
Abstract
Vedolizumab is a humanized anti-α4β7 integrin monoclonal antibody that selectively blocks trafficking of memory T cells to inflamed gut tissue by inhibiting the α4β7-mucosal addressin cell adhesion molecule-1 (MAdCAM-1) interaction. Approved for treating patients with moderately to severely active ulcerative colitis (UC) or Crohn’s disease (CD), vedolizumab is administered as a 300 mg intravenous infusion. Vedolizumab undergoes a rapid, saturable, non-linear, target-mediated elimination process at low concentrations and a slower, linear, non-specific elimination process at higher concentrations. At therapeutic concentrations, vedolizumab primarily undergoes linear elimination. From population pharmacokinetic modeling, the vedolizumab terminal elimination half-life (t ½ β) was estimated to be 25.5 days; linear clearance (CLL) was similar for patients with UC (0.159 L/day) and CD (0.155 L/day). Extreme low albumin concentrations and extreme high body weight values were potentially clinically important predictors of vedolizumab CLL. Other factors, including concomitant therapy use (methotrexate, azathioprine, mercaptopurine, or aminosalicylates) or prior tumor necrosis factor-α (TNF-α) antagonist use, had no clinically relevant effects on CLL. A positive exposure–efficacy relationship for clinical remission and clinical response was apparent for vedolizumab induction therapy in patients with UC or CD. On average, patients with higher albumin, lower fecal calprotectin (UC only), lower C-reactive protein (CD only), and no prior TNF-α antagonist use had a higher probability of remission. Off drug, 10% of patients with UC or CD were positive for anti-drug antibodies. This article provides a comprehensive review of the clinical pharmacokinetics, pharmacodynamics, exposure–efficacy relationships, and immunogenicity of vedolizumab.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Vedolizumab is a gut-selective α4β7 integrin antagonist that blocks adhesion of memory T cells to mucosal addressin cell adhesion molecule-1 (MAdCAM-1), thereby decreasing infiltration of these inflammatory cells into gut mucosal tissue and suppressing gut inflammation. |
Vedolizumab undergoes a rapid, saturable, non-linear, target-mediated elimination process at low concentrations and a slower, linear, non-specific elimination process at higher concentrations. At therapeutic concentrations, vedolizumab primarily undergoes linear elimination. |
Vedolizumab linear clearance (CLL) was similar in patients with ulcerative colitis (UC) and patients with Crohn’s disease (CD). The terminal elimination half-life (t ½ β) of vedolizumab is 25.5 days. Only extreme low albumin concentrations (<3.2 g/dL) and extreme high body weight values (>120 kg) were identified as potential clinically important predictors of vedolizumab CLL. |
Positive exposure–efficacy relationships for clinical remission and clinical response were evident for vedolizumab induction therapy, which appeared to be steeper in patients with UC than in patients with CD. |
Off drug, 10% of patients with UC or CD were positive for anti-drug antibodies (ADAs). Patients who were persistently ADA positive during treatment (positive at two or more consecutive visits) had decreased vedolizumab trough serum concentrations. |
1 Introduction
The inflammatory bowel diseases ulcerative colitis (UC) and Crohn’s disease (CD) are chronic diseases that are characterized by exacerbated inflammatory cell infiltrate into gut mucosal tissue [1, 2]. Currently available treatment options for patients with UC or CD include glucocorticosteroids, immunosuppressants (i.e., azathioprine, mercaptopurine, or methotrexate), tumor necrosis factor-α (TNF-α) antagonists, and, most recently, integrin antagonists [3,4,5]. Vedolizumab (previous versions were known as LDP-02, MLN-02, and MLN0002) is a recombinant monoclonal antibody that binds specifically to the α4β7 integrin, thereby modulating the trafficking of memory T cells into inflamed gut mucosal tissue [6, 7]. Vedolizumab is indicated for the treatment of adult patients with moderately to severely active UC or CD and is administered as a 300 mg intravenous (IV) infusion over approximately 30 min at 0, 2, and 6 weeks and every 8 weeks thereafter [7, 8]. The European prescribing information suggests that some patients with UC or CD who have experienced a decrease in their response to vedolizumab may benefit from an increase in dosing frequency to every 4 weeks [8]. The clinical profile of vedolizumab is consistent with its binding specificity, with evidence of therapeutic benefit in patients with UC or CD [9,10,11,12,13] and a lack of systemic immunosuppression [14, 15]. The aim of this article is to review the clinical pharmacokinetics, pharmacodynamics, exposure–efficacy relationships, and immunogenicity of vedolizumab.
2 Vedolizumab Structure
Vedolizumab is a recombinant humanized immunoglobulin G1 (IgG1) monoclonal antibody produced in Chinese hamster ovary cells [7]. Vedolizumab was generated by fusing the binding domains from the mouse anti-human α4β7 monoclonal antibody Act-1 to a conventional human IgG1 scaffold. In addition, two mutations were introduced into the Fc region of vedolizumab, which eliminated some Fc-mediated functions such as cytotoxicity [16]. Vedolizumab is composed of two light chains of the kappa subclass and two heavy chains linked by two disulfide bridges, forming a Y-structure that is typical of immunoglobulins. The molecular weight of vedolizumab is approximately 147 kDa [7].
3 Vedolizumab Mechanism of Action
A significant body of biochemical, preclinical, and clinical evidence supports the gut-selective anti-inflammatory mechanism of action of vedolizumab, which has been reviewed in detail elsewhere [17]. Vedolizumab binds specifically to the α4β7 integrin heterodimer, but not to the functionally distinct α4β1 or αEβ7 integrin heterodimers, thereby inhibiting the binding of α4β7-expressing cells to mucosal addressin cell adhesion molecule-1 (MAdCAM-1) and fibronectin, but not to vascular cell adhesion molecule-1 (VCAM-1) [6]. This inhibitory effect is reversible on removal of vedolizumab; binding of peripheral blood lymphocytes to MAdCAM-1 was partially restored within 24 h and fully restored after 4 days [16]. Although moderately expressed on several other types of leukocytes, such as B cells, eosinophils, and natural killer cells, the α4β7 integrin is preferentially expressed on a subset of memory CD4+ T cells and mediates binding of these cells to MAdCAM-1 expressed on endothelial cells [6, 18, 19]. The α4β7–MAdCAM-1 interaction is required for lymphocyte trafficking to the gut, and it is key to the pathology of inflammatory bowel diseases. By binding to the α4β7 integrin, vedolizumab selectively inhibits adhesion of memory T cells to MAdCAM-1 and the migration of these cells to the site of gut inflammation [6]. Vedolizumab does not activate lymphocytes, affect cytokine production by differentiated T cells, or lyse target cells by complement-dependent cytotoxicity or antibody-dependent cell-mediated cytotoxicity [16].
Blockade of the α4β7 integrin by vedolizumab has been shown to elicit gut-selective anti-inflammatory effects in primates [20, 21]. The gut selectivity of vedolizumab has also been confirmed in clinical studies. In a vaccination study in healthy volunteers, vedolizumab attenuated the response to an enteral antigen challenge (oral cholera vaccine) but did not affect the response to a parenteral antigen challenge (intramuscular hepatitis B vaccine) [14]. In contrast to natalizumab, vedolizumab had no effect on T cell trafficking to the central nervous system in preclinical and clinical studies, providing further evidence of its gut selectivity [15, 22,23,24].
4 Vedolizumab Pharmacokinetic Properties
The pharmacokinetic properties of vedolizumab were evaluated in single-dose studies in healthy volunteers and in multiple-dose studies in patients with UC or CD [11,12,13, 25,26,27,28]. In these studies, vedolizumab serum concentrations were measured using a validated sandwich enzyme-linked immunosorbent assay (ELISA), as previously described [25]. The lower limit of quantification of this assay was 1.25 ng/mL (0.125 µg/mL in undiluted serum) [25]. Pharmacokinetic parameters were determined using standard non-compartmental methods.
A population pharmacokinetic analysis was also performed to assess the effects of patient demographics and other covariates on the pharmacokinetics of vedolizumab in patients with UC or CD using extensively collected data from phase I and II studies and sparsely collected data from the phase III GEMINI studies [29]. A repeated-measures analysis was conducted using a non-linear mixed effects modeling (NONMEM) approach.
4.1 Single-Dose Pharmacokinetics in Healthy Volunteers
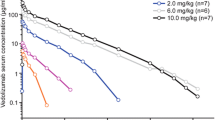
Vedolizumab was administered as a single IV infusion to healthy volunteers on a body weight-adjusted basis over the dose range of 0.2–10.0 mg/kg in a phase I study [25]. After reaching peak concentrations, vedolizumab serum concentrations declined in a generally linear fashion until concentrations reached approximately 10 µg/mL (Fig. 1). Thereafter, concentrations appeared to decline in a non-linear fashion. Vedolizumab maximum observed serum concentration (C max) was dose proportional over the dose range tested (Table 1). Greater than dose-proportional increases in the vedolizumab area under the serum concentration–time curve (AUC) from time zero to infinity (AUC0−∞) and dose-dependent decreases in clearance (CL) and increases in terminal elimination half-life (t ½ β) were observed from 0.2 to 2.0 mg/kg (Table 1). At dose levels >2.0 mg/kg, the increases in AUC0−∞ were dose proportional and CL and t ½ did not change. These data suggest that a rapid elimination process becomes saturated as the vedolizumab dose increases, and slower linear elimination processes likely account for a large fraction of vedolizumab CL at higher concentrations.

Figure reproduced from Rosario et al. [25] (© Springer International Publishing Switzerland 2016), with permission of Springer
Mean vedolizumab serum concentrations over time after a single intravenous infusion in healthy volunteers. Individuals who were persistently anti-drug antibody positive (≥2 positive samples or 1 positive sample with a titer of ≥25) were excluded from the analysis
Vedolizumab was also administered as a single IV infusion to healthy volunteers on a fixed-dose basis over the dose range of 180–750 mg. Comparison of the data across these studies revealed that vedolizumab exposure increased in a dose-proportional manner at doses >180 mg (Table 2). The apparent linear pharmacokinetics at doses >180 mg (equivalent to 2.4 mg/kg in a 75 kg individual) is consistent with the data from the healthy volunteer study with body weight-adjusted (mg/kg) dosing.
4.2 Multiple-Dose Pharmacokinetics in Patients with Ulcerative Colitis (UC) or Crohn’s Disease (CD)
The pharmacokinetics of vedolizumab following repeat IV infusions were evaluated in patients with UC or CD over the dose range of 0.5–10 mg/kg in phase II studies [26,27,28]. In patients with UC, vedolizumab concentrations increased with increasing dose over the range of 2–10 mg/kg, after an IV infusion on days 1, 15, 29, and 85 [27]. After the last dose on day 85, vedolizumab concentrations decreased in an approximately bi-exponential fashion. The pharmacokinetics of vedolizumab were dose proportional over the 2–10 mg/kg dose range based on C max and AUC during the dosing interval (AUCτ) after dosing on days 1 and 85 (Table 3).
The pharmacokinetics of vedolizumab in patients with UC or CD were also assessed in the phase III GEMINI studies using a sparse (population-based) sampling scheme. During GEMINI 1 and GEMINI 2, patients received a 300 mg IV infusion of vedolizumab at weeks 0, and 2, (induction phase) and every 4 or 8 weeks thereafter starting at week 6 (maintenance phase) [11, 12]. During GEMINI 3, patients received a 300 mg IV infusion of vedolizumab at weeks 0, 2, and 6 (induction only) [13]. At week 6, mean vedolizumab trough serum concentrations were similar in patients with UC in GEMINI 1 and in patients with CD in GEMINI 2 and GEMINI 3 (Table 4). During the maintenance phases of both GEMINI 1 and GEMINI 2, the last vedolizumab trough concentration was measured at week 46 and was used to represent the end-of-treatment, steady-state trough concentration. At week 46, the mean vedolizumab trough concentration was higher in patients who received vedolizumab every 4 weeks than in patients who received vedolizumab every 8 weeks in both studies (Table 4).
The median trough serum concentration–time profile for each treatment regimen during the maintenance phase of GEMINI 1 and GEMINI 2 has been previously reported [29]. Patients who received two doses of vedolizumab during induction and placebo during maintenance had measurable vedolizumab trough concentrations in serum until week 38.
4.3 Population Pharmacokinetic Model
From the population pharmacokinetic analysis, the pharmacokinetics of vedolizumab were best described by a two-compartment model with zero-order input and parallel linear and non-linear elimination, similar to that described for other therapeutic monoclonal antibodies [29, 30]. The non-linear elimination process was modeled using a Michaelis–Menten equation. The overall inter-individual variability was moderate to large [29]. The effects of intrinsic and extrinsic factors on the pharmacokinetics of vedolizumab were investigated using a full covariate modeling approach [29, 31]. Inferences about the clinical relevance of parameters were based on the resulting parameter estimates and measures of estimation precision [Bayesian 95% credible intervals (CDIs)] from the final model. Covariate effect sizes on linear CL (CLL) of less than ±25% from the typical population value, when evaluated over a representative range of covariate values, were not considered clinically relevant changes [29]. Pharmacokinetic parameter estimates derived from the model are described in Sects. 4.4–4.6, as are the effects of covariates on vedolizumab CLL.
4.4 Distribution
The vedolizumab volume of distribution at steady state (V ss) after a single IV infusion of 300 mg in healthy volunteers was estimated to be 4.49 L [percent coefficient of variation (%CV) 14.3%] (Table 2). From the final population pharmacokinetic model, vedolizumab V ss was estimated to be 4.84 L for both patients with UC and patients with CD [volume of distribution in the central compartment (V c) = 3.19 L plus volume of distribution in the peripheral compartment (V p) = 1.65 L] [29]. The V ss values estimated for vedolizumab are approximately equal to the volume of plasma in humans and are generally similar to those reported for other therapeutic monoclonal antibodies [32]. After a single IV infusion of vedolizumab 450 mg in healthy volunteers, vedolizumab was not detected in the cerebrospinal fluid, suggesting that vedolizumab does not penetrate the blood–brain barrier [15].
4.5 Elimination
Because of their large molecular size, therapeutic monoclonal antibodies are not directly excreted in urine; rather they are degraded to peptides and amino acids that are either reused by the body or excreted by the kidneys [32]. Several mechanisms are reportedly involved in the elimination of monoclonal antibodies, including proteolytic degradation by the liver or reticuloendothelial system, target-mediated elimination, and non-specific endocytosis [32]. Vedolizumab undergoes a rapid, saturable, non-linear, target-mediated elimination process at low concentrations and a slower, linear, non-specific elimination process at higher concentrations. From the final population pharmacokinetic model, the t ½ of vedolizumab was estimated to be 25.5 days, which only accounts for elimination through the linear process [29]. Estimates of t ½ obtained from non-compartmental analyses in the phase I and phase II studies account for elimination through both linear and non-linear processes (Tables 1, 2) and, therefore, are slightly lower than the estimate obtained from population pharmacokinetic modeling. Population estimates of CLL were 0.159 L/day for patients with UC and 0.155 L/day for patients with CD [29]. Non-linear elimination was described by a maximum elimination rate (V max) of 0.265 mg/day and concentration at half V max (K m) of 0.964 µg/mL [29]. The relationship between non-linear, linear, and total CL values across a range of vedolizumab concentrations is depicted in Fig. 2. At therapeutic concentrations, as indicated by trough concentration at steady state from dosing every 8 weeks (>10 μg/mL) [7], vedolizumab is eliminated primarily through the linear pathway.

Relationship between linear clearance (CLL), non-linear clearance (CLNL), and total clearance (CLtotal) of vedolizumab
4.6 Impact of Demographic and Other Covariates on Vedolizumab Pharmacokinetics
4.6.1 Disease Type
The population pharmacokinetic analysis investigated population differences in vedolizumab CLL and V c for patients with UC or CD [29]. Population estimates of CLL were similar for patients with UC and for patients with CD, as described in Sect. 4.5. The results also indicated that V c did not appreciably differ between the two diseases (point estimate of 1.01 and 95% CDI of 0.989–1.03) [29].
4.6.2 Sex, Age, and Race
Sex differences in pharmacokinetics have been reported for other therapeutic monoclonal antibodies. In general, volume of distribution and/or CL of monoclonal antibodies increase with body size and, thus, are higher in men than in women [33, 34]. No clinical study to date has directly assessed the effect of sex on vedolizumab pharmacokinetics. From the population pharmacokinetic model, sex was determined not to be a clinically relevant covariate after adjusting for other predictors of vedolizumab CLL; the estimates of CLL and V c were only approximately 10 and 6% lower, respectively in female patients than male patients [29].
No dedicated clinical study has assessed the effect of age on the pharmacokinetics of vedolizumab. However, population pharmacokinetic modeling indicated that age had no clinically relevant effect on vedolizumab CLL within the age range tested (18–78 years) (point estimate of −0.0346 and 95% CDI of −0.0788 to 0.01) [29].
The impact of race on vedolizumab pharmacokinetics has not been directly evaluated in a clinical study or by population pharmacokinetic modeling. From a cross-study comparison, the pharmacokinetic parameters observed in Japanese patients with UC after a 150 or 300 mg IV infusion of vedolizumab were similar to those in Western (non-Hispanic White) patients with UC after a 2–10 mg/kg dose (~150–750 mg) [27, 35].
4.6.3 Body Weight
Body weight has often been reported to be a predictor of volume of distribution and/or CL of therapeutic monoclonal antibodies, although its effect is not always considered clinically relevant. During the clinical development program, vedolizumab was administered initially on a body weight-adjusted (mg/kg) basis and subsequently on a fixed-dose basis (mg). The impact of body weight on the pharmacokinetics of vedolizumab was first assessed in a phase I study in healthy volunteers. The results of this study revealed that weight-adjusted dosing overcompensated for exposure of vedolizumab in individuals with higher weight, suggesting that weight-adjusted dosing may have little advantage over fixed doses (unpublished data). Therefore, fixed doses of vedolizumab were used in all subsequent studies, including in the phase III GEMINI studies.
The effect of body weight on vedolizumab CLL was further assessed through population pharmacokinetic modeling [29]. An allometric relationship was included in the model to describe the relationship between body weight and CLL. From this analysis, body weight was determined to be a statistically significant predictor of vedolizumab CLL, with a point estimate of 0.362 and a narrow 95% CDI of 0.299–0.427. Body weight did not appear to have a clinically relevant effect on CLL for individuals weighing <120 kg. However, the typical individual weighing 120 kg had a 19% probability of having a CLL value >25% higher than that of a patient weighing 70 kg (reference patient; albumin of 4.0 g/dL for both cases), an effect that is considered of potential clinical relevance [29].
4.6.4 Disease Activity
The effect of disease activity on the pharmacokinetics of vedolizumab was assessed through population modeling using the partial Mayo score and the Crohn’s Disease Activity Index (CDAI) score at baseline. Both covariates had no effect on vedolizumab CLL (point estimate of −0.0515 and 95% CDI of −0.14 to 0.0358 for CDAI score; point estimate of 0.0408 and 95% CDI of −0.0336 to 0.115 for partial Mayo score) [29].
For patients with UC from GEMINI 1, median measured vedolizumab trough serum concentrations at week 6 were lower in patients with higher endoscopic subscores at week 6 (more severe disease) [36]. Patients with a endoscopic score of 3, indicating severe disease, had a median trough concentration (19.6 µg/mL) below the overall median concentration (25.6 µg/mL) at week 6. A trend toward greater estimated vedolizumab CLL in patients with a higher endoscopic subscore was noted (Fig. 3) [29]. These findings are consistent with those for other therapeutic monoclonal antibodies used for the treatment of UC and CD [37, 38]. It is possible that the apparent association between higher endoscopic subscores and faster vedolizumab CLL at week 6 is not causal and could be attributed to several confounding factors, such as additional drug loss into the inflamed gastrointestinal tract (drug-losing enteropathy) [39]. The apparent relationship between vedolizumab CLL and the endoscopic subscore warrants further investigation.

(reproduced from Rosario et al. [29])
Estimated individual vedolizumab linear clearance (CLL) by Mayo endoscopic subscore at week 6 in patients with ulcerative colitis (GEMINI 1). Midlines represent medians; box limits represent the 25th and 75th percentiles; whiskers (error bars) represent the 10th and 90th percentiles; and solid circles represent datapoints outside the 10th to 90th percentile range
4.6.5 Fecal Calprotectin, C-Reactive Protein, and Albumin
The effects of fecal calprotectin and C-reactive protein (CRP) concentrations on vedolizumab CLL were evaluated in the population pharmacokinetic model as additional measures of disease severity [29]. For fecal calprotectin, the estimated effect size, although statistically significant, was small and not considered to be clinically relevant (point estimate of 0.0310 and 95% CDI of 0.0252–0.0365). An exploratory post hoc analysis suggested that, after adjusting for other predictors of vedolizumab CLL, the effect of CRP on vedolizumab CLL was also not clinically relevant and accounted for <1% of the unexplained inter-individual variability in CL [29].
Albumin has been demonstrated to be a predictor of the CL of other therapeutic monoclonal antibodies [40]. From the population pharmacokinetic model, albumin concentration was a statistically significant predictor of vedolizumab CLL, with a point estimate of −1.18 and a narrow 95% CDI of −1.24 to −1.13 [29]. The clinical relevance of the effect of albumin on CLL was assessed. Vedolizumab CLL for the typical individual with an albumin concentration of 3.2 g/dL was approximately 30% higher than that of a patient with an albumin concentration of 4 g/dL (reference patient; body weight of 70 kg in both cases), an effect that is considered of potential clinical relevance.
4.6.6 Prior Tumor Necrosis Factor-α Antagonist Therapy
From the population pharmacokinetic model, patients who had been treated with and failed prior TNF-α antagonist therapy had 4% faster CLL than patients who were naïve to TNF-α antagonist therapy (point estimate of 1.04 and 95% CDI of 1.01–1.07) [29]. Although statistically significant, this modest difference is not considered clinically relevant.
4.6.7 Anti-Drug Antibodies
In the GEMINI 1 and GEMINI 2 studies, the persistence of anti-drug antibodies (ADAs) during treatment (measurable ADA titers at two or more consecutive visits) was associated with a substantial decrease in vedolizumab serum concentrations [41]. In GEMINI 1, among the six patients who were persistently ADA positive, three patients had vedolizumab trough concentrations at steady state (week 46) that were below the limit of quantitation (BLQ; <0.125 µg/mL in undiluted serum), one patient had a trough concentration of 4.17 μg/mL, one patient had a trough concentration of 3.46 μg/mL, and one patient had a missing pharmacokinetic blood sample. In GEMINI 2, among the three patients who were persistently ADA positive, two patients had vedolizumab trough concentrations at week 46 that were BLQ and one patient had a missing sample.
From the population pharmacokinetic model, vedolizumab CLL was predicted to be 12% higher in patients who were ADA positive (either persistently or transiently) than in patients who were ADA negative (point estimate of 1.12 and 95% CDI of 1.05–1.2) [29]. Although statistically significant, this effect was not considered to be clinically relevant. However, the small number of ADA-positive patients in this analysis limit the interpretation of this result.
4.7 Drug–Drug Interactions
No clinical studies have directly assessed the impact of vedolizumab administration on the pharmacokinetics of other drugs. Classic drug–drug interactions mediated through effects on enzyme systems such as the cytochrome P450 (CYP) system are generally not expected for therapeutic monoclonal antibodies. However, monoclonal antibodies that are cytokine modulators may modify the metabolism of drugs that are CYP enzyme substrates through their effects on the regulation pathways of CYP enzymes [32, 42, 43]. Since vedolizumab is gut selective and does not modulate systemic cytokine production [16], the potential for CYP-mediated drug interactions with vedolizumab acting as the perpetrator is considered to be lower than that with drugs that systemically and directly affect cytokines.
Published data show that small-molecule immunomodulators may affect the CL of therapeutic monoclonal antibodies, possibly through an attenuating effect on immunogenicity [32, 42, 44, 45]. No clinical studies have been conducted to directly assess the effect of concomitant therapy on the pharmacokinetics of vedolizumab. However, the effect of concomitant immunomodulator (methotrexate, azathioprine, mercaptopurine) and aminosalicylate therapy on vedolizumab CLL was assessed through population pharmacokinetic modeling. The effect of each therapy was assessed individually in the model. The findings suggest that these drugs had no clinically relevant effect on vedolizumab CLL, in contrast to the apparent effect of immunomodulators on the immunogenicity of vedolizumab [29, 46].
5 Vedolizumab Pharmacokinetic–Pharmacodynamic Relationship
The extent of α4β7 integrin binding by vedolizumab on peripheral blood memory T cells has been evaluated in clinical studies using two flow cytometry binding interference assays. These assays used either the mouse monoclonal antibody Act-1, from which the binding domains of vedolizumab were derived, or MAdCAM-1, an endogenous ligand of α4β7 integrin. Act-1 was the only binding saturation biomarker used in early clinical studies, whereas a MAdCAM-1–murine Fc fusion (MAdCAM-1-Fc) was used in later clinical studies. Both assays rely on the same principle and have been described in detail elsewhere [47]. In contrast to the Act-1 assay, vedolizumab-neutralizing ADAs do not interfere with the ability of the MAdCAM-1 assay to detect vedolizumab binding to the α4β7 integrin [47].
5.1 Phase I and II Studies
In a single-dose phase I study in healthy volunteers, administration of vedolizumab over the dose range of 0.2–10.0 mg/kg resulted in rapid and near complete inhibition of Act-1 and MAdCAM-1-Fc binding to α4β7 at all timepoints at which vedolizumab was measurable in serum [25]. Once vedolizumab concentrations were no longer detectable, the inhibition of binding returned to approximately baseline levels. The duration of α4β7 receptor saturation was dose dependent.
Saturation of the α4β7 receptor was also evaluated following repeat IV infusions in patients with UC or CD over the dose range of 0.5–10.0 mg/kg in phase II studies [26,27,28]. After administration of vedolizumab 0.5 or 2.0 mg/kg on days 1 and 29 to patients with UC, saturation of α4β7 receptors on >90% of peripheral blood memory T cells was evident at both weeks 4 and 6, as determined using the Act-1 assay [26]. In a second study in patients with UC, administration of vedolizumab 2, 6, or 10 mg/kg on days 1, 15, 29, and 85 resulted in rapid and near-maximal inhibition of MAdCAM-1-Fc binding to α4β7 at all doses and timepoints at which vedolizumab was measurable in serum (Fig. 4), consistent with the single-dose healthy volunteer study [27]. MAdCAM-1-Fc binding returned to approximately baseline levels when vedolizumab concentrations were no longer detectable. The duration of α4β7 receptor saturation was dose dependent. In patients with CD, near complete saturation of α4β7 receptors was observed after administration of vedolizumab 0.5 or 2.0 mg/kg on days 1 and 29 [28]. After the dose on day 29, complete recovery of free α4β7 was evident by day 85 for the 0.5 mg/kg group and by day 180 for the 2.0 mg/kg group.

Mean percentage of CD4+CD45ROhigh memory T cells staining positive for MAdCAM-1-Fc over time after repeat intravenous infusions in patients with ulcerative colitis. Patients who were persistently anti-drug antibody positive (≥2 positive samples or 1 positive sample with a titer of ≥25) were excluded from the analysis. MAdCAM-1-Fc mucosal addressin cell adhesion molecule-1-murine Fc fusion. Figure adapted from Parikh et al. [27] ((c)Wolters Kluwer Health, Inc.), with permission of Wolters Kluwer Health, Inc
5.2 Population Pharmacokinetic–Pharmacodynamic Model
In the GEMINI 1 and GEMINI 2 studies, α4β7 receptor saturation was measured using the MAdCAM-1-Fc assay predose at weeks 0, 6, and 52. Data from these studies were pooled with data from phase I and II studies and analyzed using population pharmacokinetic–pharmacodynamic modeling [29]. The inhibition of MAdCAM-1–α4β7 binding by vedolizumab was described by a direct-effect sigmoid maximum effect (E max) model. No formal covariate modeling or model evaluation was conducted. From this model, the vedolizumab serum concentration at half-maximum effect (EC50) was estimated to be 0.093 μg/mL, suggesting that complete receptor saturation was reached at a vedolizumab serum concentration of approximately 1 μg/mL, a concentration considered subtherapeutic [29]. Exposure–efficacy data at week 6 indicate that vedolizumab concentrations of ≤17.1 µg/mL in patients with UC in GEMINI 1 and ≤16 µg/mL in patients with CD in GEMINI 2 during induction were associated with clinical remission rates similar to placebo [48]. These observations support the hypothesis that receptor saturation may be necessary but not sufficient for clinical efficacy of vedolizumab. The discrepancy between the vedolizumab concentrations required for complete receptor saturation and efficacy may be related to the fact that the MAdCAM-1-Fc assay measures α4β7 saturation on circulating T cells (as opposed to T cells in inflamed mucosal tissue). However, for another monoclonal antibody that selectively binds to the β7 subunit of the α4β7 and αEβ7 integrins, the drug serum concentration required for maximal saturation of β7 receptors on peripheral T cells was sufficient for maximal occupancy of β7 receptors in the colonic mucosa, but maximal receptor occupancy did not correlate with clinical remission [49]. The apparent lack of correlation between receptor occupancy and clinical efficacy for α4β7 and αEβ7 integrin antagonists requires further study. The MAdCAM-1 assay demonstrates α4β7 saturation from 1 µg/mL, suggesting that receptor saturation in peripheral blood lymphocytes is required for clinical efficacy but cannot achieve it alone [50].
6 Vedolizumab Exposure–Response Relationships
6.1 Patients with UC
The exposure–efficacy relationships of vedolizumab in patients with UC from GEMINI 1 have been evaluated by grouping the vedolizumab trough (predose) concentrations in quartiles and calculating the associated rates of clinical outcomes. Any confounding factors for baseline covariates were assessed using logistic regression [11, 48]. In these analyses, clinical remission was defined as a complete Mayo score ≤2 points with no individual subscore >1 point; clinical response was defined as a decrease in complete Mayo score ≥3 points and a ≥30% decrease from baseline, with a decrease in rectal bleeding subscore ≥1 point or an absolute rectal bleeding subscore ≤1 point; and mucosal healing was defined as a Mayo endoscopic subscore ≤1 point [11].
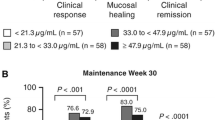
The quartile analyses revealed a positive exposure–response relationship for clinical remission, clinical response, and mucosal healing for vedolizumab induction therapy in patients with UC [11, 48]. Vedolizumab concentrations >17.1 µg/mL at week 6 were associated with higher rates of clinical remission than observed with placebo (Fig. 5a). From concentration quartile 1 to quartile 4, the absolute rate increase was approximately 31% for clinical remission, 34% for clinical response, and 43% for mucosal healing at week 6 (Fig. 5a) [48]. Consistent with these data, Mayo endoscopic scores were lower in patients with higher vedolizumab trough concentrations [36].

Percentage of patients with clinical remission by vedolizumab trough concentration quartile at week 6 or week 10 in patients with ulcerative colitis (GEMINI 1) (a) and patients with Crohn’s disease [GEMINI 2 (b) and GEMINI 3 (c, d)]. For GEMINI 1 and GEMINI 2, analyses included all patients who received vedolizumab during induction. Data for placebo-treated patients are shown for reference. CD Crohn’s disease, Q quartile, UC ulcerative colitis. Figure reproduced from Rosario et al. [48] ((c) Oxford University Press), with permission of Oxford University Press
To account for possible confounding of the unadjusted analyses by baseline covariates, the exposure–efficacy relationships were further explored using logistic regression. Results from the logistic regression analysis confirmed the positive exposure–efficacy relationships for clinical remission and response at week 6 [48]. On average, patients with higher baseline albumin concentrations, lower baseline fecal calprotectin concentrations, and no prior TNF-α antagonist use had a higher probability of clinical remission and response whether they were receiving placebo or vedolizumab. Of these covariates, prior TNF-α antagonist use had the largest effect on the probability of efficacy; the probability of clinical remission at week 6 was approximately 10% higher for patients who were naïve to TNF-α antagonists than for patients who had not responded to this treatment. Vedolizumab concentration was determined to be the strongest factor affecting clinical remission and response rates in patients with UC. After adjusting for confounding factors, increasing the cumulative average concentration through week 6 (C average) from 35 to 84 µg/mL was associated with a 15% increase in the probability of clinical remission and a 20% increase in the probability of clinical response at week 6.
Consistent with positive exposure–efficacy relationships in patients with UC, the median vedolizumab trough serum concentration at week 6 was higher in remitters [34.7 (95% confidence interval (CI) 31.7–36.6) µg/mL] than in non-remitters [23.7 (22.0–24.8) µg/mL] and also in responders [29.0 (27.3–31.7) µg/mL] than in non-responders [21.5 (20.1–23.3) µg/mL], although some overlap in concentrations was observed between the subgroups [48].
6.2 Patients with CD
The exposure–efficacy relationships of vedolizumab in patients with CD in the GEMINI 2 and GEMINI 3 studies were also assessed by analyzing the clinical remission and response rates by trough concentration quartile. Similar to the GEMINI 1 analysis, any confounding factors for baseline covariates were assessed using logistic regression [12, 25]. Clinical remission was defined as a CDAI score ≤150 points, and clinical response was defined as a decrease from baseline in CDAI score ≥70 points.
From the quartile analyses, a modest, positive exposure–response relationship was evident for vedolizumab induction therapy, which was shallower than that observed for patients with UC. Vedolizumab concentrations >16.0 µg/mL in GEMINI 2 and >17.1 µg/mL in GEMINI 3 at week 6 were associated with higher rates of clinical remission than those observed with placebo (Fig. 5b, c). From concentration quartile 1 to quartile 4, the absolute rate increase for clinical remission was approximately 14% in GEMINI 2 and only approximately 5% in GEMINI 3 at week 6 (Fig. 5b, c) [48]. However, at week 10, the absolute rate increase from quartile 1 to quartile 4 was approximately 22% for clinical remission in GEMINI 3 (Fig. 5d). These results may be a reflection of the fact that a higher percentage of patients with prior TNF-α antagonist failure were enrolled in GEMINI 3 than in GEMINI 2 (76 versus 58%) [12, 13], possibly representing a more treatment-refractory group of patients who may need a greater exposure of vedolizumab or more time to achieve remission than TNF-α antagonist-naïve patients.
From the logistic regression analysis, which accounted for baseline covariate effects, patients from GEMINI 2 with higher baseline albumin concentrations, lower CRP concentrations, and no prior TNF-α antagonist use, on average, had a higher probability of clinical remission and response whether they were receiving placebo or vedolizumab [48]. Similar to the results for patients with UC, prior TNF-α antagonist use had the largest effect on the probability of efficacy (10% higher probability of clinical remission for treatment-naïve patients). After adjusting for covariates, increasing C average from 32 to 85 µg/mL corresponded to an approximate 10% increase in the probability of clinical remission and 15% increase in the probability of clinical response at week 6.
Consistent with these results, the median vedolizumab trough concentration at week 6 was higher in remitters [26.8 (95% CI 24.9–30.1) µg/mL] than in non-remitters [23.5 (22.4–24.8) µg/mL] and also in responders [26.8 (25.2–27.7) µg/mL] than in non-responders [22.1 (20.5–23.4) µg/mL] in GEMINI 2 [48]. However, the overlap in concentrations between the subgroups was greater than for patients with UC from GEMINI 1.
Several factors may have contributed to the shallower exposure–efficacy relationships apparent in patients with CD than in patients with UC. Clinical remission rates were higher at week 10 than at week 6 in GEMINI 2 and GEMINI 3, particularly for patients who had previously not responded to TNF-α antagonist therapy [13, 51]. This observation is reflected in the European prescribing information, which states that patients with CD who have not responded to vedolizumab therapy by week 6 may be given an additional dose at week 10 before being assessed for an induction response at week 14 [8]. The exposure–efficacy relationships may have been steeper for patients with CD had a timepoint later than week 6 been evaluated. The steeper exposure–efficacy relationship for clinical remission evident for patients from GEMINI 3 at week 10 than at week 6 supports this hypothesis [48].
7 Vedolizumab Immunogenicity
The immunogenicity of vedolizumab was characterized throughout the clinical development program using screening and titration assays followed by a neutralizing assay. The screening assay was a validated bridging assay designed to detect the presence of ADAs in serum. In the presence of vedolizumab, low-titer responses were often undetectable [1 μg/mL of drug interfered with low and moderate levels of the control rabbit anti-vedolizumab antibodies (AVAs)]. For this reason, in the phase III GEMINI studies, a two-prong approach was taken to ensure all individuals with AVAs were identified. First, the assay was run using two dilutions (1:5 and 1:50) and both had to be negative to be considered negative in the assay. The dilutions were used to increase the likelihood of an interaction between the detecting reagent and the AVA, and not the drug. The second approach was to take a sample at a timepoint where drug levels had cleared below interference levels, i.e., samples were also taken when the patients were off drug. After the initial screening assay, positive samples were confirmed with a titration assay. In the GEMINI studies, samples confirmed as ADA positive were further examined for their ability to neutralize vedolizumab activity.
7.1 Rate of Anti-Drug Antibody Development
In the GEMINI 1 and GEMINI 2 combined population, 56 of 1434 (4%) patients who were treated continuously with vedolizumab for up to 52 weeks were ADA positive at any time during treatment (including the final safety visit); nine of these patients (0.6%) were persistently positive (positive at two or more consecutive visits) and 33 (2.3%) developed neutralizing antibodies [52]. None of the nine persistently positive patients achieved clinical remission at weeks 6 or 52 [7]. As frequently occurs with monoclonal antibodies, the rate of immunogenicity during vedolizumab treatment may have been underestimated. Consistent with this possibility, among the 320 patients who were treated continuously with vedolizumab for up to 52 weeks in GEMINI 1 and GEMINI 2 and who had data available at the final safety visit (week 66), 32 (10%) patients were ADA positive when off drug [52]. While there was an increase in the overall immunogenicity rate off drug compared with on drug, when we examined the impact of the immunogenicity using either the 4% population or the 10–15% population, there was no significant difference between the populations with or without AVAs, suggesting that the impact of immunogenicity was adequately captured.
The increase in the immunogenicity rate off drug compared with on drug suggests that low- to moderate-titer AVAs may have been present and unobservable. However, when comparing the results of the impact of AVAs on clinical safety, efficacy, and pharmacological parameters, only the presence of persistently positive samples resulted in any impact.
An effect of concomitant immunomodulator therapy on the rate of ADA development was evident in those patients who received two doses of vedolizumab during induction and then placebo during maintenance. In the subgroup of these patients who were using immunomodulators at baseline (regardless of other concomitant medication use), only one of 32 patients (3%) was ADA positive (persistently positive) versus 44 of 247 patients (18%) who were not using immunomodulators at baseline [30 patients (12%) were persistently positive] [41, 52]. The impact of immunomodulator coadministration on the rate of ADA development in patients who were administered vedolizumab continuously during induction and maintenance was less obvious, potentially due to a smaller percentage of patients who developed ADA in this group. A total of five of 161 patients (3%) using immunomodulators at baseline were ADA positive (none were persistently positive) versus 51 of 1273 patients (4%) not using immunomodulators at baseline [nine patients (1%) were persistently positive] [41, 52].
7.2 Safety Related to Anti-Drug Antibody Development
In the GEMINI 1 and GEMINI 2 studies, three of 61 patients (5%) who were treated with vedolizumab continuously for up to 52 weeks and who had an investigator-defined infusion-related reaction were persistently ADA positive. Three of the 56 patients (5%) who were persistently ADA positive in these studies experienced an infusion-related reaction [52].
8 Conclusions
The pharmacokinetic profile of vedolizumab is characterized by a rapid, saturable, non-linear elimination process at low concentrations, presumably attributed to target-mediated elimination via interaction with α4β7 integrin, and a slower, linear elimination process (non-specific elimination) at higher concentrations (>10 μg/mL). At therapeutic concentrations, vedolizumab is primarily eliminated through the linear pathway. The distribution pharmacokinetics of vedolizumab was best described by a two-compartment model. The t ½ of vedolizumab was estimated to be 25.5 days. The pharmacokinetic properties of vedolizumab were similar in patients with UC and patients with CD. Of the covariates tested, extreme low serum albumin (<3.2 g/dL) and extreme high body weight (>120 kg) values were identified as potential clinically important predictors of vedolizumab CLL.
Vedolizumab reversibly and maximally inhibited MAdCAM-1-Fc binding to α4β7 integrin on peripheral blood memory T cells at all doses and timepoints at which vedolizumab was measurable in serum. Once vedolizumab was no longer detectable in serum, the inhibition of MAdCAM-1-Fc binding returned to baseline levels.
Positive exposure–efficacy relationships for vedolizumab induction therapy were evident for clinical remission and clinical response in patients with UC or CD. The relationships were steeper for patients with UC than for patients with CD. On average, patients with higher albumin, lower fecal calprotectin (UC only), lower CRP (CD only), and no prior TNF-α antagonist use had a higher probability of remission. Prior TNF-α antagonist use had the largest effect on the probability of efficacy in both diseases (10% higher probability of clinical remission for treatment-naïve patients). After adjusting for covariates, an increase in C average from 32 to 85 µg/mL corresponded to increases of approximately 15 and 10% in the probability of clinical remission in patients with UC and patients with CD, respectively. No strong conclusions can be drawn from the exposure–efficacy relationship because no dose-ranging studies have been performed. However, some patients may benefit from an extra dose at week 10. The GEMINI 1 and 2 studies reported greater than 95% α4β7 saturation on memory T cells in peripheral blood [11, 12], placing doubt regarding the viability of therapeutic drug monitoring for vedolizumab [50]. A proposal for a therapeutic window for vedolizumab can only be addressed and validated by a prospective study.
The rate of ADA development off drug was 10%. During treatment, a meaningful decrease in vedolizumab exposure was only observed in patients who were persistently ADA positive.
References
Ordás I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet. 2012;380(9853):1606–19.
Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012;380(9853):1590–605.
Bryant RV, Brain O, Travis SP. Conventional drug therapy for inflammatory bowel disease. Scand J Gastroenterol. 2015;50(1):90–112.
Cheifetz AS. Management of active Crohn disease. JAMA. 2013;309(20):2150–8.
Danese S, Fiorino G, Peyrin-Biroulet L, Lucenteforte E, Virgili G, Moja L, et al. Biological agents for moderately to severely active ulcerative colitis: a systematic review and network meta-analysis. Ann Intern Med. 2014;160(10):704–11.
Soler D, Chapman T, Yang LL, Wyant T, Egan R, Fedyk ER. The binding specificity and selective antagonism of vedolizumab, an anti-α4β7 integrin therapeutic antibody in development for inflammatory bowel diseases. J Pharmacol Exp Ther. 2009;330(3):864–75.
ENTYVIO (vedolizumab) package insert. Deerfield: Takeda Pharmaceuticals America, Inc.; 2014.
ENTYVIO (vedolizumab) summary of product characteristics. Taastrup: Takeda Pharma A/S; 2016.
Dulai PS, Mosli M, Khanna R, Levesque BG, Sandborn WJ, Feagan BG. Vedolizumab for the treatment of moderately to severely active ulcerative colitis. Pharmacotherapy. 2015;35(4):412–23.
Garnock-Jones KP. Vedolizumab: a review of its use in adult patients with moderately to severely active ulcerative colitis or Crohn’s disease. BioDrugs. 2015;29(1):57–67.
Feagan BG, Rutgeerts P, Sands BE, Hanauer S, Colombel JF, Sandborn WJ, GEMINI 1 Study Group, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369(8):699–710.
Sandborn WJ, Feagan BG, Rutgeerts P, Hanauer S, Colombel JF, Sands BE, GEMINI 2 Study Group, et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2013;369(8):711–21.
Sands BE, Feagan BG, Rutgeerts P, Colombel JF, Sandborn WJ, Sy R, et al. Effects of vedolizumab induction therapy for patients with Crohn’s disease in whom tumor necrosis factor antagonist treatment failed. Gastroenterology. 2014;147(3):618–27.
Wyant T, Leach T, Sankoh S, Wang Y, Paolino J, Pasetti MF, et al. Vedolizumab affects antibody responses to immunisation selectively in the gastrointestinal tract: randomised controlled trial results. Gut. 2015;64(1):77–83.
Milch C, Wyant T, Xu J, Parikh A, Kent W, Fox I, et al. Vedolizumab, a monoclonal antibody to the gut homing α4β7 integrin, does not affect cerebrospinal fluid T-lymphocyte immunophenotype. J Neuroimmunol. 2013;264(1–2):123–6.
Wyant T, Yang L, Fedyk E. In vitro assessment of the effects of vedolizumab binding on peripheral blood lymphocytes. MAbs. 2013;5(6):842–50.
Wyant T, Fedyk E, Abhyankar B. An overview of the mechanism of action of the monoclonal antibody vedolizumab. J Crohns Colitis. 2016;10(12):1437–44.
Berlin C, Berg EL, Briskin MJ, Andrew DP, Kilshaw PJ, Holzmann B, et al. α4β7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 1993;74(1):185–95.
Wang C, Hanly EK, Wheeler LW, Kaur M, McDonald KG, Newberry RD. Effect of α4β7 blockade on intestinal lymphocyte subsets and lymphoid tissue development. Inflamm Bowel Dis. 2010;16(10):1751–62.
Hesterberg PE, Winsor-Hines D, Briskin MJ, Soler-Ferran D, Merrill C, Mackay CR, et al. Rapid resolution of chronic colitis in the cotton-top tamarin with an antibody to a gut-homing integrin α4β7. Gastroenterology. 1996;111(5):1373–80.
Fedyk ER, Wyant T, Yang LL, Csizmadia V, Burke K, Yang H, et al. Exclusive antagonism of the α4β7 integrin by vedolizumab confirms the gut-selectivity of this pathway in primates. Inflamm Bowel Dis. 2012;18(11):2107–19.
Haanstra KG, Hofman SO, Lopes Estêvão DM, Blezer EL, Bauer J, Yang LL, et al. Antagonizing the α4β1 integrin, but not α4β7, inhibits leukocytic infiltration of the central nervous system in rhesus monkey experimental autoimmune encephalomyelitis. J Immunol. 2013;190(5):1961–73.
Stüve O, Marra CM, Bar-Or A, Niino M, Cravens PD, Cepok S, et al. Altered CD4+/CD8+ T-cell ratios in cerebrospinal fluid of natalizumab-treated patients with multiple sclerosis. Arch Neurol. 2006;63(10):1383–7.
Stüve O, Marra CM, Jerome KR, Cook L, Cravens PD, Cepok S, et al. Immune surveillance in multiple sclerosis patients treated with natalizumab. Ann Neurol. 2006;59(5):743–7.
Rosario M, Wyant T, Leach T, Sankoh S, Scholz C, Parikh A, et al. Vedolizumab pharmacokinetics, pharmacodynamics, safety, and tolerability following administration of a single, ascending, intravenous dose to healthy volunteers. Clin Drug Investig. 2016;36(11):913–23.
Feagan BG, Greenberg GR, Wild G, Fedorak RN, Paré P, McDonald JW, et al. Treatment of ulcerative colitis with a humanized antibody to the α4β7 integrin. N Engl J Med. 2005;352(24):2499–507.
Parikh A, Leach T, Wyant T, Scholz C, Sankoh S, Mould DR, et al. Vedolizumab for the treatment of active ulcerative colitis: a randomized controlled phase 2 dose-ranging study. Inflamm Bowel Dis. 2012;18(8):1470–9.
Feagan BG, Greenberg GR, Wild G, Fedorak RN, Paré P, McDonald JW, et al. Treatment of active Crohn’s disease with MLN0002, a humanized antibody to the α4β7 integrin. Clin Gastroenterol Hepatol. 2008;6(12):1370–7.
Rosario M, Dirks NL, Gastonguay MR, Fasanmade AA, Wyant T, Parikh A, et al. Population pharmacokinetics-pharmacodynamics of vedolizumab in patients with ulcerative colitis and Crohn’s disease. Aliment Pharmacol Ther. 2015;42(2):188–202.
Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(10):633–59.
Gastonguay MR. Full covariate models as an alternative to methods relying on statistical significance for inferences about covariate effects: a review of methodology and 42 case studies [poster]. In: 20th Annual Meeting of the Population Approach Group in Europe, 7–10 Jun 2011, Athens.
Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493–507.
Ordás I, Mould DR, Feagan BG, Sandborn WJ. Anti-TNF monoclonal antibodies in inflammatory bowel disease: pharmacokinetics-based dosing paradigms. Clin Pharmacol Ther. 2012;91(4):635–46.
Ternant D, Aubourg A, Magdelaine-Beuzelin C, Degenne D, Watier H, Picon L, et al. Infliximab pharmacokinetics in inflammatory bowel disease patients. Ther Drug Monit. 2008;30(4):523–9.
Kobayashi K, Suzuki Y, Watanabe K, Mukae M, Yamada A, Yamagami H, et al. A phase I study of vedolizumab, humanized antibody to α4β7 integrin in patients with ulcerative colitis in Japan [poster]. In Japan Digestive Disease Week, 9–12 Oct 2013, Tokyo.
Rosario M, Abhyankar B, Sankoh S, Dirks NL, Lasch K, Sandborn WJ. Relationship between vedolizumab pharmacokinetics and endoscopic outcomes in patients with ulcerative colitis [poster]. In: Congress of European Crohn’s and Colitis Organisation, 18–21 Feb 2015, Barcelona.
Brandse JF, Mathât RA, van der Kleij D, Rispens T, Ashruf Y, Jansen JM, et al. Pharmacokinetic features and presence of antidrug antibodies associate with response to infliximab induction therapy in patients with moderate to severe ulcerative colitis. Clin Gastroenterol Hepatol. 2016;14(2):251–8.
Colombel JF, Sandborn WJ, Allez M, Dupas JL, Dewit O, D’Haens G, et al. Association between plasma concentrations of certolizumab pegol and endoscopic outcomes of patients with Crohn’s disease. Clin Gastroenterol Hepatol. 2014;12(3):423–31.
Brandse JF, van den Brink GR, Wildenberg ME, van der Kleij D, Rispens T, Jansen JM, et al. Loss of infliximab into feces is associated with lack of response to therapy in patients with severe ulcerative colitis. Gastroenterology. 2015;149(2):350–5.
Fasanmade AA, Adedokun OJ, Olson A, Strauss R, Davis HM. Serum albumin concentration: a predictive factor of infliximab pharmacokinetics and clinical response in patients with ulcerative colitis. Int J Clin Pharmacol Ther. 2010;48(5):297–308.
Rosario M, Fox IH, Milch C, Parikh A, Feagan BG, Sandborn WJ, et al. Pharmacokinetic/pharmacodynamic relationship and immunogenicity of vedolizumab in adults with inflammatory bowel disease: additional results from GEMINI 1 and 2 [poster]. In: Crohn’s and Colitis Foundation Advances in Inflammatory Bowel Diseases, 12–14 Dec 2013, Hollywood.
Seitz K, Zhou H. Pharmacokinetic drug-drug interaction potentials for therapeutic monoclonal antibodies: reality check. J Clin Pharmacol. 2007;47(9):1104–18.
Ferri N, Bellosta S, Baldessin L, Boccia D, Racagni G, Corsini A. Pharmacokinetics interactions of monoclonal antibodies. Pharmacol Res. 2016;111:592–9.
Velagapudi RB, Noertersheuser PA, Awni WM. Effect of methotrexate (MTX) coadministration on pharmacokinetics (PK) of adalimumab (HUMIRA, Abbott) following a single intravenous (iv) injection. Arthritis Rheum. 2003;48(9 Suppl):S141.
Zhuang Y, Xu Z, Frederick B, de Vries DE, Ford J, Keen M. Golimumab pharmacokinetics after repeated subcutaneous and intravenous administration in patients with rheumatoid arthritis and the effect of concomitant methotrexate: an open-label, randomized study. Clin Ther. 2012;34(1):77–90.
Earp J, Fang L, Ma L, Wang YM, Rosario MC, Dirks NL, et al. Assessing labeling claims for drug interactions using a population PK approach: vedolizumab. J Pharmacokinet Pharmacodyn. 2014;41(Suppl 1):S39.
Wyant T, Estevam J, Yang L, Rosario M. Development and validation of receptor occupancy pharmacodynamic assays used in the clinical development of the monoclonal antibody vedolizumab. Cytometry B Clin Cytom. 2016;90(2):168–76.
Rosario M, French J, Dirks N, Sankoh S, Parikh A, Yang H, et al. Exposure–efficacy relationships for vedolizumab induction therapy in patients with ulcerative colitis or Crohn’s disease. J Crohns Colitis. 2017. doi:10.1093/ecco-jcc/jjx021.
Vermeire S, O’Byrne S, Keir M, Williams M, Lu TT, Mansfield JC, et al. Etrolizumab as induction therapy for ulcerative colitis: a randomised, controlled, phase 2 trial. Lancet. 2014;384(9940):309–18.
Dreesen E, Gils A. Blocking the α4β7 integrin through vedolizumab: necessary but not sufficient? J Crohns Colitis. 2017. doi:10.1093/ecco-jcc/jjx033.
Sandborn WJ, Rutgeerts P, Xu J, Abhyankar B, Fox I. Efficacy of induction treatment with vedolizumab for patients with Crohn’s disease who have experienced tumour necrosis factor antagonist failure or are tumour necrosis factor antagonist naive [poster no. DOP073]. J Crohns Colitis. 2014;8(Suppl 1):S50.
Colombel JF, Sands BE, Rutgeerts P, Sandborn W, Danese S, D’Haens G, et al. The safety of vedolizumab for ulcerative colitis and Crohn’s disease. Gut. 2017;66(5):839–51.
Acknowledgements
The authors would like to thank Sanhita Abrol, Ph.D., for providing assistance with the figures.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The studies discussed in this review were funded by Takeda Pharmaceutical Company Limited. Medical writing assistance was provided by Elisabeth R. Wann, PhD, Wann Medical Communications, LLC, Wilmette, IL, USA, and was supported by Takeda Development Center Americas, Inc., Deerfield, IL, USA.
Conflict of interest
Maria Rosario, Catherine Milch, Asit Parikh, Michael Bargfrede, and Irving Fox are employees of Takeda Development Center Americas, Inc., Cambridge, MA, USA. Nathanael L. Dirks is an employee of Metrum Research Group LLC, Tariffville, CT, USA, and has been a paid consultant to Takeda. Timothy Wyant was an employee of Takeda Development Center Americas, Inc., Cambridge, MA, USA, at the time that this research was conducted. Eric Fedyk is an employee of Takeda Pharmaceuticals International, Inc., Deerfield, IL, USA.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rosario, M., Dirks, N.L., Milch, C. et al. A Review of the Clinical Pharmacokinetics, Pharmacodynamics, and Immunogenicity of Vedolizumab. Clin Pharmacokinet 56, 1287–1301 (2017). https://doi.org/10.1007/s40262-017-0546-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-017-0546-0