
Abstract
Background
Drug–drug interactions (DDIs) and drug–gene interactions (DGIs) pose a serious health risk that can be avoided by dose adaptation. These interactions are investigated in strictly controlled setups, quantifying the effect of one perpetrator drug or polymorphism at a time, but in real life patients frequently take more than two medications and are very heterogenous regarding their genetic background.
Objectives
The first objective of this study was to provide whole-body physiologically based pharmacokinetic (PBPK) models of important cytochrome P450 (CYP) 2C8 perpetrator and victim drugs, built and evaluated for DDI and DGI studies. The second objective was to apply these models to describe complex interactions with more than two interacting partners.
Methods
PBPK models of the CYP2C8 and organic-anion-transporting polypeptide (OATP) 1B1 perpetrator drug gemfibrozil (parent–metabolite model) and the CYP2C8 victim drugs repaglinide (also an OATP1B1 substrate) and pioglitazone were developed using a total of 103 clinical studies. For evaluation, these models were applied to predict 34 different DDI studies, establishing a CYP2C8 and OATP1B1 PBPK DDI modeling network.
Results
The newly developed models show a good performance, accurately describing plasma concentration–time profiles, area under the plasma concentration–time curve (AUC) and maximum plasma concentration (Cmax) values, DDI studies as well as DGI studies. All 34 of the modeled DDI AUC ratios (AUC during DDI/AUC control) and DDI Cmax ratios (Cmax during DDI/Cmax control) are within twofold of the observed values.
Conclusions
Whole-body PBPK models of gemfibrozil, repaglinide, and pioglitazone have been built and qualified for DDI and DGI prediction. PBPK modeling is applicable to investigate complex interactions between multiple drugs and genetic polymorphisms.
Similar content being viewed by others

1 Introduction
From epidemiological data, it is estimated that 5–20% of adverse drug events resulting in hospital admission are caused by drug–drug interactions (DDIs), with an especially high risk for elderly patients due to polypharmacy [1]. Indeed, data show that in the USA, 67% of the adults older than 62 years take more than five medications. As a result, about one in six older adults may be at risk for a major DDI [2] resulting in decreased efficacy, increased risk for adverse drug reactions, and increased healthcare costs. A second important aspect is that genetic polymorphisms in drug transporters or metabolizing enzymes may result in drug–gene interactions (DGIs). Similarly to DDIs, these DGIs can result in significantly altered drug exposure. In current clinical practice, DDIs and DGIs are considered separate entities. However, they are interconnected and ignoring drug–drug–gene interactions (DDGIs) can jeopardize patient safety. Ideally, guidelines on how to manage DDIs and DDGIs should be based on results from clinical trials.
However, in reality, most DDGIs cannot be investigated in clinical trials for many reasons, including ethical and feasibility restrictions due to their complexity. Usually, classic DDI studies are performed as typical phase I studies in healthy volunteers using so-called index substances to characterize a certain DDI potential. The study participants are mostly young, healthy, take only two drugs at the same time, and are genetically homogenous, and, consequently, they do not mimic real-life multimorbid patients with polypharmacy and genetic polymorphisms [3]. Thus, there is a translational challenge to assess and manage complex multifactorial DDGIs in real-life patients.
One possibility to loosen this Gordian knot might be the application of whole-body physiologically based pharmacokinetic (PBPK) modeling. PBPK models are increasingly used to evaluate the effects of patient factors on drug exposure [4] and they are excellent tools to predict the DDGI potential of drugs in silico and allow development of alternative dosing regimens for patients. The interest in PBPK modeling is continuously rising in academia and the pharmaceutical industry. Regulatory agencies (European Medicines Agency [EMA], U.S. Food and Drug Administration [FDA]) recommend PBPK modeling for the assessment of DDI potential, the development of alternative dosing regimens, and, in some cases, even to waive clinical studies [5, 6]. To project the reality of patients, complex DDI networks and thoroughly developed PBPK models are required. Even though many perpetrator and victim drug models have been developed and published so far [7], there is still a need for further models and more comprehensive DDI networks.
The main focus of the presented work is the description of cytochrome P450 (CYP) 2C8- and organic-anion-transporting polypeptide (OATP) 1B1-based DDIs, using PBPK models of the perpetrator drug gemfibrozil (strong CYP2C8 index inhibitor and inhibitor of OATP1B1) and of the two victim drugs repaglinide (sensitive CYP2C8 index substrate and substrate of OATP1B1) and pioglitazone (moderate sensitive CYP2C8 substrate) [6, 8]. Gemfibrozil, repaglinide, and pioglitazone are all recommended by the FDA for use in clinical DDI studies [8]. As clinically relevant genetic polymorphisms of CYP2C8 and solute carrier organic anion transporter family member (SLCO) 1B1 (OATP1B1) are reported to impact the pharmacokinetics of repaglinide and pioglitazone, the effects of the CYP2C8*3 and SLCO1B1 521T>C alleles were considered and implemented into the respective models.
The aims of this study were (1) to develop a PBPK DDI network [9] for CYP2C8 and thereby to expand the library of publicly available models for DDI prediction with verified whole-body PBPK models of gemfibrozil, repaglinide, and pioglitazone, and to apply these models to (2) describe and predict DDIs including complex DDIs with more than two drugs, (3) describe and predict DGIs with two or more drugs, and (4) exemplarily develop victim drug dose adaptations for patients with genetic polymorphisms and coadministration of two perpetrator drugs. The supplementary document to this paper (Electronic Supplementary Material [ESM]) is compiled as a transparent and comprehensive documentation and reference manual, providing detailed information on all PBPK models and modeled DDI studies. Model files are freely available in the Open Systems Pharmacology (OSP) repository (https://www.open-systems-pharmacology.org).
2 Methods
2.1 Software
PBPK models were developed using PK-Sim® and MoBi® modeling software (version 7.3.0, part of the OSP suite). Parameter optimizations (Monte-Carlo-algorithm) and sensitivity analyses were performed with PK-Sim®. Clinical study data was digitized using GetData Graph Digitizer (version 2.26.0.20 ©, S. Fedorov). Pharmacokinetic parameter analyses were performed and plots were created with R (version 3.4.4, R Foundation for Statistical Computing, Vienna, Austria) and R Studio (version 1.1.423, RStudio, Inc., Boston, MA, USA).
2.2 Physiologically Based Pharmacokinetic (PBPK) Model Building
PBPK model building was started with an extensive literature search to gain information on the physicochemical properties and absorption, distribution, metabolism, and excretion (ADME) processes of the drugs of interest as well as to obtain clinical studies (healthy individuals) of intravenous and oral administration in single and multiple doses. The plasma concentration–time profiles of the clinical studies were digitized and divided into an internal dataset used for model building and parameter optimization, and an external dataset used for model evaluation. Parameters that could not be informed from the literature were optimized, fitting the model simultaneously to the observed data of all studies assigned to the internal dataset.
The mean individuals used to simulate the different studies were modeled according to the respective study reports, with corresponding age, weight, height, sex, and ethnicity. If no information on these demographics was found, a 30-year-old male European was assumed, with the mean weight and height characteristics given in the PK-Sim® database. The creation of virtual populations to compare predicted and observed population plasma concentration–time profiles is described in the ESM (Sect. 3.9).
Protein expression of enzymes and transporters was implemented according to the literature, using the PK-Sim® database [10]. For more details, see ESM Table S3.9.1.
2.3 PBPK Model Evaluation
The PBPK models were evaluated by comparison of predicted population plasma concentration–time profiles to observed data. The observed data were mostly reported as arithmetic or geometric mean plasma concentration–time profiles with standard deviations. To compare the variability of predicted to observed profiles, 68% population prediction intervals were plotted, as this interval corresponds to the range of ± 1 standard deviation around the mean if normal distribution is assumed.
Furthermore, the predicted plasma concentration values were compared to their respective observed values in goodness-of-fit plots and the model performance was evaluated by comparison of predicted to observed area under the plasma concentration–time curve (AUC), maximum plasma concentration (Cmax), apparent oral clearance (CL/F), and half-life values. For model evaluation, all AUC values were calculated from time zero to infinity (AUC∞).
As quantitative measures of the model performance, mean relative deviations (MRDs) of the predicted plasma concentrations (see ESM Eq. 3.1) and geometric mean fold errors (GMFEs) of the AUC∞, Cmax, CL/F, and half-life values (see ESM Eq. 3.2) were calculated.
2.4 Drug–Drug Interaction (DDI) Network Modeling
In addition to the evaluation methods described in Sect. 2.3, a CYP2C8 DDI network was built to evaluate the DDI performance of the developed models (Fig. 1). Gemfibrozil with its metabolite gemfibrozil 1-O-β-glucuronide was used as the CYP2C8 and OATP1B1 inhibitor, repaglinide as the CYP2C8 and OATP1B1 victim, and pioglitazone as the CYP2C8 victim drug. In addition, repaglinide is also a substrate of CYP3A4 and OATP1B3, adding DDI potential during coadministration of CYP3A4 and OATP1B3 perpetrators, such as itraconazole, clarithromycin, and rifampicin. Rifampicin was used as the inducer and competitive inhibitor of CYP2C8, CYP3A4, OATP1B1, and OATP1B3, interacting with repaglinide and pioglitazone. Interaction processes are modeled using the equations given in the ESM (Sect. 1).

The developed drug–drug interaction (DDI) network with gemfibrozil and gemfibrozil 1-O-β-glucuronide as cytochrome P450 (CYP) 2C8 and organic-anion-transporting polypeptide (OATP) 1B1, itraconazole as CYP3A4, OATP1B1, and OATP1B3, rifampicin as CYP2C8, CYP3A4, OATP1B1, and OATP1B3, and clarithromycin as CYP3A4, OATP1B1, and OATP1B3 perpetrator drugs (upper part); and repaglinide as CYP2C8, CYP3A4, OATP1B1, and OATP1B3 and pioglitazone as CYP2C8 victim drugs (lower part). Physiologically based pharmacokinetic models of itraconazole, rifampicin and clarithromycin were adopted from Hanke et al. [9]. Metabolism and transport are illustrated as black arrows. Solid red lines indicate reversible inhibition processes, dashed bold red lines indicate mechanism-based inactivation. Dashed violet lines indicate interaction processes by rifampicin consisting of inhibition as well as induction processes. The postulated physicochemical interactions are shown as dotted black lines
2.5 DDI Network Evaluation
The quality of the DDI modeling was evaluated by comparison of predicted to observed victim drug plasma concentration–time profiles when administered alone and during coadministration, DDI AUC ratios (Eq. 1) and DDI Cmax ratios (Eq. 2). For DDI evaluation, all AUC values were calculated from time zero to the time of the last concentration measurement (AUClast).
To assess the DDI modeling performance, the GMFEs of the predicted DDI AUC ratios and DDI Cmax ratios were calculated according to ESM Eq. 3.2.
2.6 Sensitivity Analysis
Sensitivity analyses were performed on the gemfibrozil, repaglinide, and pioglitazone models to investigate the impact of single model parameters on the predicted AUC at steady state given the highest recommended dose. Parameters were included into the analysis if they have been optimized, if they could have a strong influence on the pharmacokinetics due to their use in calculation of permeabilities or partition coefficients (e.g., fraction unbound), or if they had significant impact in formerly developed models (e.g., blood/plasma concentration ratio).
Sensitivity is calculated as the ratio of the relative change of the simulated AUC to the relative variation of the tested parameter around the parameter value used in the model, according to Eq. 3:
where S is the sensitivity of the AUC to the tested model parameter, ΔAUC is the change of the AUC, AUC is the AUC calculated with the original model parameter value, p is the original model parameter value, and Δp is the change of the tested model parameter value.
Sensitivity analyses were performed using a relative perturbation of 100%. The threshold value for sensitivity was set at 0.5. A sensitivity value of + 0.5 indicates that a 100% increase of the model parameter value causes a 50% increase of the predicted AUC. In addition, the parameters were varied within a 0.03- to 30-fold range and the resulting fold changes of AUC were investigated in spider plots.
3 Results
3.1 PBPK Model Building and Evaluation
Whole-body PBPK models of gemfibrozil with gemfibrozil 1-O-β-glucuronide (parent–metabolite model), repaglinide, and pioglitazone have been successfully developed. A total number of 103 studies showing plasma concentration–time profiles were used for model building and evaluation; all of these are presented in the ESM.
A detailed description of each model, including Tables listing the drug-dependent parameters, is given in ESM Sects. 3.3 (gemfibrozil and gemfibrozil 1-O-β-glucuronide model), 3.4 (repaglinide model), and 3.5 (pioglitazone model). System-dependent parameters were taken directly from the PK-Sim® database or, if not available, they were gathered from literature, as summarized in ESM Table S3.9.1. The good model performance for both internal and external datasets is demonstrated by comparison of population predicted to observed plasma concentration–time profiles in Fig. 2 (representative studies for each compound) and in ESM Figs. S3.3.1, S3.3.2, S3.3.3, S3.3.4, S3.4.1, S3.4.2, S3.4.3, S3.4.4, S3.5.1, and S3.5.2 (all studies, semilogarithmic and linear plots). Goodness-of-fit plots are presented in ESM Figs. S3.3.5, S3.4.5, and S3.5.3 and MRD values for all studies are given in ESM Tables S3.3.3, S3.4.3, and S3.5.3. Correlation of predicted to observed AUC and Cmax values is presented in ESM Figs. S3.3.6, S3.4.6, and S3.5.4, and the corresponding values are given in ESM Tables S3.3.4, S3.4.4, and S3.5.4, including calculated model GMFE values. Correlation of predicted to observed CL/F and half-life values is shown in ESM Figs. S3.3.7, S3.4.7, and S3.5.5 and the corresponding values are given in ESM Tables S3.3.5, S3.4.5, and S3.5.5. The results of sensitivity analyses are described in ESM Sects. 3.3, 3.4, and 3.5 and Figs. S3.3.8, S3.3.9, S3.3.10, S3.4.8, S3.4.9, S3.5.6, and S3.5.7.

Gemfibrozil (a, b), gemfibrozil 1-O-β-glucuronide (gemfi-glu) (a, b), repaglinide (c–f), and pioglitazone (g–i) plasma concentration–time profiles. Observed data are shown as triangles ± standard deviation [29,30,31,32,33,34,35,36,37]. Population simulation arithmetic means or geometric means (a) are shown as black (gemfibrozil), red (gemfibrozil 1-O-β-glucuronide), green (repaglinide), or blue (pioglitazone) lines. The shaded areas represent the respective 68% population prediction intervals. Detailed information about dosing regimens and study populations is given in Electronic Supplementary Material (ESM) Tables S3.3.1, S3.4.1, and S3.5.1. Predicted and observed area under the plasma concentration–time curve (AUC) and maximum plasma concentration (Cmax) values are compared in ESM Tables S3.3.4, S3.4.4, and S3.5.4. b.i.d. twice daily, conc concentration, CYP cytochrome P450, po oral, q.d. once daily, s.d. single dose, SLCO solute carrier organic anion transporter family member, t.i.d. three times daily
For the development of the gemfibrozil parent–metabolite PBPK model, plasma concentration–time profiles of 23 studies (oral administration), including ten studies showing gemfibrozil 1-O-β-glucuronide concentrations (after oral administration of gemfibrozil) were used. Gemfibrozil 1-O-β-glucuronide was incorporated into the model because it is the major circulating metabolite of gemfibrozil and a mechanism-based inactivator of CYP2C8 [11], contributing significantly to the strong CYP2C8 inhibition by gemfibrozil. The gemfibrozil model applies an unspecified active uptake of gemfibrozil into hepatocytes [12], metabolism by uridine 5ʹ-diphospho-glucuronosyltransferase (UGT) 2B7 to form gemfibrozil 1-O-β-glucuronide and glomerular filtration. The metabolite model applies a hepatic uptake transport by OATP1B1 [12], an efflux transport into bile via multidrug resistance-associated protein (MRP) 2 and glomerular filtration.
For the development of the repaglinide model, plasma concentration–time profiles of 56 studies (intravenous and oral administration) as well as the fraction metabolized via CYP2C8 information were used. The model applies hepatic uptake via OATP1B1 and OATP1B3, metabolism by CYP2C8 and CYP3A4, and glomerular filtration. An unbound Michaelis-Menten contant (KM) value determined for repaglinide uptake into untransfected primary human hepatocytes was used as the KM value for the OATP1B1 and OATP1B3 transport processes [13]. For the metabolism via CYP2C8, a fraction metabolized of 89% was assumed, according to a clinical DDI study with gemfibrozil [14]. The remaining repaglinide is metabolized via CYP3A4 [15]. The model slightly overpredicts the low repaglinide plasma concentrations at later times after dosing (ESM Figs. S3.4.1, S3.4.2, and S3.4.5). This could be due to a misspecification of the CYP2C8 or OATP1B1/1B3 KM values taken from the literature, or to an unknown mechanism that impacts the pharmacokinetics of repaglinide but is missing in the model. This limitation of the model affects the prediction of low repaglinide plasma concentrations, but Cmax and AUC values are well-predicted (ESM Fig S3.4.6). For the investigation of DGIs, the model adequately describes the pharmacokinetics of repaglinide in carriers of the CYP2C8*3, SLCO1B1 521C and SLCO1B1 –11187A alleles. The implementation of polymorphic CYP2C8 and OATP1B1 is described in ESM Sects. 2.2, 2.3, and 3.4.
For the development of the pioglitazone model, plasma concentration–time profiles of 13 studies (oral administration), one study describing the fraction of pioglitazone excreted to urine, as well as the fraction metabolized via CYP2C8 information were used. Pioglitazone is reported to be predominantly metabolized by CYP2C8 [16], with no consistent information on the identity of other metabolic enzymes involved. Therefore, the model applies metabolism by CYP2C8, an unspecific hepatic clearance and glomerular filtration. For the metabolism via CYP2C8, a fraction metabolized of 70–75% was assumed [17]. For the investigation of DGIs, the model adequately describes the pharmacokinetics of pioglitazone in carriers of the CYP2C8*3 allele. The implementation of polymorphic CYP2C8 is described in ESM Sects. 2.2 and 3.5.
3.2 DDI Network Modeling
For the DDI network modeling, a total number of 34 DDI studies were available in literature and used to evaluate the modeled interactions (Fig. 1). Thereof, 23 studies describe the gemfibrozil–repaglinide DDI, four the gemfibrozil–pioglitazone DDI, and one study each the itraconazole–repaglinide DDI, the itraconazole–pioglitazone DDI, the gemfibrozil–itraconazole–repaglinide DDI, the gemfibrozil–itraconazole–pioglitazone DDI, the rifampicin–repaglinide DDI, the rifampicin–pioglitazone DDI, and the clarithromycin–repaglinide DDI. The previously developed models of itraconazole, rifampicin, and clarithromycin [9] were used without changes other than the addition of interaction parameters to model the CYP2C8 and OATP1B1/1B3 DDIs. A full description of the DDI modeling is given in ESM Sect. 4, including all interaction parameters, administration protocols, and study population demographics.
All DDI victim drug plasma concentration–time profiles are well-predicted using interaction parameters taken from the literature (listed in ESM Tables S3.3.2, S3.6.1, S3.7.1, and S3.8.1 summarizing the drug-dependent parameters of the perpetrator drugs), except for the hydroxy-itraconazole OATP1B1 and OATP1B3 inhibitory constant (Ki) values, which had to be optimized. To describe the DDIs with itraconazole, the solubility value of the previously developed itraconazole model was adjusted (see ESM Figs. S3.6.1 and S3.6.2). Itraconazole is a poorly soluble compound (8.0 mg/L in fasted state simulated intestinal fluid [18]), leading to variable absorption and, therefore, to large inter-individual differences in itraconazole plasma concentration–time profiles. In the gemfibrozil–itraconazole–pioglitazone interaction study [19], itraconazole concentration–time profiles before and during gemfibrozil coadministration are reported, in addition to the DDI victim drug concentrations. For itraconazole and hydroxy-itraconazole, a reduction of the AUCs is described if gemfibrozil is added to the itraconazole–pioglitazone DDI. Possible explanations for these finding proposed by Jaakkola et al. [19] and Niemi et al. [20] are the displacement of itraconazole from plasma proteins or a reduction of itraconazole bioavailability by gemfibrozil. Both hypotheses were tested with the models by either increasing the fraction unbound of itraconazole during the DDI or by reducing its solubility (to describe reduced bioavailability). The observed plasma concentration–time profiles of itraconazole and hydroxy-itraconazole could not be described properly by a change in fraction unbound, but the change of itraconazole solubility resulted in a good description of the observed data. To accurately model the observed itraconazole plasma concentrations of the gemfibrozil–itraconazole–pioglitazone interaction study [19], the solubility of itraconazole (capsule formulation, fasted state) was optimized to 14.5 mg/L at pH 6.5 in the absence of gemfibrozil and to 0.69 mg/L at pH 6.5 in the presence of gemfibrozil (shown in ESM Sect. 3.6), indicating a physicochemical DDI between gemfibrozil and itraconazole.
The second assumed physicochemical DDI affects pioglitazone during coadministration of both gemfibrozil and itraconazole (see Figs. 3c and ESM Fig. S4.7.1). Like itraconazole, pioglitazone is poorly soluble, with a reported solubility of 16.8 mg/L at pH 6.5 [21]. To accurately describe the pioglitazone plasma concentrations during gemfibrozil–itraconazole–pioglitazone coadministration, pioglitazone solubility was adjusted to 1.59 mg/L at pH 6.5 (shown in ESM Sect. 4.7).

Pioglitazone plasma concentration–time profiles during the gemfibrozil–pioglitazone (a), itraconazole–pioglitazone (b), gemfibrozil–itraconazole–pioglitazone (c), and rifampicin–pioglitazone (d) drug–drug interaction (DDI). Observed data are shown as triangles ± standard deviation (dark blue: control, light blue: with perpetrator drug) [19, 38]. Pioglitazone population simulation arithmetic means are shown as lines (dark blue: control, light blue: with perpetrator drug), the dashed line (c) shows the prediction of the gemfibrozil–itraconazole–pioglitazone without pioglitazone solubility adjustment. The shaded areas represent the respective 68% population prediction intervals. Detailed information about dosing regimens and study populations is given in Electronic Supplementary Material (ESM) Tables S4.3.1, S4.5.1, S4.7.1, and S4.9.1. Predicted and observed DDI area under the plasma concentration–time curve (AUC) ratios and DDI maximum plasma concentration (Cmax) ratios are compared in ESM Tables S4.3.2, S4.5.2, S4.7.2, and S4.9.2. b.i.d. twice daily, conc concentration, po oral, q.d. once daily, s.d. single dose
The good DDI performance of the models is demonstrated in predicted compared to observed victim drug plasma concentration–time profiles before and during perpetrator drug coadministration in Figs. 3 and 4 (representative studies for each DDI) and in ESM Figs. S4.2.1, S4.2.2, S4.2.3, S4.2.4, S4.2.5, S4.2.6, S4.3.1, S4.3.2, S4.4.1, S4.5.1, S4.6.1, S4.7.1, S4.8.1, S4.9.1, and S4.10.1 (all studies, semilogarithmic and linear plots). For the gemfibrozil–repaglinide DDI, the time-dependency (Fig. 4 and ESM Figs. S4.2.3 and S4.2.4) and dose-dependency (ESM Figs. S4.2.5 and S4.2.6) of the interaction were also modeled and compared to clinical data.

Repaglinide plasma concentration–time profiles during the gemfibrozil–repaglinide (a), itraconazole–repaglinide (b), gemfibrozil–itraconazole–repaglinide (c), rifampicin–repaglinide (d), and clarithromycin–repaglinide (e) drug–drug interaction (DDI). Observed data are shown as triangles, crosses, or stars ± standard deviation (dark green: control, light green: with perpetrator drug) [20, 31, 39, 40]. Repaglinide population simulation arithmetic means are shown as lines (dark green: control, light green: with perpetrator drug). The shaded areas represent the respective 68% population prediction intervals. Detailed information about dosing regimens and study populations is given in Electronic Supplementary Material (ESM) Tables S4.2.1, S4.4.1, S4.6.1, S4.8.1, and S4.10.1. Predicted and observed DDI area under the plasma concentration–time curve (AUC) ratios and DDI maximum plasma concentration (Cmax) ratios are compared in ESM Tables S4.2.2, S4.4.2, S4.6.2, S4.8.2, and S4.10.2. b.i.d. twice daily, conc concentration, po oral, q.d. once daily, s.d. single dose
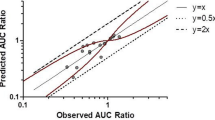
As further evaluation of the performance of the entire DDI network, the correlation of predicted to observed DDI AUC ratios and DDI Cmax ratios of all modeled DDI studies is shown in Fig. 5. The corresponding values are listed in ESM Tables S4.2.2, S4.3.2, S4.4.2, S4.5.2, S4.6.2, S4.7.2, S4.8.2, S4.9.2, and S4.10.2, including GMFE values calculated for each perpetrator–victim drug combination.

Correlation of predicted and observed drug–drug interaction (DDI) area under the plasma concentration–time curve (AUC) ratios and DDI maximum plasma concentration (Cmax) ratios of all studies. The upper panel illustrates DDI AUC ratios (a) and DDI Cmax ratios (b) of the gemfibrozil–repaglinide, itraconazole–repaglinide, gemfibrozil–itraconazole–repaglinide, rifampicin–repaglinide, or clarithromycin–repaglinide DDIs. The lower panel illustrates DDI AUC ratios (c) and DDI Cmax ratios (d) of the gemfibrozil–pioglitazone, itraconazole–pioglitazone, gemfibrozil–itraconazole–pioglitazone, or rifampicin–pioglitazone DDIs. The colors represent different perpetrator drugs and the symbols the victim drugs repaglinide (dots) and pioglitazone (triangles). The straight black line marks the line of identity. Light grey lines indicate 0.8- to 1.25-fold and dark grey lines indicate 0.5- to 2-fold acceptance limits. The curved black lines show the prediction success limits suggested by Guest et al. [41]. Detailed information about dosing regimens and study populations is given in Electronic Supplementary Material (ESM) Tables S4.2.1, S4.3.1, S4.4.1, S4.5.1, S4.6.1, S4.7.1, S4.8.1, S4.9.1, and S4.10.1. The plotted DDI AUC ratios and Cmax ratios are listed in ESM Tables S4.2.2, S4.3.2, S4.4.2, S4.5.2, S4.6.2, S4.7.2, S4.8.2, S4.9.2, and S4.10.2
3.3 Dose Adaptation Considerations
To show the utility of the models to individualize and improve drug therapy, dose adaptations for different DGI scenarios were calculated. Exemplarily, victim drug plasma concentration–time profiles in subjects with polymorphisms, and with polymorphisms during perpetrator drug coadministration, were simulated and compared to those of a CYP2C8 and SLCO1B1 wild-type individual without DDI (control). Then, potential dose adaptations were calculated, aiming to match control exposure (i.e., AUC in steady state). Figure 6 shows the different simulated scenarios, the extrapolated iso-exposure doses (% of control), simulations of plasma concentration–time profiles with the same dose for all individuals, and simulations of plasma concentration–time profiles with the extrapolated adjusted doses.

Dose adjustments developed with the physiologically based pharmacokinetic models for repaglinide (upper panel) and pioglitazone (lower panel). Predicted plasma concentration–time profiles are shown for European male CYP2C8 and SLCO1B1 wild-type individuals (red lines) as well as for CYP2C8*3/*3 or SLCO1B1 521CC individuals (repaglinide: green lines, pioglitazone: blue lines) before or during perpetrator drug coadministration. The left-hand plots show predicted plasma concentrations without dose adjustment; the right-hand plots show predicted plasma concentrations with dose adjustment. conc concentration, CYP cytochrome P450, n.a. not applicable, SLCO solute carrier organic anion transporter family member
As expected, the worst-case scenario is the repaglinide administration to a patient carrying the CYP2C8 wild-type sequence and simultaneously homozygously the SLCO1B1 521C allele who is co-medicated with gemfibrozil and itraconazole, leading to a 49-fold increase in the repaglinide steady state AUC. According to the simulations, a repaglinide dose reduction by nearly 98% would be necessary to produce the same drug exposure in this individual as in the control person given the normal dose. A dose adjustment to simultaneously match both AUC and Cmax values to the control profiles was not possible, as the drug half-life is changed due to the polymorphisms and the DDIs. The safety and efficacy of increased or decreased Cmax values or the drug half-life following dose adaptations that were calculated to match the AUC in steady state cannot be predicted from the models.
4 Discussion
In this study, whole-body PBPK models of gemfibrozil (parent–metabolite model of gemfibrozil and gemfibrozil 1-O-β-glucuronide), repaglinide, and pioglitazone for the investigation and prediction of DDIs and DGIs have been successfully built and evaluated. All models reliably describe and predict plasma concentration–time profiles over a broad dosing range and for single- and multiple-dose administration. Their good performance has been demonstrated by (1) comparison of predicted to observed plasma concentration–time profiles; (2) goodness-of-fit plots; (3) the calculation of MRD values; (4) the comparison of predicted to observed AUC and Cmax values; (5) the calculation of model GMFEs; and (6) their good predictions within a CYP2C8 PBPK DDI modeling network.
The itraconazole, rifampicin, and clarithromycin PBPK models applied in this study have been evaluated in a previously described CYP3A4 DDI network [9, 22]. This network has now been expanded with the presented gemfibrozil, repaglinide, and pioglitazone models and their interactions via CYP2C8, OATP1B1, OATP1B3, and CYP3A4. To use the previously established itraconazole, rifampicin, and clarithromycin models for CYP2C8 and OATP1B1/1B3 DDI modeling, interaction constants describing their induction and inhibition of CYP2C8, OATP1B1, and OATP1B3 have been added, but otherwise no drug- or system-dependent parameters have been changed. To model the induction of CYP2C8 by rifampicin, the maximum effect value (Emax CYP2C8 = 3.2, ESM Table S3.7.1) was taken from literature [23]. Concerning the induction of OATP1B1 and OATP1B3 by rifampicin, only a modest and variable induction was found in primary human hepatocytes [24, 25]. The Emax OATP1B1 = 0.38 (≙ 1.38-fold) identified during the rifampicin model development [9] was assumed to be adequate to model the induction of both OATP1B1 and OATP1B3. Except for the hydroxy-itraconazole OATP1B1 and OATP1B3 Ki values (optimized, see ESM Sect. 4.4), all interaction parameter values were taken from previous experimental or modeling studies.
During the modeling of the complex DDIs with simultaneous administration of the two perpetrator drugs gemfibrozil and itraconazole, the clinical data show effects that cannot be explained with the expected CYP enzyme or drug transporter inhibition [19]. First, a reduction of itraconazole and hydroxy-itraconazole AUCs was described during coadministration of itraconazole and pioglitazone if gemfibrozil is added to this regimen as the third drug [19]. As itraconazole and hydroxy-itraconazole are both solely metabolized by CYP3A4 and gemfibrozil is not reported to affect CYP3A4, we propose the possibility of a physicochemical DDI between gemfibrozil and itraconazole. Simultaneous administration of gemfibrozil might further decrease the poor solubility of itraconazole, causing a decrease in absorption and thereby a decrease of the plasma concentrations of itraconazole and hydroxy-itraconazole. To test this hypothesis with the PBPK models, a reduction of itraconazole solubility during gemfibrozil coadministration was performed (from 14.5 to 0.69 mg/L), leading to a good description of the itraconazole exposure of this study (ESM Figs. S3.6.1 and S3.6.2). The phenomenon of decreased itraconazole plasma concentrations during co-treatment with gemfibrozil has been discussed by Jaakkola et al. [19] and Niemi et al. [20]. They proposed a displacement of itraconazole from plasma proteins by gemfibrozil as a possible explanation, which was also tested with our models but did not describe the observed effects, or an effect of gemfibrozil on itraconazole bioavailability, which is in full agreement with our hypothesis of reduced itraconazole solubility. This example demonstrates the great value of perpetrator drug plasma concentration assessment in clinical DDI studies.
Furthermore, a reduction of the pioglitazone AUC was observed during coadministration of gemfibrozil and pioglitazone if itraconazole was added to this regimen as the third drug [19]. As itraconazole alone has no effect on the pioglitazone pharmacokinetics (ESM Fig. S4.5.1), the effect of the combination of gemfibrozil and itraconazole on the pioglitazone plasma concentrations is expected to be similar to the effect of gemfibrozil alone (2.5-fold increase of AUC, no change of Cmax). Surprisingly, during the combination of all three drugs, the pioglitazone AUC is increased only 2.2-fold and the Cmax is reduced to 75% of the Cmax in control conditions. To explain the considerable decrease of the pioglitazone Cmax, we again postulate a physicochemical DDI decreasing the solubility of pioglitazone and thereby its absorption and plasma concentrations. To test this hypothesis applying the PBPK models, a reduction of pioglitazone solubility during gemfibrozil plus itraconazole coadministration was performed (from 16.8 to 1.59 mg/L), leading to a good description of the pioglitazone exposure during this complex DDI (ESM Fig. S4.7.1). These two examples illustrate that PBPK modeling is a valuable tool to develop and test hypotheses for unexpected clinical findings. Furthermore, they certainly raised our awareness of the possibility of solubility interactions in the gut, given the great number of poorly soluble drugs. These solubility interactions could be mediated either by the coadministered drugs themselves or by coadministered solubilizing agents that are used in the marketed formulations of Biopharmaceutics Classification System (BCS) class II drugs. However, these hypotheses need to be further investigated experimentally to verify or reject them. Additionally, inhibition of transporters or enzymes by excipients has been described in literature [26]. A change in bioavailability due to such an interaction cannot be excluded.
In addition to prediction of DDIs, the presented PBPK models of repaglinide and pioglitazone adequately describe the effects of the CYP2C8*3 and SLCO1B1 521C alleles on repaglinide pharmacokinetics and the effect of the CYP2C8*3 allele on pioglitazone pharmacokinetics. Considering the 13% frequency of the CYP2C8*3 allele [27] and 14.3% frequency of the SLCO1B1 521T>C SNP in Caucasians [28], these polymorphisms are clinically relevant. With our models, a dose adjustment in case of complex DDGIs and drug–drug–drug–gene interactions was exemplarily performed to show how PBPK modeling could support drug therapy and labeling for complex scenarios.
5 Conclusion
In summary, comprehensive whole-body PBPK models of gemfibrozil (parent–metabolite model), repaglinide, and pioglitazone have been carefully built and evaluated in a CYP2C8 DDI network. The network described in this study represents an extension of a previously developed network of CYP3A4-interacting drugs [9], and the new models were challenged and verified in DDI predictions with different perpetrator and victim drugs, causing different kinds of interactions such as competitive inhibition, mechanism-based inactivation, and induction. Furthermore, the presented models are able to describe DGIs of repaglinide and pioglitazone, complex DDIs during coadministration of more than two interacting drugs, and were used to postulate DDIs on a physicochemical level. All models are transparently documented and model files are available in the OSP repository. They can be applied to investigate the DDI or DGI potential of drugs, inform the design of clinical trials, or simulate complex interactions (e.g., drug–drug–drug–gene interactions).
References
Laatikainen O, Sneck S, Bloigu R, Lahtinen M, Lauri T, Turpeinen M. Hospitalizations due to adverse drug events in the elderly—a retrospective register study. Front Pharmacol. 2016;7:358.
Qato DM, Wilder J, Schumm LP, Gillet V, Alexander GC. Changes in prescription and over-the-counter medication and dietary supplement use among older adults in the United States, 2005 vs 2011. JAMA Intern Med. 2016;176:473–82.
Lau SWJ, Schlender JF, Abernethy DR, Burckart GJ, Golden A, Slattum PW, et al. Improving therapeutics to better care for older adults and the young: report from the American College of Clinical Pharmacology workshop. J Clin Pharmacol. 2017;58:277–80.
Jamei M. Recent advances in development and application of physiologically-based pharmacokinetic (PBPK) models: a transition from academic curiosity to regulatory acceptance. Curr Pharmacol Rep. 2016;2:161–9.
European Medicines Agency. Guideline on the investigation of drug interactions. London: EMA; 2015. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions_en.pdf. Accessed 1 Mar 2019.
U.S. Food and Drug Administration. Clinical drug interaction studies—study design, data analysis, and clinical implications. Draft Guidance for Industry. 2017. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-drug-interaction-studies-study-design-data-analysis-and-clinical-implications-guidance. Accessed 1 Mar 2019.
Sager JE, Yu J, Ragueneau-Majlessi I, Isoherranen N. Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab Dispos. 2015;43:1823–37.
U.S. Food and Drug Administration. Drug development and drug interactions: table of substrates, inhibitors and inducers. 2017. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm. Accessed 19 Oct 2018.
Hanke N, Frechen S, Moj D, Britz H, Eissing T, Wendl T, et al. PBPK models for CYP3A4 and P-gp DDI prediction: a modeling network of rifampicin, itraconazole, clarithromycin, midazolam, alfentanil, and digoxin. CPT Pharmacometrics Syst Pharmacol. 2018;7:647–59.
PK-Sim® Ontogeny Database Documentation, Version 7.3. 2017. https://github.com/Open-Systems-Pharmacology/OSPSuite.Documentation/blob/master/PK-Sim%20Ontogeny%20Database%20Version%207.3.pdf. Accessed 1 Mar 2019.
Ogilvie BW, Zhang D, Li W, Rodrigues AD, Gipson AE, Holsapple J, et al. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: implications for drug-drug interactions. Drug Metab Dispos. 2006;34:191–7.
Kimoto E, Li R, Scialis RJ, Lai Y, Varma MVS. Hepatic disposition of gemfibrozil and its major metabolite gemfibrozil 1-O-β-glucuronide. Mol Pharm. 2015;12:3943–52.
Ménochet K, Kenworthy KE, Houston JB, Galetin A. Use of mechanistic modeling to assess interindividual variability and interspecies differences in active uptake in human and rat hepatocytes. Drug Metab Dispos. 2012;40:1744–56.
Honkalammi J, Niemi M, Neuvonen PJ, Backman JT. Dose-dependent interaction between gemfibrozil and repaglinide in humans: strong inhibition of CYP2C8 with subtherapeutic gemfibrozil doses. Drug Metab Dispos. 2011;39:1977–86.
Bidstrup TB, Bjørnsdottir I, Sidelmann UG, Thomsen MS, Hansen KT. CYP2C8 and CYP3A4 are the principal enzymes involved in the human in vitro biotransformation of the insulin secretagogue repaglinide. Br J Clin Pharmacol. 2003;56:305–14.
Jaakkola T, Laitila J, Neuvonen PJ, Backman JT. Pioglitazone is metabolised by CYP2C8 and CYP3A4 in vitro: potential for interactions with CYP2C8 inhibitors. Basic Clin Pharmacol Toxicol. 2006;99:44–51.
Itkonen MK, Tornio A, Neuvonen M, Neuvonen PJ, Niemi M, Backman JT. Clopidogrel markedly increases plasma concentrations of CYP2C8 substrate pioglitazone. Drug Metab Dispos. 2016;44:1364–71.
Taupitz T, Dressman JB, Buchanan CM, Klein S. Cyclodextrin-water soluble polymer ternary complexes enhance the solubility and dissolution behaviour of poorly soluble drugs. Case example: itraconazole. Eur J Pharm Biopharm. 2013;83:378–87.
Jaakkola T, Backman JT, Neuvonen M, Neuvonen PJ. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics of pioglitazone. Clin Pharmacol Ther. 2005;77:404–14.
Niemi M, Backman JT, Neuvonen M, Neuvonen PJ. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia. 2003;46:347–51.
Tsume Y, Amidon GL, Takeuchi S. Dissolution effect of gastric and intestinal pH for a BCS class II drug, pioglitazone: new in vitro dissolution system to predict in vivo dissolution. J Bioequiv Availab. 2013;5:224–7.
Moj D, Hanke N, Britz H, Frechen S, Kanacher T, Wendl T, et al. Clarithromycin, midazolam, and digoxin: application of PBPK modeling to gain new insights into drug-drug interactions and co-medication regimens. AAPS J. 2017;19:298–312.
Buckley DB, Wiegand CM, Prentiss PL, Fahmi OA. Time-course of cytochrome P450 (CYP450) induction in cultured human hepatocytes: evaluation of activity and mRNA expression profiles for six inducible CYP450 enzymes [poster no. P186]. 10th International ISSX Meeting; 29 Sep–3 Oct 2013; Toronto.
Dixit V, Hariparsad N, Li F, Desai P, Thummel KE, Unadkat JD. Cytochrome P450 enzymes and transporters induced by anti-human immunodeficiency virus protease inhibitors in human hepatocytes: implications for predicting clinical drug interactions. Drug Metab Dispos. 2007;35:1853–9.
Williamson B, Dooley KE, Zhang Y, Back DJ, Owen A. Induction of influx and efflux transporters and cytochrome P450 3A4 in primary human hepatocytes by rifampin, rifabutin, and rifapentine. Antimicrob Agents Chemother. 2013;57:6366–9.
Zou L, Ni Z, Tsakalozou E, Giacomini KM. Impact of pharmaceutical excipients on oral drug absorption: a focus on intestinal drug transporters. Clin Pharmacol Ther. 2019;105:323–5.
Dai D, Zeldin DC, Blaisdell JA, Chanas B, Coulter SJ, Ghanayem BI, et al. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics. 2001;11:597–607.
Nies AT, Niemi M, Burk O, Winter S, Zanger UM, Stieger B, et al. Genetics is a major determinant of expression of the human hepatic uptake transporter OATP1B1, but not of OATP1B3 and OATP2B1. Genome Med. 2013;5:1.
Honkalammi J, Niemi M, Neuvonen PJ, Backman JT. Mechanism-based inactivation of CYP2C8 by gemfibrozil occurs rapidly in humans. Clin Pharmacol Ther. 2011;89:579–86.
Backman JT, Honkalammi J, Neuvonen M, Kurkinen KJ, Tornio A, Niemi M, et al. CYP2C8 activity recovers within 96 hours after gemfibrozil dosing: estimation of CYP2C8 half-life using repaglinide as an in vivo probe. Drug Metab Dispos. 2009;37:2359–66.
Tornio A, Niemi M, Neuvonen M, Laitila J, Kalliokoski A, Neuvonen PJ, et al. The effect of gemfibrozil on repaglinide pharmacokinetics persists for at least 12 h after the dose: evidence for mechanism-based inhibition of CYP2C8 in vivo. Clin Pharmacol Ther. 2008;84:403–11.
Hatorp V, Huang WC, Strange P. Repaglinide pharmacokinetics in healthy young adult and elderly subjects. Clin Ther. 1999;21:702–10.
Kalliokoski A, Backman JT, Kurkinen KJ, Neuvonen PJ, Niemi M. Effects of gemfibrozil and atorvastatin on the pharmacokinetics of repaglinide in relation to SLCO1B1 polymorphism. Clin Pharmacol Ther. 2008;84:488–96.
Niemi M, Leathart JB, Neuvonen M, Backman JT, Daly AK, Neuvonen PJ. Polymorphism in CYP2C8 is associated with reduced plasma concentrations of repaglinide. Clin Pharmacol Ther. 2003;74:380–7.
Deng LJ, Wang F, De Li H. Effect of gemfibrozil on the pharmacokinetics of pioglitazone. Eur J Clin Pharmacol. 2005;61:831–6.
Manitpisitkul P, Curtin CR, Shalayda K, Wang SS, Ford L, Heald D. Pharmacokinetic interactions between topiramate and pioglitazone and metformin. Epilepsy Res. 2014;108:1519–32.
Tornio A, Niemi M, Neuvonen PJ, Backman JT. Trimethoprim and the CYP2C8*3 allele have opposite effects on the pharmacokinetics of pioglitazone. Drug Metab Dispos. 2007;36:73–80.
Jaakkola T, Backman JT, Neuvonen M, Laitila J, Neuvonen PJ. Effect of rifampicin on the pharmacokinetics of pioglitazone. Br J Clin Pharmacol. 2006;61:70–8.
Niemi M, Backman JT, Neuvonen M, Neuvonen PJ, Kivistö KT. Rifampin decreases the plasma concentrations and effects of repaglinide. Clin Pharmacol Ther. 2000;68:495–500.
Niemi M, Neuvonen PJ, Kivistö KT. The cytochrome P4503A4 inhibitor clarithromycin increases the plasma concentrations and effects of repaglinide. Clin Pharmacol Ther. 2001;70:58–65.
Guest EJ, Aarons L, Houston JB, Rostami-Hodjegan A, Galetin A. Critique of the two-fold measure of prediction success for ratios: application for the assessment of drug-drug interactions. Drug Metab Dispos. 2011;39:170–3.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This project has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under Grant agreement no. 668353. Matthias Schwab was supported by the Robert Bosch Stiftung, Stuttgart, Germany.
Conflict of Interest
Sebastian Frechen, Thomas Eissing, and Thomas Wendl are employees and potential shareholders of Bayer AG. No potential conflicts of interest were disclosed by the other authors (Denise Türk, Nina Hanke, Sarah Wolf, Matthias Schwab, and Thorsten Lehr).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary document:
Model information and evaluation (PDF 17255 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Türk, D., Hanke, N., Wolf, S. et al. Physiologically Based Pharmacokinetic Models for Prediction of Complex CYP2C8 and OATP1B1 (SLCO1B1) Drug–Drug–Gene Interactions: A Modeling Network of Gemfibrozil, Repaglinide, Pioglitazone, Rifampicin, Clarithromycin and Itraconazole. Clin Pharmacokinet 58, 1595–1607 (2019). https://doi.org/10.1007/s40262-019-00777-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00777-x