated in the EPRs permits dose-specific monitoring of adverse drug reactions (ADRs) and observations of patients affected by ADRs in the actual clinical setting. |
High throughput analysis of the data already collected in EPRs by healthcare providers can be used to verify suspected correlations, as well as suggest novel likely correlations between drugs and adverse events. |
1 Introduction
Despite increasing knowledge and prevention methods, adverse drug reactions (ADRs) continue to impact a large part of the patient population [1, 2], leading to hospitalisations and substantial healthcare expenses [3, 4]. Several of these noxious and unintended responses, occurring at normal doses [5], have been identified after approved drugs were introduced on the market, in some cases resulting in serious harm and ultimately in drugs being withdrawn [6]. This highlights that drug safety cannot be fully assessed solely through the preclinical [7] and clinical stages [8]. Clinical trials only expose a limited number of subjects in selected groups, are often underpowered to identify all ADRs, and the limited time window often prevents identification of ADRs with late onset [8]. Thus post-approval surveillance is an essential part of every drug’s lifecycle. Additionally, observational data, where co-morbidities and concomitant drug use are present, has been suggested to represent a better measurement of harms experienced by actual patients in comparison to data collected from clinical trials [9].
For the past 40 years, medical product agencies have relied heavily on spontaneous reporting systems for post-approval drug safety surveillance [10–12]. Adverse events (AEs) occurring during treatment are collected and analysed in databases such as the US FDA Adverse Event Reporting System (FAERS) and the WHO VigiBase [13]. The aim of these analyses is to generate hypotheses of causal relationships between drugs and AEs, referred to as signal detection [14]. However, there are several recognised shortcomings with this scheme. The main issues are the widespread underreporting and reporting bias largely caused by submitters prioritising other tasks, selectively reporting severe less-known AEs, and data quality issues such as report duplication [8, 11, 15]. This is despite efforts to improve reporting in several countries through tightened legislation making suspected AE reporting mandatory [10]. Alternatively, AE relationships can be tracked in health registries. The FDA Sentinel Initiative [12], the EU-ADR project [16] and the Observational Medical Outcomes Partnership [17] aim at large-scale aggregation and integration of data captured in databases. However, far from all AE evidence is reported or captured in coded formats. It is thus desirable to monitor AEs from the detailed clinical observations already recorded by healthcare providers in electronic patient records (EPRs), in particular the clinical narratives. This is in contrast to the data stored in an electronic health record (EHR), which consist of computer-readable data represented according to relevant controlled vocabularies [18].
In this study we use temporal data mining of an EPR system to link AEs to drugs, in a multi-diseased and polypharmacy population.
2 Methods
2.1 Study Population
We gained full access to the EPR system of a tertiary Danish psychiatric hospital with free text and structured data from 3,394 patients prescribed at least one drug between 1998 and 2010. The patient population (electronic supplementary material [ESM] 1) ranged from 17 to 86 years of age, mean age was 43.6 years (standard deviation 12.5 years) at the midpoint of treatment, counting all admissions for each patient as one single period. The male:female ratio was 2:1. The average total clinical narrative was 66,000 words and 1,400 notes per patient; this high number is related to the nature of this patient population. Communication plays an even more vital role in diagnosis and treatment planning within psychiatry compared with other medical specialties.
2.2 Temporal Data Mining
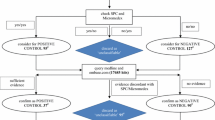
The overall strategy of our approach is to combine structured prescription data with AEs extracted from clinical notes to identify AEs occurring between drug introduction and discontinuation (Fig. 1). The approach is enabled by the time-stamped arrangement of both the structured and unstructured data. All AEs were passed through filters to discard AEs within text passages with words suggesting that the mentioned AE did not happen, affected someone else or happened in the past [19]. To eliminate pre-existing conditions, we filtered out all AEs present prior to drug introduction or coincided with indications of the drug. Only AEs retained after filtering were used for the subsequent analyses.

AE extraction and data integration. AEs were extracted between drug introduction and discontinuation, where we filtered out AEs if the text in the clinical note suggests it did not happen, affected someone else or happened in the past. Additionally, we filtered all indications of the drug and ADRs related to additional drugs. Finally, all pre-existing conditions were removed. Remaining AEs were sorted into ADRs and possible ADRs; the latter was presented for manual review. ADR adverse drug reaction, AE adverse event, EPR electronic patient record, SPC Summary of Product Characteristics
2.3 Drug Prescriptions
The structured prescription-data contains all orders made in the hospital, meaning that all prescriptions are stored in the electronic records. We extracted the drug identity, the total daily dose, as well as the times of introduction and discontinuation for each prescription, including premature discontinuation of a prescribed drug not necessarily being the same as the last treatment day on the original prescription. The total daily dose was calculated for each treatment day, including all prescriptions and allowing changes in dosing to be accounted for. Depot formulations were considered continuously over the whole dosing period.
2.4 Adverse Event (AE) and Drug Name Identification
It has previously been shown that investigation of clinical narratives can give an up to fourfold boost in the number of unique diagnoses associated with each patient [20], when comparing to assigned International Classification of Diseases (ICD) 10 codes. Considering that only a subset of the data is available in coded formats and that many code formats are not designed for AE investigation, it is desirable to detect AE in clinical narratives.
We have previously developed a method for identification of possible AEs in unstructured clinical narrative text [19]. The method is based on a named entity recognition (NER) tagger, which identifies matches in the clinical text to a dictionary of drug-related AEs. It takes into account alternative spelling, synonyms and inflectional variants, which are merged and treated as the same AE. Moreover, anatomical structures are linked, so that cell types and tissues are merged to larger anatomical structures, e.g. hepatocellular damage and liver damage. Together these features allow for appropriate grouping of effects. The current version has been improved to handle more than 4 × 1012 different ways of writing descriptions of drug related AEs. A negation and other negatives filtering step removes disqualified concepts. This includes negations or mentions of other subjects. Furthermore, information about undesirable effects given from the prescriber or AE descriptions from the past that are disqualified for temporal reasons as these occurred at some other time point were filtered in a similar manner.
Understanding the complexity of AE descriptions requires interpretation of clinical narratives. We therefore merged terms that are not necessarily synonyms but have the same medical implication, e.g. kidney failure and elevated creatinine (ESM 2). We also extended the method with trade names and generic names of all drugs authorised in Denmark, including likely spelling variants and misspellings based on the interchangeable characters, phonetic similarity, and endings. For all subsequent analyses we considered drugs with the same active substance as a single drug. We used the Anatomical Therapeutic Chemical (ATC) classification system [21] to group drugs into classes. To assess how well this method performed, we manually curated 200 randomly selected clinical notes, according to a previously described method [19]. On this set, the method identified possible AE words and drugs with a 75 % recall (sensitivity) and 95 % precision (positive predictive value).
2.5 AE Filtering
When an AE is mentioned in the clinical narrative it does not necessarily imply that the patient experienced the AE. Prescribers are required to document that they have informed patients about the ADRs they may experience. Also, AEs and indications overlap, meaning that the mentioned AE may be the very reason why the patient received the drug.
We used a set of filters to address these and other complications (Fig. 1). First, we considered only AEs if they were mentioned at least 1 day after drug introduction. The only exception was anaphylaxis, which we recorded on the introduction day. The first day was excluded in order to reduce errors in AE detection originating from the prescribers’ information about the treatment risks. Second, we used linguistic filters described elsewhere [19] to disqualify AEs in text passages with negations, words suggesting that the mentioned AE did not happen, affected other subjects, described previously experienced events or information about ADRs. In this way we were able to filter out AEs recorded on a particular date, taking into account previously experienced AEs or AEs that might occur in the future. We similarly disregarded AEs from sentences that mentioned two or more drugs as this implied information was given to the patient from a healthcare provider or implied a medical history about the patient. Third, we filtered out indications listed in the official product documentation required within the EU, namely the Summary of Product Characteristics (SPCs) [22, 23]. We used the SPCs as of 4 November 2011, combined indications for all drugs containing the same active substance to eliminate manufacturer-specific differences, and manually added certain off-label uses. Additionally, the filtering was extended to disqualify contraindications and secondary effects of the indication to the drug in question. Fourth, we filtered AEs on the first treatment day of any additional drug introduced coinciding with its ADRs. Finally, we also filtered out pre-existing conditions that were not indications of the drug, but were mentioned within the last 14 days before the drug was prescribed. Different time intervals between 1 to 4 weeks were analysed.
2.6 Statistical Analyses, Frequencies and Adverse Drug Reaction (ADR) Characteristics
Next, we used a statistical approach to identify possible causal links between drugs and AEs, building upon counting the extracted and merged AEs once per patient and drug.
Two confidence levels were used for drug–AE pairs. The lower level included all AEs identified throughout the entire treatment period. The higher level further requires within-sentence co-occurrence of the AE and the same drug as is in the prescription, and was used in all calculations except for dose–ADR relationship.
All AEs were assigned to a 2 × 2 contingency table [14], consisting of patients on drug X or not, and patients experiencing AE Y or not. We tested each drug–AE pair for statistically significant overrepresentation using one-sided Fisher’s exact test, and used the Benjamini–Hochberg procedure to control the false discovery rate (FDR) using a threshold of 5 %. Furthermore, the odds ratio (OR) and the proportional reporting ratio (PRR) were calculated. Additionally, a value corrected by matching for gender and age was calculated for all drug–AE pairs. Each of the statistically significant drug–AE pairs was compared with the ADRs listed in the ‘undesirable effects’ section of the SPC of the drug. In case of multiple drugs with the same active substances, we included the ADRs from all the SPCs, thereby eliminating product differences. AEs not identified as known ADRs, and therefore not known for the drug in question, were classified as possible ADRs for further manual review (Fig. 1). Here, comorbidities, concomitant drugs and other factors such as biological plausibility can be assessed.
Absolute frequencies, defined as the proportion of patients treated with a certain drug who are affected by a specific AE, were calculated from structured drug-prescription data and AEs identified in the clinical narratives. We also calculated the relative frequencies, defined as the proportion that a specific AE makes up of all reported AEs for a drug.
From the statistically significantly overrepresented ADRs, we calculated the ADR profile similarity for any two drugs using the Jaccard similarity coefficient. This resulted in a drug network based on ADR profile similarity identified in our study population.
To investigate dose–ADR relationships, we included events in the first continuous steady dosage interval, between introduction and discontinuation, in patients prescribed the drug for at least 5 and 10 days (also including patients in the 5-day observation period). Only standing oral orders were considered and patients on depot formulations were excluded. The dose distributions prescribed non-sedated and sedated patients for each interval were analysed for statistically significant differences using the two-sided t-test.
3 Results
By integrating text mining results and structured data in a temporal manner, we were able to identify AEs documented in the hospital’s EPR system and link these AEs with the drugs used.
3.1 Drug Fingerprint and AEs Linked to Drugs
The distribution and prescription patterns were examined and revealed a median usage of 14.5 drug substances over the study period (Fig. 2a). Only ten drugs were prescribed to more than 1,000 patients. To give an overview, we summarise the drug prescriptions as a network that captures how often drugs from a given class are prescribed and how often drugs from two different classes are prescribed to the same patient (Fig. 2b).

Patient population. a Distribution of unique recorded drug substance prescriptions per patient and average proportions of first-level ATC group prescription. The total number of drug substances prescribed to a patient throughout the study (x-axis) and the proportions of drug classes prescribed to the patients prescribed this number of drug substances (y-axis). Anesthetics (N01) and analgesics (N02) are separated from nervous system (N), permitting illustration of psychiatric drug prescription. b Within-patient drug prescription between second-level ATC groups. Nodes represent the second-level ATC, sizes, the amount prescribed and colour of the anatomical main group. Edges show within-patient drug prescription between second-level ATC groups, while darker edges indicate more frequent co-administration or within-patient prescription. ATC Anatomical Therapeutic Chemical
Of the 3,394 patients prescribed at least one drug, our method found AEs in 2,402 patients (70.8 %). In total, of the 43,528 patient-specific drug substances prescribed in the study we identified 14,736 (33.9 %) AEs linked to a drug. Of these, 47 % were linked to antipsychotics (N05A), 13 % to drugs used in addictive disorders (N07B), and 9 % to antiepileptics (N03A). The classes with the highest average AEs per prescription were psychostimulants, agents used for ADHD and nootropics (N06B) [1.13 AEs/prescription], antipsychotics (N05A) [0.88 AEs/prescription] and drugs used in addictive disorders (N07B) [0.76 AEs/prescription]. In total 2,104 statistically significantly overrepresented drug–AE pairs were extracted, and 1,252 corrected drug–AE pairs were significant.
We further analysed the data for sensitive patients. Of the patients where we found an AE, 26 (1.1 %) patients experienced the same AE for three or more drugs. In the analysis we found 48 (2.0 %) patients were associated with more than ten AEs to a single drug.
3.2 Extracted ADR Frequencies
Based on the extracted ADRs from the clinical texts and the structured prescription data, we calculated the frequency with which a specific ADR occurred. We compared the absolute frequencies of the 150 most statistically overrepresented extracted ADRs prescribed to more than ten patients with the manufacturer-stated frequencies (Fig. 3). Only 11 were omitted as they had frequencies <1/1,000 according to the SPC, but in our data prescribed to 328 patients or less. Compared with the SPCs, we estimate common ADRs to occur at similar frequencies, very common ADRs slightly lower, and uncommon ADRs slightly higher.

ADR frequency comparison. Absolute frequency comparison between the most statistically significant extracted ADRs at least prescribed to ten patients and the manufacturer listed. ADR adverse drug reaction, EPR electronic patient record
We further compared the findings of our method with suspected AEs that had been reported to the Danish Medicines Agency (now the Danish Health and Medicines Authority) [ESM 3], showing we can extract more AEs statistically significantly associated with a particular drug in a single hospital than reports in the database for the same drug.
In addition to the comparison between AE–drug pairs identified by our method and ADRs stated in the SPCs used to identify known correlations, we also compared our results with unlikely associations [24]. This second comparison showed that none of the ‘negative control’ associations were identified by our method.
3.3 ADR Profiles
Placing the drugs in a network based on their actual ADR profiles demonstrated that drugs with similar indications tend to group together, and have similar ADR profiles (Fig. 4), which was expected because in many cases they also have common targets [25]. For example, this is the case for psycholeptics (N05A) and psychoanaleptics (N06A). Drugs with similar active substances but different indications also group together (e.g. opioids). Finally, there are examples of drugs that have similar ADR profiles despite neither having similar indication nor being chemically similar, e.g. laxatives (A06A) and antibacterials for systemic use (J01). The contrast being antiepileptics (N03) displaying diverse profiles, not unexpected since antiepileptics are known to vary in terms of ADRs [26].

ADR-profile network. Condensed force-directed ADR-profile network, represented by the 500 largest Jaccard similarity coefficient values. Nodes depict drug substances, identified by the fifth-level ATC code, sizes indicate the number of ADRs detected. Subgroups of the ATC group Nervous system (N), are given separate colours, except for grey, which symbolises all other drug substances. Edges display ADR profile similarity, while darker edge indicates higher similarity. ADR adverse drug reaction, ATC Anatomical Therapeutic Chemical classification system
3.4 Dose–ADR Relationship
To test if events where patients experiencing a certain ADR were related to drug dosage, we focused on the most frequently identified AE, namely sedation, which was associated with 940 patients. As many antipsychotics cause sedation [27–29], we selected the ten most prescribed antipsychotics in the cohort for investigating dose–ADR relationships. For the first steady dosage exceeding 5 and 10 days, the dose distribution of non-sedated and sedated patients were compared (Table 1). For all drugs except haloperidol, the groups that experienced sedation had a higher mean dose compared with the groups that did not. The 5-day period displayed a statistically significant difference in four drugs, which rose to five drugs in the 10-day period.
3.5 Cases of Severe and Likely ADRs
The vast majority of the ADRs we identified and quantified were of a less severe nature, but we also associated drugs with potentially fatal AEs. We identified three patient cases (0.8 %) of methadone-associated QT interval prolongation [30, 31] of the 392 patients in methadone treatment (p = 0.008; FDR = 0.02), verified by manual chart review.
The most significant, not documented correlation (p < 0.001; FDR < 0.001) was between levomepromazine and nightmares, which is not listed as an ADR in the SPC but still represents a plausible and probable effect of antipsychotics [32]. We found nightmares occurring in 4 of 523 patients (0.8 %), verified by manual chart review. In this analysis, we excluded any nightmares occurring during concurrent treatment with any known nightmare-inducing drug. These drugs were extracted from the Danish SPCs. The existence of such correlation was previously suspected by some of the hospital’s clinical psychiatry staff. Baseline patient characteristics are retained from the cohort, which is essentially heavily treated with psychiatric drugs.
4 Discussion
Through this analysis we were able to identify thousands of described ADRs, to suggest non-described likely ADR correlations, and to recapitulate many of the known characteristics of drugs and ADRs in a patient-specific manner. The described method demonstrates an individualised model for drug safety monitoring based on the rich data already collected by healthcare providers. In contrast to earlier work [33], for the first time we identified ADRs in a dose-specific manner by integrating prescribed exposure from structured data and unstructured clinical narrative text. A key feature that makes this possible is that we merge terms that have the same anatomical location or medical implication, even if they are not merely alternative spelling variants. We demonstrate the potential on a psychiatric cohort, a population with higher frequency of ADRs than other specialties [34], but our method is not specific for this domain and therefore also extends into other medical specialties. In addition, neither is the approach language-specific as the ADR dictionaries may be replaced with dictionaries based on other languages.
The links between drugs and AEs are uncovered in a high-throughput manner from raw clinical information that requires essentially no extra work for professionals in the healthcare system and could eventually reduce the burden of submitting spontaneous reports. Additionally, time and phenotypic data loss is minimised as no information has to be rewritten, transferred, transformed or coded into a separate database [13, 16, 35]. The linking is done by investigating actual clinical data, which differ many times from the artificial conditions of a clinical trial, where co-morbidities and concomitant drug treatments are normally excluded.
Due to the limited number of subjects in the cohort and the large number of different drugs prescribed, the number of individuals prescribed each drug only sometimes reaches the number of participants in a phase III clinical trial [13]. Despite this, we were able to identify thousands of statistically significant links between drugs and AEs, and in many cases even estimate their frequencies close to those listed in the SPCs. We extracted lower frequencies of very common ADRs, possibly explained by these being so common the clinician never records them. On the other hand, we estimate slightly higher frequencies for the most significant rare (≥1/10,000 to <1/1,000) and very rare (<1/10,000) ADRs. We cannot conclude if the frequencies listed in the SPC should be higher because too few patients were prescribed these drugs to compare them with the frequencies listed in the SPCs. Furthermore, the observed difference is possibly explained by the fact that the study population includes chronic and hospitalised patients, sometimes with extreme polypharmacy and doses. With that said, our method might show the clinical reality better and therefore the frequencies differ from the carefully selected patients included in a clinical trial and presented in the SPCs.
Our method could also be used for discovering very rare ADRs, which are notoriously hard to identify in smaller studies such as most randomised clinical trials [36]. In this relation, nationwide EPR systems could be of interest, indisputably providing a larger study population, but still permit individual drug response observations. Moreover, the method has no constraints preventing integration into efforts such as the FDA Sentinel Initiative [12] where sensitive data is not transferred outside the originating organisation.
The dose difference between groups unaffected and affected by a particular ADR could possibly point at dose limits that should not be exceeded if it is desirable to prevent a certain ADR, thereby individualising the treatment for each patient. Haloperidol, which has been associated with less sedative effects [27], is the only drug where the non-sedated proportion is given a larger mean dose. This is possibly explained by the prominent extrapyramidal symptoms associated with an increase in haloperidol dose, resulting in a reduction in attention to sedation compared with the other notable effects, as these increased with dose increments. Comparing our results with the tolerability of antipsychotics, similar patterns are revealed [37]. Substances displaying a higher proportion of sedated patients, such as ziprasidone, also display more sedation compared with placebo; similarly haloperidol and aripiprazole show a lower proportion of sedated patients in our study, as well as less sedation compared with placebo in the study by Leucht et al. [37] The sedative profile of chlorprothixene seen in the investigation might be the real indication for many prescriptions of this drug. The decision to prescribe chlorprothixene very likely involves its sedative effects, despite it not being stated in the prescription decision. This is showing another quality of the method.
It is most likely that further fine-tuning of the extraction of information would present further possibilities of correcting for additional confounding factors to a future analysis. This also applies to the construction and usage of EPR systems, which are constantly becoming more advanced, interconnected and information-rich.
Any drug–AE pair the method identifies as statistically overrepresented and which is not previously known to be associated, is presented for manual review. This output occasionally contains trivial correlations and we recognise the value of further improvements limiting the output, potentially by a similar method, as suggested by Tatonetti et al. [38]. Alternatively, the output could also simply be analysed in a similar way as spontaneous reports, generating signals validated in a separate investigation [5, 10, 13].
One aspect we are not investigating is patient self-administered drugs and drugs used prior to admission, as we do not have information on this. These drugs can potentially cause effects that we interpret as AEs and link to another drug, although they are unlikely to give rise to statistically significantly overrepresented ADRs. Another limitation is the non-perfect extraction from the narratives, where more advanced linguistic techniques would likely improve the performance of the method. There is also a risk of AEs never being recorded in the first place, but this type of error would most likely affect less severe AEs. Due to the filtering of identified possible AEs, we are not able to find AEs identical to the indication and are thus not able to identify deterioration in the original treatment cause. We are currently filtering sentences containing two or more drugs, which limits the risk of falsely linking an AE to a drug. It also reduces the possibility of detecting drug–drug interactions, which in any case is not our aim with this study. Furthermore, we do not investigate late effects appearing after drug discontinuation.
5 Conclusions
Not only do we present a high-throughput method to identify AE correlations in noisy real clinical data, but we also display the clinical research potential of the underexploited asset EPRs represent. For the first time, temporal data mining of an EPR system is used to identify ADRs and enable drug safety monitoring in a dose-specific manner by integrating structured exposure data and unstructured clinical narrative text. Our method utilises an individualised model to harness highly detailed phenotypic data already collected by healthcare providers. This type of method may limit the critical time before a serious ADR is identified after market introduction, thereby preventing morbidity and mortality in the population. The described method, in combination with future advances of clinical tools, could provide the prescriber with real-time decision support, highlighting possible AEs a patient is having or has experienced.
References
Taché SV, Sönnichsen A, Ashcroft DM. Prevalence of adverse drug events in ambulatory care: a systematic review. Ann Pharmacother. 2011;45:977–89.
Van der Hooft CS, Sturkenboom MCJM, van Grootheest K, Kingma HJ, Stricker BHC. Adverse drug reaction-related hospitalisations: a nationwide study in The Netherlands. Drug Saf. 2006;29:161–8.
Rottenkolber D, Schmiedl S, Rottenkolber M, Farker K, Saljé K, Mueller S, et al. Adverse drug reactions in Germany: direct costs of internal medicine hospitalizations. Pharmacoepidemiol Drug Saf. 2011;20:626–34.
Gautier S, Bachelet H, Bordet R, Caron J. The cost of adverse drug reactions. Expert Opin Pharmacother. 2003;4:319–26.
World Health Organization. The importance of pharmacovigilance. Geneva: World Health Organization; 2002.
Avorn J. Two centuries of assessing drug risks. N Engl J Med. 2012;367:193–7.
Kramer JA, Sagartz JE, Morris DL. The application of discovery toxicology and pathology towards the design of safer pharmaceutical lead candidates. Nat Rev Drug Discov. 2007;6:636–49.
Ray WA. Population-based studies of adverse drug effects. N Engl J Med. 2003;23(349):1592–4.
Vandenbroucke JP, Psaty BM. Benefits and risks of drug treatments how to combine the best evidence on benefits with the best data about adverse effects. JAMA. 2008;300:2417–9.
Mann RD, Andrews EB, editors. Pharmacovigilance. 2nd ed. Chichester: Wiley; 2007.
Gibbons RD, Amatya AK, Brown CH, Hur K, Sue M, Bhaumik DK, et al. Post-approval drug safety surveillance. Annu Rev Public Health. 2010;31:419-37.
Robb MA, Racoosin JA, Sherman RE, Gross TP, Ball R, Reichman ME, et al. The US Food and Drug Administration’s Sentinel Initiative: expanding the horizons of medical product safety. Pharmacoepidemiol Drug Saf. 2012;21:9–11.
Strom BL, Kimmel SE, Hennessy S. Pharmacoepidemiology. 5th ed. Chichester: Wiley; 2012.
Bate A, Evans SJW. Quantitative signal detection using spontaneous ADR reporting. Pharmacoepidemiol Drug Saf. 2009;18:427–36.
Hazell L, Shakir SAW. Under-reporting of adverse drug reactions a systematic review. Drug Saf. 2006;29:385–96.
Coloma PM, Schuemie MJ, Trifiro G, Gini R, Herings R, Hippisley-Cox J, et al. Combining electronic healthcare databases in Europe to allow for large-scale drug safety monitoring: the EU-ADR project. Pharmacoepidemiol Drug Saf. 2011;20:1–11.
Stang PE, Ryan PB, Racoosin JA, Overhage JM, Hartzema AG, Reich C, et al. Advancing the science for active surveillance: rationale and design for the Observational Medical Outcomes Partnership. Ann Intern Med. 2010;2(153):600–6.
Jensen PB, Jensen LJ, Brunak S. Mining electronic health records: towards better research applications and clinical care. Nat Rev Genet. 2012;13:395–405.
Eriksson R, Jensen PB, Frankild S, Jensen LJ, Brunak S. Dictionary construction and identification of possible adverse drug events in Danish clinical narrative text. J Am Med Inf Assoc. 2013;1(20):947–53.
Roque FS, Jensen PB, Schmock H, Dalgaard M, Andreatta M, Hansen T, et al. Using electronic patient records to discover disease correlations and stratify patient cohorts. PLoS Comput Biol. 2011;7:e1002141.
WHO Collaborating Centre for Drug Statistics Methodology. Guidelines for ATC classification and DDD assignment, 2013. 15th ed. Norway: Oslo; 2012.
European Medicines Agency. Human medicines. 2012. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp. Accessed 27 Sep 2013
Lægemiddelstyrelsen. Produktresuméer—human. 2012. http://www.produktresume.dk/docushare/dsweb/View/Collection-96. Accessed 27 Sep 2013
Coloma PM, Avillach P, Salvo F, Schuemie MJ, Ferrajolo C, Pariente A, et al. A reference standard for evaluation of methods for drug safety signal detection using electronic healthcare record databases. Drug Saf. 2013;36:13–23.
Garcia-Serna R, Mestres J. Anticipating drug side effects by comparative pharmacology. Expert Opin Drug Metab Toxicol. 2010;6:1253–63.
Perucca P, Gilliam FG. Adverse effects of antiepileptic drugs. Lancet Neurol. 2012;11:792–802.
Rang PH, Dale MM, Ritter JM, Flower RJ, Henderson G. Antipsychotic drugs. Rang and Dale’s pharmacology. 7th ed. London: Elsevier; 2011. p. 553–63.
Meltzer H. Antipsychotic agents & lithium. In: Katzung BG, Masters S, Trevor AJ, editors. Basic and clinical pharmacology. 11th ed. New York: Lange Medical Books/McGraw Hill; 2009. p. 487–507.
Stanniland C, Taylor D. Tolerability of atypical antipsychotics. Drug Saf. 2000;22:195–214.
Keller GA, Ponte ML, Di Girolamo G. Other drugs acting on nervous system associated with QT-interval prolongation. Curr Drug Saf. 2010;5:105–11.
Stringer J, Welsh C, Tommasello A. Methadone-associated Q-T interval prolongation and torsades de pointes. Am J Heal Syst Pharm. 2009;1(66):825–33.
Pagel JF, Helfter P. Drug induced nightmares: an etiology based review. Hum Psychopharmacol. 2003;18:59–67.
Lependu P, Iyer SV, Bauer-Mehren A, Harpaz R, Mortensen JM, Podchiyska T, et al. Pharmacovigilance using clinical notes. Clin Pharmacol Ther. 2013;4(93):547–55.
Rothschild JM, Mann K, Keohane CA, Williams DH, Foskett C, Rosen SL, et al. Medication safety in a psychiatric hospital. Gen Hosp Psychiatry. 2007;29:156–62.
Emmendorfer T, Glassman PA, Moore V, Leadholm TC, Good CB, Cunningham F. Monitoring adverse drug reactions across a nationwide health care system using information technology. Am J Health Syst Pharm. 2012;15(69):321–8.
Goldsmith S, Pellmar T, Kleinman A, Bunney W. Reducing suicide: a national imperative. Washington, DC: National Academies Press; 2002. p. 376.
Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013;14(382):951–62.
Tatonetti NP, Ye PP, Daneshjou R, Altman RB. Data-driven prediction of drug effects and interactions. Sci Transl Med. 2012;4:125ra31.
Acknowledgments
The authors would like to thank Henriette Schmock and Sabrina Ellesøe for valuable input and critical reading of the manuscript.
Ethical approval
The project was ethically approved by the Danish National Board of Health (7-604-04-2/33/EHE).
Author Contributions
Study concept and design: Robert Eriksson, Lars Juhl Jensen and Søren Brunak. Acquisition of data: Thomas Werge and Søren Brunak. Analysis and interpretation of data: Robert Eriksson. Drafting of the manuscript: Robert Eriksson. Critical revision of the manuscript for important intellectual content: Robert Eriksson, Thomas Werge, Lars Juhl Jensen and Søren Brunak. Statistical analysis: Robert Eriksson and Lars Juhl Jensen. Obtained funding: Thomas Werge and Søren Brunak. Administrative, technical, or material support: Lars Juhl Jensen and Søren Brunak. Study supervision: Thomas Werge, Lars Juhl Jensen and Søren Brunak.
Conflict of Interest
Thomas Werge has previously received consulting fees from H. Lundbeck A/S. Robert Eriksson, Thomas Werge, Lars Juhl Jensen and Søren Brunak have no other conflicts of interest that are directly relevant to the content of this article.
Funding and Support
The project was supported by grants from the Danish Council for Strategic Research (Grant No. 0603-00321B), the Novo Nordisk Foundation, the Villum Foundation. The sponsors had no role in the design and conduct of the study; collection, management, analysis and interpretation of the data; and preparation, review or approval of the manuscript.
Availability of Data and Materials
Due to the sensitive nature of the original data, which has not been de-identified, no original data will be made available by the authors. The dictionary and algorithm will be made available to readers under a materials transfer agreement or purchase.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Eriksson, R., Werge, T., Jensen, L.J. et al. Dose-Specific Adverse Drug Reaction Identification in Electronic Patient Records: Temporal Data Mining in an Inpatient Psychiatric Population. Drug Saf 37, 237–247 (2014). https://doi.org/10.1007/s40264-014-0145-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-014-0145-z