
Abstract
Background
Three comparative clinical studies assessed the pharmacokinetics (PK), pharmacodynamics (PD), immunogenicity and safety of PF-06881893 (filgrastim-aafi; Nivestym™), a filgrastim biosimilar, versus US-licensed reference product (filgrastim; US-Neupogen®) in healthy volunteers (HVs).
Methods
Two separate open-label, crossover-design PK/PD studies were conducted: a single-dose study (n = 24) and a multiple-dose study (n = 60). In each study, HVs were randomized to Nivestym followed by US-Neupogen, or vice versa. Study drug (5 μg/kg) was administered subcutaneously as a single injection or as five consecutive daily injections. Primary PK and PD endpoints were area under the filgrastim serum concentration–time curve, maximum observed concentration, area under the effect curve (AUEC) for absolute neutrophil count (ANC), maximum observed ANC, AUEC for cluster of differentiation (CD)-34+ count, and maximum observed CD34+ count. In an open-label, parallel-design, non-inferiority, comparative immunogenicity study, HVs were randomized (n = 128/treatment) to Nivestym or US-Neupogen. The primary endpoint was the proportion of subjects with a negative baseline antidrug antibody (ADA) test result and one or more confirmed post-dose positive ADA result.
Results
Overall demographics were as follows: female (n = 162/340); White (n = 274/340), Black (n = 58/340), and other (n = 8/340); age (18–65 years); and weight (50.8–96.5 kg). All primary PK and PD endpoints met the pre-specified criteria for PK and PD equivalence. The primary endpoint in the comparative immunogenicity study met pre-specified criteria for non-inferiority.
Conclusions
Nivestym demonstrated PK and PD equivalence in single and multiple subcutaneous-dose settings and non-inferiority for immunogenicity to US-Neupogen, with a comparable safety profile, supporting the demonstration of biosimilarity.
Trial registration
ClinicalTrials.gov C1121002 (NCT02766647); C1121003 (NCT02766634); C1121012 (NCT02923791).
Similar content being viewed by others
Single-dose and multiple-dose pharmacokinetics and pharmacodynamics of PF-06881893 (filgrastim-aafi; Nivestym™), a filgrastim biosimilar, were equivalent to those of the US-licensed reference product (filgrastim; US-Neupogen®) in healthy volunteers (HVs). The immunogenicity profile of Nivestym is non-inferior to that of US-Neupogen in HVs. |
There were no clinically meaningful differences in safety profiles between Nivestym and US-Neupogen. |
These findings support the demonstration of biosimilarity of Nivestym to US-Neupogen, leading to regulatory approval of Nivestym by the US FDA in July 2018. |
1 Introduction
Granulocyte colony-stimulating factor (G-CSF) promotes the survival, proliferation, differentiation, and maturation of neutrophil progenitor cells and the survival and functional stimulation of mature neutrophils [1]. Filgrastim is a recombinant methionyl human G-CSF (rhG-CSF), and licensed reference/originator filgrastim is marketed under the trade name Neupogen® in the USA and EU [2, 3]. Because of its effects on neutrophil progenitors and neutrophils, Neupogen is approved for use across multiple indications. Common indications for Neupogen in the USA and EU include patients with cancer who are receiving myelosuppressive chemotherapy or undergoing myeloablative chemotherapy followed by bone marrow transplant and patients with severe congenital, cyclic, or idiopathic neutropenia [2, 3]. In addition, Neupogen is approved for the mobilization of peripheral blood progenitor cells (PBPCs) [2, 3].
PF-06881893 (filgrastim-aafi; Nivestym™; Hospira Inc., a Pfizer Company, Lake Forest, IL, USA) was approved by the US FDA as biosimilar to US-licensed Neupogen (Amgen Inc, Thousand Oaks, CA, USA), hereafter referred to as US-Neupogen [4]. Biosimilarity is defined to mean “that the biological [biosimilar] product is highly similar to the reference product notwithstanding minor differences in clinically inactive components and that there are no clinically meaningful differences between the biological [biosimilar] product and the reference product in terms of the safety, purity, and potency of the product” [5]. The FDA recommends a stepwise approach to demonstrating biosimilarity that begins with comparative analytical (structural and functional) assessments of the proposed biosimilar and the reference product, followed by non-clinical testing (if necessary), and clinical studies [5].
Rigorous structural and functional characterization demonstrated fingerprint-like analytical similarity between Nivestym and US-Neupogen [6]. This analytical similarity assessment was completed using a comprehensive set of physicochemical and biological methods to fully characterize the molecule, including structure, product-related substances and impurities, protein concentration, isoelectric point, and sub-visible particle concentration [6]. Three methods were used to compare the biological activity of Nivestym versus US-Neupogen. A cell-based bioassay measured the ability of filgrastim products to cause proliferation of a neutrophil precursor cell line in a dose-dependent manner [6]. This measurement is directly relevant to filgrastim’s mechanism of action in inducing neutrophil production. A second bioassay measured the ability of Nivestym versus US-Neupogen to displace labeled filgrastim bound to rhG-CSF receptor [6]. Human G-CSF receptor engagement is necessary for the maturation and proliferation of neutrophil precursors, and for activation of function [7]. In addition, confirmation of similarity of binding kinetics and affinity for the interaction of filgrastim products with rhG-CSF receptor was determined using a highly specific and sensitive single-cycle kinetic surface plasmon resonance method [6]. Nivestym and US-Neupogen also demonstrated similar pharmacology and toxicology profiles in a non-clinical pharmacokinetic (PK)/toxicokinetic study in rats [6].
In line with FDA guidelines for demonstrating biosimilarity to a reference product, the clinical development program must include a comparative clinical pharmacology study and a clinical immunogenicity assessment, conducted as part of a comparative clinical study or as a comparative parallel-design immunogenicity study [5]. Clinical pharmacology studies should be conducted using a population, dose(s), and route of administration that are sensitive to detecting potential differences between the proposed biosimilar and the reference product, should they exist [5, 8]. Healthy volunteers (HVs) are considered to be the most sensitive population for evaluating similarity in a clinical pharmacology study because they are likely to exhibit less variability in PK and pharmacodynamic (PD) responses than patients with potential confounding factors (e.g., underlying and/or concomitant disease and concomitant medications) [8]. In addition, HVs are the most immunocompetent population, particularly in the setting of supportive therapy with leukocyte growth factors, where relevant patients may be immunocompromised because of disease state or myelosuppressive therapeutic intervention. Small differences in PK, PD, or immunogenicity responses (if present) would more likely be detected in HVs than in patients with underlying disease. Once biosimilarity has been established in HVs, biosimilarity to the reference product is expected in all patient populations indicated for use.
With the concurrence of the FDA, three clinical studies were conducted in HVs to assess PK/PD, immunogenicity, and safety of Nivestym and US-Neupogen following subcutaneous dose administration: a single-dose, crossover-design PK/PD equivalence study in support of the neutropenia indications; a multiple-dose, crossover-design PK/PD equivalence study in support of the PBPC mobilization indication; and a parallel-design, non-inferiority, comparative immunogenicity study. These clinical studies were performed as part of the overall US biosimilar development program for Nivestym and contribute to the totality of the evidence demonstrating biosimilarity of Nivestym to US-Neupogen.
2 Methods
2.1 Study Design
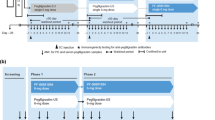
C1121002 (NCT02766647) was a randomized, open-label, single-dose, crossover-design PK/PD equivalence study in HVs conducted at a single center in the USA. The study consisted of a 21-day screening period followed by two 28-day treatment periods (TPs), TP1 and TP2, with ≥ 28 days’ washout between treatments (Fig. 1a). Eligible subjects were randomized 1:1 to one of two treatment sequence groups: Nivestym followed by US-Neupogen (sequence group 1) or US-Neupogen followed by Nivestym (sequence group 2). In each TP, subjects received a single 5 μg/kg dose of study drug as a subcutaneous injection in the deltoid region on day 1 and were confined to the study site from the evening before study drug administration through completion of scheduled study procedures on day 3. Thereafter, subjects returned to the study site on an outpatient basis for scheduled study assessments.

Nivestym biosimilar clinical development program in healthy volunteers. a C1121002: single-dose PK/PD study of Nivestym vs. US-Neupogena. b C1121003: multiple-dose PK/PD study of Nivestym vs. US-Neupogenb. c C1121012: comparative immunogenicity study of Nivestym vs. US-Neupogenc. aPK samples were collected 1 h before and 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 10, 12, 16, 24, and 48 h after dose administration in each TP. PD samples were collected 1 h before and 0.5, 1, 2, 4, 6, 8, 24, 48, 72, 96, and 120 h after dose administration in each TP. Immunogenicity samples were collected before dose administration on day 1 and on day 12 ± 2 days of each TP, on day 28 of TP1, and at the final visit (day 28 of TP2). Day − 1 is technically part of the screening period. However, there are day − 1 activities that are related to the TP. Therefore, in the study design, day − 1 is included in the TP rather than in the screening period. bPK samples were collected 1 h before dose administration on days 1–5 and at 0.5, 1, 2, 3, 4, 6, 9, 12, 16, and 24 h after dose administration on day 5 of each TP. PD samples were collected 1 h before dose administration on days 1-5 and at 24, 48, 72, 96, and 120 h after dose administration on day 5 of each TP. Immunogenicity samples were collected before dose administration on day 1 and on day 12 of each TP, on day 33 of TP1, and at the final visit (day 33 of TP2). Day − 1 is technically part of the screening period. However, there are day − 1 activities that are related to the TP. Therefore, in the study design, day − 1 is included in the TP rather than in the screening period. cImmunogenicity samples were collected on days 0 and 10 of each TP, on day 26 (± 1) of TP1, and at the final visit (day 31 ± 2 of TP2). PD pharmacodynamic, PK pharmacokinetic, SC subcutaneous, TP treatment period, US-Neupogen filgrastim reference product licensed in the USA
C1121003 (NCT02766634) was a randomized, open-label, multiple-dose, crossover-design PK/PD equivalence study in HVs conducted at a single center in the USA. The study consisted of a 14-day screening period followed by two 33-day TPs, TP1 and TP2, with ≥ 28 days’ washout between the last dose of TP1 and the first dose of TP2 (Fig. 1b). Eligible subjects were randomized 1:1 to sequence group 1 or sequence group 2, as in C1121002. In each TP, subjects received five consecutive daily 5 μg/kg doses of study drug as subcutaneous injections in the deltoid region over days 1–5 and were confined to the study site from the evening before study drug administration through completion of scheduled study procedures on day 6. Thereafter, subjects returned to the study site on an outpatient basis for scheduled study assessments.
Study C1121012 (NCT02923791) was a randomized, open-label, multiple-dose, parallel-design, non-inferiority, comparative immunogenicity study in HVs conducted at two centers in the USA. The study consisted of a 28-day screening period followed by two TPs, TP1 (26 ± 1 days) and TP2 (31 ± 2 days), with approximately 1 month of washout between day 1 of TP1 and day 1 of TP2 (Fig. 1c). Eligible subjects were randomized 1:1 to Nivestym or US-Neupogen and administered five consecutive daily 5 μg/kg doses of study drug as subcutaneous injections in the deltoid region over days 1–5 of TP1, with a single subcutaneous dose of the same study drug received in TP1 on day 1 of TP2.
2.2 Participants
Study entry criteria were consistent across studies, except as noted. Eligible participants included non-smoking male or non-pregnant/non-lactating female HVs, aged 18–65 years, with a body mass index of 19–30 kg/m2 and body weight of 50–100 kg (single-dose and multiple-dose PK/PD studies) or 50–95 kg (comparative immunogenicity study). Subjects with any active systemic or immunologic disease or condition (e.g., cardiovascular/pulmonary, hepatorenal, or systemic infection); hematologic laboratory abnormalities; clinically significant vital sign, chest X-ray (single-dose and multiple-dose PK/PD studies), or 12-lead electrocardiogram (ECG) abnormality; or inadequate hepatic or renal reserve, or a known history of glomerulonephritis (comparative immunogenicity study only) were excluded.
Additional key exclusion criteria included a history of splenic rupture or asplenia, pulmonary infiltrate or pneumonia, sickle-cell disease, chronic neutropenia, thrombocytopenia, vasculitis, drug or alcohol abuse, and biological growth factor exposure; recent blood loss or donation; hereditary fructose intolerance; drug sensitivity or known hypersensitivity to Escherichia coli-derived proteins and any rhG-CSF or product component thereof; and positive serology (HIV or hepatitis). Concurrent use of prescription or over-the-counter medications (excluding hormonal contraceptives and vitamins/calcium ≤ 100% daily value) was not permitted. Likewise, subjects were not permitted to consume alcohol-containing beverages throughout the duration of the PK/PD studies or for ≥ 48 h before day 1 and throughout days 1–5 of each TP in the comparative immunogenicity study.
2.3 Objectives, Endpoints, and Assessments
Primary objectives of the single-dose PK/PD study were to assess the PK and PD equivalence of Nivestym and US-Neupogen based on filgrastim serum concentration and absolute neutrophil count (ANC), respectively. Secondary objectives were to evaluate safety, including immunogenicity. Primary PK endpoints were area under the filgrastim serum concentration–time curve (AUC) from time zero to infinity (AUC0–inf) following dose administration and the maximum observed serum filgrastim concentration (Cmax). Primary PD endpoints were area under the effect curve (AUEC) for ANC (AUECANC) from time zero to 120 h post-dose and the maximum observed ANC (ANCmax).
The primary objective of the multiple-dose PK/PD study was to assess the PD equivalence of Nivestym and US-Neupogen based on cluster of differentiation (CD)-34+ count. Secondary objectives were to assess PK equivalence and to evaluate safety, including immunogenicity. Primary PD endpoints were AUEC for CD34+ count (AUECCD34+) from day 1 through 120 h post-dose day 5 and maximum observed CD34+ count (CD34+max). The primary PK endpoints were AUC from time zero (pre-dose) to 24 h (AUC0–24) and Cmax, both post-dose on day 5.
The primary objective of the comparative immunogenicity study was to assess non-inferiority for immunogenicity of Nivestym and US-Neupogen based on the incidence of antidrug antibodies (ADAs) against filgrastim. The secondary objective was to assess safety. The primary endpoint was the proportion of subjects with a negative baseline ADA test result and one or more confirmed post-dose positive ADA test result at any time during the study. The secondary endpoint was the proportion of subjects with a negative baseline ADA test result and a post-dose positive neutralizing antibody (NAb) test result at any time during the study.
The three clinical studies used a tiered approach for assessing immunogenicity, consistent with FDA guidelines for demonstrating biosimilarity and draft guidelines for immunogenicity testing [5, 9]. A validated electrochemiluminescence (ECL)-based bridging assay was used to test samples for the presence of ADAs against filgrastim (tier 1 testing). Samples that tested positive in the initial screening assay were further tested in a confirmatory ECL assay (tier 2 testing) to assess filgrastim specificity. Confirmed ADA-positive samples were further characterized (tier 3 testing) by measurement of antibody titer (ECL assay) and for the presence of NAbs (cell-based assay). Immunogenicity assays used in the single-dose and multiple-dose PK/PD studies were developed and validated according to industry and regulatory expectations current at the time [10,11,12]. Immunogenicity assays used in the comparative immunogenicity study were validated according to current FDA requirements [9]. Further details of the immunogenicity assay used in these studies are provided in the ‘Supplemental Methods’ in the Electronic Supplementary Material [ESM].
PK, PD, and immunogenicity sampling schemes are summarized in Fig. 1. Safety variables included adverse events (AEs), AEs of special interest (AESIs), clinical laboratory tests (hematology, clinical chemistry, and urinalysis), vital signs, physical examination, 12-lead ECG, chest X-ray (single-dose and multiple-dose PK/PD studies), prior and concomitant medications, and local injection-site reaction (ISR; a formalized assessment of ISRs was only performed in the comparative immunogenicity study). AEs were coded using the Medical Dictionary for Drug Regulatory Activities (MedDRA) version 18.0 or later. The following events were considered AESIs for filgrastim and were summarized by the medical concept (narrow standardized MedDRA query [SMQ] and preferred term, as appropriate): potential allergic reactions, splenomegaly, splenic rupture, acute respiratory distress syndrome, alveolar hemorrhage, hemoptysis, leukocytosis, thrombocytopenia, capillary leak syndrome, cytokine release syndrome, cutaneous vasculitis, and glomerulonephritis. The AESIs were identified prospectively based on the safety profile of the reference product [2] and any preclinical or clinical data for the EU-approved filgrastim biosimilar, Nivestim (filgrastim; Hospira UK Ltd., Maidenhead, UK) [13].
2.4 Statistical Analysis and Determination of Sample Size
Analysis populations for the single-dose and multiple-dose PK/PD studies included the PK population (all subjects who received both treatments and had sufficient data to calculate primary PK endpoints), the PD population (all subjects who received both treatments and had sufficient data to calculate primary PD endpoints), and the safety population (all subjects who received one or more dose of study drug). Primary PK and PD analyses were conducted using the PK and PD populations, respectively. Sensitivity analyses were performed using the PK and PD populations excluding subjects with confirmed positive ADAs, and using the safety population. Immunogenicity and safety analyses were conducted using the safety population.
PK and PD parameters were calculated using non-compartmental analysis in Phoenix® WinNonlin®, version 6.4 (Certara USA, Inc., Princeton, NJ, USA), and data were log-transformed before statistical analysis. PK and PD equivalence was concluded in the single-dose study if the 90% confidence intervals (CIs) for the geometric mean ratios (GMRs) of Nivestym/US-Neupogen for both PK (AUC0–inf and Cmax) and both PD (AUECANC and ANCmax) primary endpoints were completely contained within the acceptance limits of 0.80-1.25. Likewise, PD and PK equivalence was concluded in the multiple-dose study if the 90% CIs for the GMRs (Nivestym/US-Neupogen) for both PD (AUECCD34+ and CD34+max) and both PK (AUC0–24 and Cmax post-dose on day 5) primary endpoints were completely contained within the acceptance limits (0.80–1.25). The PK parameters were analyzed with analysis of variance, with treatment, TP, subject within sequence group, and sequence group as effects. The PD parameters were analyzed with analysis of covariance, with baseline ANC (single-dose PK/PD study) or baseline CD34+ (multiple-dose PK/PD study) as a covariate, and with treatment, TP, subject within sequence group, and sequence group as effects.
For the single-dose PK/PD study, a total of 20 subjects were required for > 90% power at a 5% significance level to demonstrate equivalence between treatments assuming the true ratio of the means was 1.0, between-subject coefficient of variation (CV) was 23%, within-subject CV was 17%, and equivalence limits of the mean ratio were 0.80–1.25. For the multiple-dose PK/PD study, a total of 52 subjects was required for 90% power at a 5% significance level to demonstrate equivalence between treatments assuming the true ratio of the means was 1.0, the CV was 35%, and the equivalence limits of the mean ratio were 0.80–1.25.
Analysis populations for the comparative immunogenicity study included the full analysis set (FAS; all subjects who were randomized and received one or more dose of study drug) and the safety analysis set (SAS; all subjects who received one or more dose of study drug). Immunogenicity data were analyzed using the FAS and by the treatment to which subjects were randomized. Safety data were analyzed using the SAS and by the treatment subjects received. A 90% CI for the test (Nivestym) minus reference (US-Neupogen) difference between groups for the primary endpoint was constructed using the exact method of Chan and Zhang [14]. Non-inferiority for immunogenicity was demonstrated if the upper bound of the 90% CI of risk difference between groups was ≤ 10%. A sample size of 125 subjects per group provided > 95% power to demonstrate non-inferiority between treatments. A non-inferiority test was not applied to the secondary endpoint.
3 Results
3.1 Subject Disposition and Baseline Demographic and Other Characteristics
In total, 24 subjects (12 per sequence group) were randomized in the single-dose PK/PD study (Fig. S1a in the ESM). One subject who prematurely discontinued from the study (lost to follow-up) was included in the total safety population (n = 24) and excluded from the PK and PD populations (n = 23 each). In total, 60 subjects (30 per sequence group) were randomized in the multiple-dose PK/PD study (Fig. S1b in the ESM). Four subjects who prematurely discontinued from the study (lost to follow-up, protocol deviation, withdrew consent, and non-compliance with outpatient visits; n = 1 each) were included in the total safety population (n = 60) and excluded from the PK and PD populations (n = 56 each).
A total of 256 subjects were randomized in the comparative immunogenicity study (Fig. S1c in the ESM). One subject assigned to US-Neupogen experienced a personal emergency and so did not receive the dose; therefore, 255/256 (Nivestym, n = 128; US-Neupogen, n = 127) randomized subjects were treated. A total of 244/256 (Nivestym, n = 121; US-Neupogen, n = 123) randomized subjects were evaluated for non-inferiority for immunogenicity. Nine (Nivestym) and six (US-Neupogen) subjects discontinued from the study for the following reasons: AEs (n = 2 and 1, respectively), lost to follow-up (n = 2 per group), protocol deviation (n = 1 per group), withdrew consent (n = 4 and 1, respectively) and other (n = 0 and 1, respectively).
Demographic and baseline characteristics for the safety populations for single-dose and multiple-dose PK/PD studies were generally well-matched between sequence groups (Table 1). Demographic and baseline characteristics were comparable between the two treatment groups in the comparative immunogenicity study (Table 1).
3.2 Pharmacokinetics and Pharmacodynamics
The 90% CIs for the GMRs for primary PK (AUC0–inf and Cmax) and PD (AUECANC and ANCmax) endpoints in the single-dose study, and for primary PD (AUECCD34+ and CD34+max) and PK (AUC0–24 and Cmax post-dose on day 5) endpoints in the multiple-dose study, were completely contained within the statutory acceptance limit of 0.80–1.25 (Table 2). The PK and PD parameter-over-time profiles were similar between Nivestym and US-Neupogen (Fig. 2). Sensitivity analyses using the PK and PD populations excluding subjects with confirmed positive ADAs demonstrated results consistent with those for the primary PK and PD populations (Table S1 in the ESM). Secondary PK and PD parameters were also generally similar between Nivestym and US-Neupogen (Tables S2 and S3 in the ESM).

PK and PD parameter-over-time profiles for Nivestym and US-Neupogena. a Single-dose PK/PD study: mean ± SD filgrastim serum concentration-over-time profiles (semi-log) for Nivestym and US-Neupogenb. b Single-dose PK/PD study: mean ± SD ANC-over-time profiles (linear) for Nivestym and US-Neupogen. c Multiple-dose PK/PD study: mean ± SD CD34+-over-time profiles (linear) for Nivestym and US-Neupogen. d Multiple-dose PK/PD study: mean ± SD filgrastim serum concentration-over-time profiles (linear) for Nivestym and US-Neupogen post-dose administration on day 5. aPK and PD analyses were conducted using the PK and PD populations, respectively. bConcentrations below BQ occurring after Tmax were treated as missing values and BQ before Tmax (including pre-dose concentrations) were set to zero. ANC absolute neutrophil count, BQ lower limit of quantitation, CD cluster of differentiation, PD pharmacodynamic, PK pharmacokinetic, SD standard deviation, Tmax time to maximum observed concentration, US-Neupogen filgrastim reference product licensed in the USA
3.3 Immunogenicity
In the comparative immunogenicity study, 9/121 (7.4%) subjects in the Nivestym group and 6/123 (4.9%) in the US-Neupogen group had a negative baseline ADA test result and at least one confirmed post-dose positive ADA test result at any visit during the study (Table 3). The upper bound of the 90% CI (− 2.717%, 8.096%) for the risk difference (2.560%) between treatments was within the non-inferiority margin of ≤ 10%. The ADAs were generally low titer, in the range of < 2–4. A maximum titer of 16 was observed for one subject (US-Neupogen). For ADA-positive subjects, antibody titer did not increase with additional study drug, including during TP2, which followed a break to allow an opportunity for antigen re-challenge.
In the single-dose PK/PD study, 3/24 (12.5%) subjects, all randomized to sequence group 2 (US-Neupogen in TP1 followed by Nivestym in TP2), tested positive for ADA at least once during the study (Tables S4 and S5 in the ESM). Two of the three subjects tested ADA positive before first administration of US-Neupogen in TP1. The third subject tested ADA negative before first administration of US-Neupogen in TP1 and ADA positive on day 12 of TP1 and at all subsequent assessments.
In the multiple-dose PK/PD study, 1/60 (1.7%) subjects, who was randomized to sequence group 1 (Nivestym in TP1 followed by US-Neupogen in TP2), tested ADA positive at one time point (day 33 of TP1) during the study. Examination of the individual PK and PD parameters for the ADA-positive subjects in both PK/PD studies suggested that these parameters were not influenced by the presence of ADA.
In addition, all ADA-positive subjects in the PK/PD studies were clinically stable, with no AEs reported throughout the studies. No subject in the Nivestym biosimilar clinical development program developed NAbs.
3.4 Safety
In the single-dose and multiple-dose PK/PD studies, similar percentages of subjects reported at least one treatment-emergent AE (TEAE) following administration of Nivestym and US-Neupogen (Tables 4, 5). The most common (more than one subject regardless of treatment) TEAEs were headache (n = 6 subjects) in the single-dose PK/PD study (no other TEAE occurred in more than one subject following either treatment). In the multiple-dose PK/PD study, the most common TEAEs were back pain (n = 12 subjects), pyrexia (n = 8), headache (n = 7), cough (n = 6), oropharyngeal pain (n = 3), and chills (n = 2). All TEAEs reported in the single-dose and multiple-dose PK/PD studies were considered mild (14/15 [93.3%] and 61/63 [96.8%], respectively) or moderate (1/15 [6.7%] and 2/63 [3.2%], respectively) in intensity.
No subject in the single-dose or multiple-dose PK/PD studies experienced a TEAE that led to discontinuation of study drug or from study. One subject in the single-dose PK/PD study, who was randomized to sequence group 1 (Nivestym in TP1 followed by US-Neupogen in TP2), reported an AESI of allergic rhinitis in the SMQ of hypersensitivity under the category of potential allergic reaction; the event was considered by the investigator to be mild in intensity, non-serious, and unrelated to study drug. One subject in the multiple-dose PK/PD study, who was randomized to sequence group 1, reported an AESI of dermatitis in the SMQ of hypersensitivity under the category of potential allergic reaction; the event was considered by the investigator to be mild in intensity, non-serious, and related to study drug.
In the comparative immunogenicity study, similar percentages of subjects in the Nivestym and US-Neupogen groups experienced TEAEs (Table 6). The most frequently reported (≥ 5% of subjects in either group) TEAEs by preferred term were back pain, headache, pain in extremity, and injection-site hemorrhage. The majority of TEAEs were mild (220/248 [88.7%]) or moderate (26/248 [10.5%]) in severity. Two (1.6%) subjects in the Nivestym group experienced three TEAEs reported as severe. Of these, one subject reported severe TEAEs of diverticular perforation and wound infection; both events were considered by the investigator to be unrelated to study drug. The remaining subject experienced dyspnea, which was also considered by the investigator to be unrelated to study drug; severity of this event was unknown and imputed as severe. No subjects in the US-Neupogen group experienced severe TEAEs. One subject in the Nivestym group discontinued from study because of the previously described severe TEAE of diverticular perforation. One additional subject in the Nivestym group and one (0.8%) subject in the US-Neupogen group discontinued from study after experiencing a TEAE (back pain; moderate, related to study drug).
One subject (Nivestym group) reported two AESIs of dermatitis in the SMQ of hypersensitivity under the category of potential allergic reaction; both events were considered by the investigator to be moderate in severity, non-serious, and unrelated to study drug (due to soap sensitivity). This subject was ADA negative at baseline and at all post-dose assessments. No subjects in the US-Neupogen group reported an AESI. No TEAEs were considered to be associated with positive ADA results in any of the subjects with a confirmed post-dose positive ADA test result at any visit during the study. There was no correlation between potential immunogenicity-associated TEAEs and ADA development. Two (1.6%) subjects in the Nivestym group and 13 (10.2%) in the US-Neupogen group reported ISRs. Of these, one subject (US-Neupogen group) tested ADA positive. However, the occurrence of an ISR in this subject was not considered to be related to the ADA findings.
As mentioned, one subject (0.8%) in the Nivestym group in the comparative immunogenicity study experienced two severe TEAEs (diverticular perforation and wound infection). Both events were reported as serious AEs and considered by the investigator to be unrelated to study drug. Pathology assessment indicated that the event of diverticular perforation was likely related to ingestion of a foreign material. No other serious AEs or deaths were reported during any of the three clinical studies. There were no clinically meaningful changes from baseline in any of the laboratory parameters or in vital signs and no clinically meaningful differences in these safety outcomes between treatment groups. No clinically significant ECG, physical examination, or chest X-ray results were reported for any subject.
4 Discussion
Biosimilarity is established by the totality of the evidence from analytical, non-clinical, and clinical studies to demonstrate that the proposed biosimilar product is highly similar in structure and function to—and has no clinically meaningful differences in safety, purity, and potency from—the reference product [5]. The objective of a biosimilar clinical development program is to address any residual uncertainties regarding biosimilarity that remain after analytical assessments and, where relevant, non-clinical evaluations [5]. Comprehensive and robust analytical studies, together with clinical PK/PD studies in HVs, may be adequate to support regulatory approval of some biosimilars without the need to conduct a comparative safety and efficacy study in patients. This HVs-only approach is part of the evolving regulatory landscape for biosimilars [15]. With the concurrence of the FDA, three clinical studies were conducted following this HVs-only approach to support regulatory assessments of Nivestym for the filgrastim indications of neutropenia and PBPC mobilization for all target populations of use.
The clinical pharmacology studies reported here evaluated PK and PD equivalence between Nivestym and US-Neupogen, following single-dose or multiple-dose subcutaneous administration. Nivestym demonstrated PK and PD equivalence to US-Neupogen in the single-dose setting based on measures of filgrastim concentration (AUC0–inf and Cmax) and ANC response (AUECANC and ANCmax), respectively. Likewise, Nivestym demonstrated PD and PK equivalence to US-Neupogen in the multiple-dose setting based on measures of CD34+ count (AUECCD34+ and CD34+max) and filgrastim concentration (AUC0–24 and Cmax post-dose on day 5), respectively. Secondary PK and PD parameters were also generally similar between treatments, supporting the primary results of PK and PD equivalence between Nivestym and US-Neupogen. Results from sensitivity analyses of the PK and PD populations, excluding ADA-positive subjects, were consistent with those of the primary populations and demonstrated robustness of the primary PK and PD analyses.
FDA biosimilarity guidance recommends that clinical pharmacology studies employ either a crossover design or a parallel design to evaluate PK and PD similarity and that study design is selected based on product half-life duration as well as the time courses of PD response and immunogenicity [8]. The single-dose and multiple-dose PK/PD studies used a crossover design, and the choice of study design was appropriate for several reasons. First, a crossover design is generally preferred and is recommended for products, such as filgrastim, “with a short half-life (e.g., shorter than 5 days), a rapid PD response (e.g., the time of onset, maximal effect, and disappearance in conjunction with drug exposure), and a low anticipated incidence of immunogenicity” [5, 8]. In addition, a crossover design is considered the most sensitive in evaluating PK similarity [8]. Furthermore, crossover-design studies can provide reliable estimates of differences in exposure with smaller sample sizes [8].
With respect to study population, regulatory guidelines recommend that “clinical PK and PD studies should be conducted in healthy subjects if the product can be safely administered to them” [8]. The clinical PK/PD studies reported here were conducted in HVs, which is appropriate since the safety of US-Neupogen in healthy subjects has been evaluated [16, 17] and because the mechanism of action and pharmacological properties of filgrastim products are similar in healthy subjects and in patients [2, 6]. Furthermore, HVs are likely to exhibit less PK and PD variability than patients with potential confounding factors (e.g., underlying and/or concomitant disease and concomitant medications) [8], and therefore constitute the most sensitive study population for evaluating differences in clinical pharmacology profiles between Nivestym and US-Neupogen.
Regulatory guidelines also recommend that clinical pharmacology studies should be conducted using the most sensitive dose and route of administration for detecting and evaluating differences in PK and PD profiles between the proposed biosimilar product and the reference product [8]. US-Neupogen is approved for intravenous or subcutaneous injection at doses of 5–10 μg/kg/day [2]. The clinical PK/PD studies were conducted using only a subcutaneous dose of 5 μg/kg via prefilled syringes. The selected dose was appropriate because it provided adequate exposure to assess the pharmacologic properties of Nivestym and US-Neupogen; it is on the steep part of the exposure-response curve, which is appropriate for PD evaluations or for studies conducted in HVs [8]; and it is consistent with dosing of Neupogen for its approved indications [2]. Furthermore, the subcutaneous route of administration is considered most sensitive for detecting clinically meaningful differences because it can discern potential PK differences in drug absorption, in addition to drug distribution and elimination [8].
The selected primary PD endpoints based on ANC response and CD34+ count are relevant to clinical outcomes. ANC has been studied and validated as a PD marker for G-CSF activity, and the PD of filgrastim as measured by ANC response has been well-characterized following subcutaneous administration to HVs [16, 18, 19]. In addition, measurement of CD34+ count in the blood provides an assessment of CD34+ PBPC mobilization by filgrastim, and a 5-day regimen of filgrastim is sufficient to produce peak CD34+ levels in HVs [20,21,22]. Furthermore, sampling schemes were sufficiently dense to characterize the primary PD and PK endpoints.
The comparative immunogenicity study demonstrated non-inferiority for immunogenicity of Nivestym versus US-Neupogen. Use of immunocompetent HVs provides the most sensitive population for evaluating immunogenicity because they are more likely to develop immune responses than patients who are candidates for filgrastim therapy and have compromised immunity, such as patients with cancer or treated with myelosuppressive therapies. The selected dosing regimen (subcutaneous 5 μg/kg/day for 5 consecutive days) is consistent with dosing of Neupogen for its approved indications [2]. Furthermore, subcutaneous administration and repeated dosing followed by a break allowed for antigen re-challenge and a higher chance of observing immunogenicity, if it occurred.
Clinical immunogenicity of Nivestym and US-Neupogen was also evaluated in the clinical PK/PD studies. Examination of the individual PD and PK parameters for ADA-positive subjects in the single-dose and multiple-dose PK/PD studies suggests they were not influenced by the presence of ADA. Differences in ADA incidence between the PK/PD studies and the comparative immunogenicity study, as well as higher incidence than historically reported for the US-Neupogen reference product, may be attributed to more stringent requirements of 2016 draft FDA guidance for immunogenicity testing [9] that were used in the comparative immunogenicity study. The confirmatory assay criterion for positive samples (i.e., cut point) in 2016 FDA guidance was based on a 1% false-positive rate [9], which should increase the number of positive samples compared with the previous 0.1% false-positive criterion. Across all three clinical studies, safety profiles were comparable, with no clinically meaningful differences between Nivestym and US-Neupogen.
5 Conclusion
Nivestym demonstrated PK and PD equivalence, in single and multiple subcutaneous dose study settings, and non-inferiority for immunogenicity to US-Neupogen. Additionally, safety profiles were comparable, with no clinically meaningful differences between Nivestym and US-Neupogen. These data support the demonstration of biosimilarity of Nivestym to US-Neupogen, leading to regulatory approval of Nivestym by the FDA in July 2018.
References
Metcalf D. The colony-stimulating factors and cancer. Cancer Immunol Res. 2013;1(6):351–6.
Amgen Inc. Neupogen (filgrastim) US prescribing information. 2018. http://pi.amgen.com/united_states/neupogen/neupogen_pi_hcp_english.pdf. Accessed 17 Aug 2018.
Amgen Ltd. Neupogen (filgrastim) 30MU and 48MU solution for injection: summary of product characteristics. 2018. https://www.medicines.org.uk/emc/search?q=neupogen. Accessed 26 Sept 2018.
Hospira Inc. Nivestym (filgrastim-aafi) US prescribing information. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761080s000lbl.pdf. Accessed 17 Aug 2018.
US Food and Drug Administration. Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. Guidance for Industry. 2015. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed 9 May 2018.
Przepiorka D. Cross-discipline team leader review: BLA 761080 Nivestym (filgrastim-aafi). 2018. https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DevelopmentResources/UCM617299.pdf. Accessed 28 Aug 2018.
Dwivedi P, Greis KD. Granulocyte colony-stimulating factor receptor signaling in severe congenital neutropenia, chronic neutrophilic leukemia, and related malignancies. Exp Hematol. 2017;46:9–20.
US Food and Drug Administration. Clinical pharmacology data to support a demonstration of biosimilarity to a reference product. Guidance for industry. 2016. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM397017.pdf. Accessed 9 May 2018.
US Food and Drug Administration. Assay development and validation for immunogenicity testing of therapeutic protein products (draft guidance). 2016. http://www.nihs.go.jp/dbcb/immunogenicity/FDA_2016_Assay%20Development%20and%20Validation%20for%20Immunogenicity%20Testing%20of%20Therapeutic%20Protein%20Products.pdf. Accessed 8 May 2018.
Shankar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Finco-Kent D, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48(5):1267–81.
US Food and Drug Administration. Assay development for immunogenicity testing of therapeutic proteins (draft guidance). 2009. https://www.fdanews.com/ext/resources/files/archives/8/8495dft12_09.pdf. Accessed 8 May 2018.
US Food and Drug Administration. Immunogenicity Assessment for Therapeutic Protein Products. Guidance for Industry. 2014. https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm338856.pdf. Accessed 8 May 2018.
Hospira UK Ltd. Nivestim (filgrastim) 12 MU/0.2 ml, 30 MU/0.5 ml and 48MU/0.5 ml solution for injection/infusion: summary of product characteristics. 2018. https://www.medicines.org.uk/emc/product/575/smpc. Accessed 26 Sept 2018.
Chan IS, Zhang Z. Test-based exact confidence intervals for the difference of two binomial proportions. Biometrics. 1999;55(4):1202–9.
Frapaise FX. The end of phase 3 Clinical trials in biosimilars development? BioDrugs. 2018;32(4):319–24.
Borleffs JC, Bosschaert M, Vrehen HM, Schneider MM, van Strijp J, Small MK, et al. Effect of escalating doses of recombinant human granulocyte colony-stimulating factor (filgrastim) on circulating neutrophils in healthy subjects. Clin Ther. 1998;20(4):722–36.
van Der Auwera P, Platzer E, Xu ZX, Schulz R, Feugeas O, Capdeville R, et al. Pharmacodynamics and pharmacokinetics of single doses of subcutaneous pegylated human G-CSF mutant (Ro 25-8315) in healthy volunteers: comparison with single and multiple daily doses of filgrastim. Am J Hematol. 2001;66(4):245–51.
Gascon P, Fuhr U, Sorgel F, Kinzig-Schippers M, Makhson A, Balser S, et al. Development of a new G-CSF product based on biosimilarity assessment. Ann Oncol. 2010;21(7):1419–29.
Lubenau H, Bias P, Maly AK, Siegler KE, Mehltretter K. Pharmacokinetic and pharmacodynamic profile of new biosimilar filgrastim XM02 equivalent to marketed filgrastim Neupogen: single-blind, randomized, crossover trial. BioDrugs. 2009;23(1):43–51.
Eidenschink L, DiZerega G, Rodgers K, Bartlett M, Wells DA, Loken MR. Basal levels of CD34 positive cells in peripheral blood differ between individuals and are stable for 18 months. Cytom B Clin Cytom. 2012;82(1):18–25.
Grigg AP, Roberts AW, Raunow H, Houghton S, Layton JE, Boyd AW, et al. Optimizing dose and scheduling of filgrastim (granulocyte colony-stimulating factor) for mobilization and collection of peripheral blood progenitor cells in normal volunteers. Blood. 1995;86(12):4437–45.
Stroncek DF, Clay ME, Petzoldt ML, Smith J, Jaszcz W, Oldham FB, et al. Treatment of normal individuals with granulocyte-colony-stimulating factor: donor experiences and the effects on peripheral blood CD34+ cell counts and on the collection of peripheral blood stem cells. Transfusion. 1996;36(7):601–10.
Acknowledgements
Medical writing support was provided by Elyse Smith, PhD, of Engage Scientific Solutions and was funded by Pfizer Inc. The authors thank Sarah Kiehna, PhD, of Pfizer Inc. for providing a description of the bioanalytical assessments that were performed for Nivestym.
Author information
Authors and Affiliations
Contributions
TB, SH, JL, TBM, FDO, and JZ contributed to the conception or design of the work. TB, SH, and JL were involved in data acquisition. TBM (immunogenicity data), SM (safety data), H-MY (pharmacology data), and JZ (statistical analyses) were involved in the data analysis. All authors were involved in data interpretation and developing the manuscript and/or revising it critically for important intellectual content and provided final approval of the submitted manuscript. H-MY is the guarantor for the overall content.
Corresponding author
Ethics declarations
Funding
This work was funded by Hospira Inc, which was acquired by Pfizer Inc in September 2015, and by Pfizer Inc. Open access publication of this article was funded by Pfizer Inc.
Conflict of interest
TBM, SM, MS, H-MY, and JZ are employees of and own stock or options in Pfizer Inc. FDO was an employee of and held stock or options in Pfizer Inc at the time of study conduct. TB, SH, and JL are employees of Quotient Sciences, formerly Seaview Research Inc., which received funding from Pfizer Inc to perform the studies described in the manuscript.
Ethical approval and informed consent
The protocol and all amendments for each study were approved by the institutional review board at each of the investigational centers participating in the studies. All studies were conducted in compliance with the protocol, International Conference on Harmonization guidelines, all applicable regulatory requirements, and the Declaration of Helsinki. Subjects provided written informed consent prior to the performance of any study-specific procedures. Volunteers were free to withdraw from the study at any time.
Data availability
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the USA and/or EU or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yao, HM., Ottery, F.D., Borema, T. et al. PF-06881893 (Nivestym™), a Filgrastim Biosimilar, Versus US-Licensed Filgrastim Reference Product (US-Neupogen®): Pharmacokinetics, Pharmacodynamics, Immunogenicity, and Safety of Single or Multiple Subcutaneous Doses in Healthy Volunteers. BioDrugs 33, 207–220 (2019). https://doi.org/10.1007/s40259-019-00343-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-019-00343-8