
Abstract
The coronavirus disease of 2019 (COVID-19) is caused by the severe acute respiratory syndrome coronavirus-2 (SARS CoV-2), that already appeared as a global pandemic. Presentation of the disease often includes upper respiratory symptoms like dry cough, dyspnea, chest pain, and rhinorrhea that can develop to respiratory failure, needing intubation. Furthermore, the occurrence of acute and subacute neurological manifestations such as stroke, encephalitis, headache, and seizures are frequently stated in patients with COVID-19. One of the reported neurological complications of severe COVID-19 is the demolition of the myelin sheath. Indeed, the complex immunological dysfunction provides a substrate for the development of demyelination. Nevertheless, few published reports in the literature describe demyelination in subjects with COVID-19. In this short narrative review, we discuss probable pathological mechanisms that may trigger demyelination in patients with SARS‐CoV‐2 infection and summarize the clinical evidence, confirming SARS-CoV-2 condition as a risk factor for the destruction of myelin.
Similar content being viewed by others
Introduction
The outbreak of novel coronavirus disease known as severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), first identified in Wuhan, China, in December 2019. Previously, other coronaviruses were recognized to cause severe respiratory disease in humans and were associated with epidemic prevalence, such as the severe acute respiratory syndrome coronavirus (SARS-CoV) and the Middle East respiratory syndrome coronavirus (MERS-CoV), both enhancing the risk of mortality [1]. The average diameter of the COVID virus is almost 100 nm and is spherical or oval in shape. The large spikes of membrane glycoproteins of the virus are located on the viral surface, and these negatively stained virus particles are expressed as typical crown-like shapes under electron microscopy [2]. The viral genome encodes several important structural proteins, including spike, membrane, envelope, and nucleocapsid proteins [3]. Similar to the previously identified SARS and MERS diseases, the main indications of COVID-19 are one or a combination of fever, fatigue, dry cough, and shortness of breath, followed by other symptoms like headache, nasal congestion, sore throat, myalgia, or arthralgia [4, 5]. However, extra respiratory complications such as neurological findings have been reported. Neurological manifestations, including headache, disturbed consciousness, paresthesia, and seizures are appeared in almost 36.4% (78/214) of patients suffering from COVID-19 [6]. On March 4, 2020, Beijing Ditan Hospital, for the first time, reported a patient with viral encephalitis instigated by a novel coronavirus attacking the central nervous system (CNS). This study illustrated the potential effect of COVID-19 in triggering damage to the nervous system [7]. According to accumulating evidence, the different types of Coronaviruses can induce neurological disorders such as polyneuropathy, encephalopathy, ischemic stroke, and demyelinating lesions, like Multiple sclerosis (MS) and Guillain-Barré syndrome (GBS) [8, 9]. The neurological problems could be presented delayed to respiratory signs [8], and severely affected patients are more vulnerable to neurological manifestations related to patients with mild or moderate disease [10]. Amongst neurological impairments, demolition of myelin appears as a visible complication in patients with severe COVID-19 [6]. MS is an acquired inflammatory immune-mediated disorder of the CNS, characterized by the focal demyelination associated with various degrees of neurodegeneration [11]. GBS, as another demyelinating disease, is an acute/subacute inflammatory polyneuropathy and immune-mediated disease triggered by a preceding bacterial or viral infection [12]. To support this assumption, the existence of demyelination in cases with COVID-19 has been reported, as well as particles of the SARS-CoV virus and genome sequences have been found in autopsy reports [13, 14]. Therefore, this review discusses the involvement of COVID-19 in triggering immunological host response and demyelination in central and peripheral nervous systems.
The entrance of SARS-CoV-2 virus into the CNS
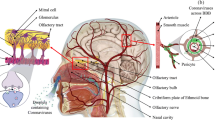
Similar to other Coronaviruses, two possibilities appear for penetration of SARS-CoV-2 into the CNS, including (1) hematogenous dissemination of SARS-CoV-2 and rapidly spreading through the cerebral circulation, where the slower blood flow accelerates the capillary endothelium damages and makes the brain accessible, and (2) gaining entry through the cribriform plate and olfactory bulb [15]. Here, infectious agents like coronaviruses can spread through cranial nerves, including the olfactory, the trigeminal, the glossopharyngeal, and the vague nerves, via retrograde axonal transport [16]. Notably, the entrance of SARS-CoV-2 into the brain through the olfactory nerves infects neurons that control breathing [17]. Similar to SARS-CoV, the SARS-CoV-2 and a host receptor, Angiotensin-converting enzyme 2 (ACE2) have a stronger binding capacity [18]. ACE2, as a cardio-cerebral vascular protection factor, is capable of converting the angiotensin (Ang) I to Ang II that activates Ang II receptor type1 (AT1R), promoting the blood pressure, inflammation, and even neurodegeneration [19]. This receptor is present in human endothelial cells, neurons, respiratory epithelia, pulmonary parenchyma, small bowel cells, renal cells, and nerve cells, enabling the dispersal of SARS-CoV-2 into the host cells [20, 21]. Thus, the wide expression of the ACE2 receptor on nerve cells and capillary endothelium allows the direct access of SARS-CoV-2 to the CNS [22,23,24], where the virus binds to nerve cells via attachment between the spike glycoprotein and the existent ACE2 receptors [25]. On the other hand, the interaction of SARS-CoV-2 with ACE-2 receptors in the vascular endothelial cells may be conductive of the blood–brain-barrier (BBB) destruction, leading to the direct virus entry into the brain [26, 27]. In addition to ACE2, basigin (BSG; CD147) and neuropilin-1 (NRP1) act as docking receptors for SARS-CoV-2 virus, while several proteases including transmembrane protease serine 2 (TMPRSS2), cathepsin B and L (CatB/L), and furin are implicated in the virus entry to cell and replication [28]. NRP1, a transmembrane receptor lacking a cytosolic protein kinase domain, is highly expressed in the olfactory epithelium and improves the SARS-CoV-2 entry into the brain [29]. CD147 (also known as Basigin is an alternative receptor for SARS-CoV-2 invasion into the brain, which is present on neural cells [30]. Furthermore, other endosomal cysteine proteases, namely CatB/L contribute in priming of the spike protein of SARS-CoV-2 virus [31]. Hoffmann et al. demonstrated that SARS-CoV-2 uses ACE2 for entry and needs the TMPRSS2 for S protein priming to efficiently enter the cell, and TMPRSS2 inhibitor can block virus entry to exert a treatment effect [32]. TMPRSS2 is expressed throughout olfactory epithelial cells and choroid plexus of both mouse and human [33, 34]. The other results indicated that TMPRSS2 and lysosomal cathepsins are capable of activating SARS-CoV-2 entry; moreover, furin has cumulative effects on this process [35].
Furthermore, another avenue for the accession of SARS-CoV-2 to the host cells is binding to integrins. These receptors are present on the surface of different cells in the body and contribute in triggering several signaling pathways [36].
Evidence of Covid-19 virus involvement in demyelination
Accumulating evidence confirms the SARS-CoV-2 viral infection as a risk factor of demyelination both in the peripheral and central nervous systems. In agreement with this idea, Zanin et al. reported a case of COVID-19 admitted for interstitial pneumonia and seizures. Brain MRI indicated newly diagnosed demyelination injuries. However, treatment with high-dose steroid resulted in respiratory and neurological recovery. They postulated that SARS-CoV-2-induced delayed immune response was a causative factor for the demolition of the myelin sheath [37].
In another report, Mehta et al. detected demyelinating lesions associated with the neurological damage in a case with COVID-19. The brain and spine MRI of the patient exhibited a new onset of multiple, non-enhancing demyelinating lesions. They assumed that following SARS-CoV-2 infection, the pro-inflammatory environment induced by the cytokine storm might be responsible for the activation of glial cells with subsequent demyelination [38]. Furthermore, Zoghi et al. reported a 21-year-old male with encephalomyelitis after experiencing intermittent 4 days vomiting and malaise that showed upper respiratory indications 2 weeks before this presentation. Brain MRI revealed bilateral posterior internal capsule lesions expanding to the ventral part of the pons and a marbled splenium hyperintensity pattern. In their study, Cervical and thoracic MRI showed the sizable transverse myelitis [39]. In another report, Gilles et al. stated a 54-year-old woman affected to SARS-CoV-2 with brain lesions representing acute demyelination. The brain CT scan of the patient confirmed hypodense lesions, involving supratentorial white matter and pallidum bilaterally. A brain MRI showed lesions with restricted diffusion without any hemorrhage or enhancement. The thalamus, the striatum, and the posterior fossa were spared. A follow-up MRI displayed no new lesions, and the spinal cord MRI was without abnormalities [40].
In another case report, A 33-year-old male suffering from morbid obesity, diabetes mellitus, and hypoxemic showed a respiratory failure secondary to COVID-19 affection. MRI presented widespread foci of the demolition of the myelin with restricted diffusion, most located in the corpus callosum and the pericallosal white matter [41]. Furthermore, Karapanayiotides et al. described a 57-year-old male affected by COVID-19 along with particular localization and a concentric demyelination pattern. MRI documented the occurrence of hemosiderin deposits concurrent with a concentric demyelination pattern [42].
A recent study described a COVID-19 patient with isolated symmetrical demyelinating lesions of bilateral posterior internal capsules that presented stroke-like clinical demonstration. The lack of grey matter damage and absence of hemorrhage or cavitation confirmed demyelination, rather than encephalomyelitis [43].
In support of the involvement of SARS-CoV-2 infection in GBS and consequent demyelination, Bracaglia described a 66-year-old woman with acute demyelinating polyneuritis related to asymptomatic SARS-CoV-2 infection. She was unable to walk, speech and swallow, as well as tendon reflexes were abolished. The patient showed normal vital signs, adverse medical history and no report of the previous month infection. They assumed GBS and conducted nerve conduction studies consistent with demyelinating polyneuropathy [44].
Immunological host response against SARS-CoV-2 infection
It is worth noting that the severe form of COVID-19 causes not only the viral infection, but also leads to an abnormal and aggravated immunological host response, resulting in severe systemic damage [45]. SARS-CoV-2 infection is capable of triggering innate and adaptive immune responses. Uninhibited inflammatory innate responses and debilitated adaptive immune responses may cause harmful locally and systemically tissue damage [46]. Accordingly, unpredictable effects of acute viral infection on the host immune response is probably an important reason for nervous tissue damage [47].
The immunopathological pathway resulting in COVID-19 mortality contains three stages, that progress rapidly from onset of disease to death over 16–18.5 days. Stage 1 is the initiation phase, with early signs of systemic inflammation, early induction of predominant chemokines, and decreased peripheral lymphocyte counts. Stage 2 is the amplification phase, with a massive production of inflammatory cells, including macrophages, monocytes, and neutrophils to intensify the immunopathological process. Stage 3 is the consummation phase. In this stage, the peripheral neutrophil counts further increase, while lymphopenia worsens with continuous enhancement of inflammatory mediators and cytokine storm, leading to the widespread organ damages [48].
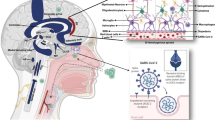
Most patients with COVID-19 display normal or decreased leukocyte count [49,50,51], and lymphopenia [52]. lymphopenia is a common feature in patients with severe COVID-19, but not in patients with a mild infection [53]. In this condition, the numbers of CD4 + T cells, CD8 + T cells, B cells, and natural killer (NK) cells subsets are extremely reduced in the lymph nodes and spleen of the patients (Fig. 1) [54]. Besides, in stage 2, the percentage of monocytes and macrophages is increased following viral infection, which leads to the release of a large amount of pro-inflammatory cytokines, referred to as cytokine storm [55, 56]. This phenomenon is an enhancement of serum levels of pro-inflammatory cytokines including IL-6 and IL-1β, as well as IL-2, IL-8, IL-17, G-CSF, GM-CSF, IP10, MCP1, MIP1α (also known as CCL3), and TNF, which has found in most patients with severe COVID-19 [57]. Accordingly, a dysfunctional and disharmonic immune response in severe COVID-19 cases initiates an extensive lung and systemic inflammation by triggering cytokine storm, that possibly worsens infection in the brain [58]. The cytokine storm targets the CNS because the released cytokines are able to cross the BBB by disrupting the integrity of this structure [59]. This process allows the penetration of monocytes, macrophages, and T-lymphocytes into the brain [60]. The cytokine storm induced by SARS-CoV-2 infection, at least in part caused by the toll-like receptors (TLRs) involvement in the immune responses in the CNS (Fig. 2). In the normal innate immune system, various pattern recognition receptors (PRRs) are expressed in macrophages, monocytes, dendritic cells, and neutrophils that recognize pathogen-associated molecular patterns (PAMPs), which are expressed by infectious agents. PRRs are the fundamental players in mediating innate immune response and comprise TLRs and nod-like receptors (NLRs) [61]. It has postulated that TLRs are the main PRRs, taking part in the induction of cytokine storm in the COVID-19-infected patients [61, 62]. Ten TLRs, TLR1-10 have been recognized in humans, while TLR1-13, with the exemption of TLR10 are present in rodents [63]. It is remarkable that IL-6 and TNF-α, the main cytokines that participated in severe COVID-19 cases, are downstream of the TLR4 signaling pathway [64]. TLRs are expressed by monocytes, astrocytes, macrophages, and dendritic cells, which are the primary operators of the innate immune system and take part in host defense and the recognition of invading pathogens, suggesting the role of these receptors in MS pathophysiology [65]. Interestingly, these receptors identify viral particles and are involved in the moderation of the immune response in MS patients. Apparently, there may be an association between viral infections, such as coronavirus, and the growth of demyelination [65]. Two necessary proteins that activate these receptors are the cluster of differentiation 14 (CD14) and myeloid differential protein-2 (MD-2), triggering two internal signaling pathways, including the myeloid differentiating primary response gene 88 (MyD88)-dependent and the MyD88-independent pathways, which is known as toll/interleukin-1 receptor (TIR)-domain-containing adapter-inducing interferon-β (TRIF)-dependent signaling pathway (Fig. 2) [66]. With the exemption of TLR3, all other TLRs utilize the MyD88-dependent signaling pathway. TLR3 uses the MyD88-independent signaling pathway [67]. TLR4 is the only TLR that employs both the MyD88-dependent and the TRIF-dependent signaling pathways. TLRs trigger the internal signaling pathways to activate some transcription factors responsible for the generation of pro-inflammatory cytokines, like nuclear factor-kβ (NF- kβ), as well as interferon regulatory factors (IRF) that mediates the type I interferon (IFNs)-dependent antiviral response [62, 68]. Activation of TLRs initiates a cell signaling cascade, leading to the release of several interleukins, interferons, and other pro-inflammatory cytokines [62]. Activation of diverse TLRs can lead to a robust enhancement in cytokine release, inducing the cytokine storm in the CNS [69]. The main cytokines involved in the cytokine storm in the CNS are IL-6, interferons and IL-10 that result from astrocyte and microglia TLR signaling. Microglial cells and astrocytes are CNS immune cells and the cytokine storm that mostly produced by microglia can increase BBB permeability and may be responsible for neurological manifestations of COVID-19 [70, 71]. Some of the produced cytokines lead to the death of neurons and oligodendrocytes and, subsequently demyelination [71]. They also could increase glial activation and promote the expression of TLR in neural cells, exacerbating the cytokine storm and further inflammatory response [72]. Moreover, these signals are capable of attracting macrophages, NK cells, mast cells, and etc., which eventually may release reactive oxygen species (ROS) and reactive nitrogen species (RNS) [73]. An additional mechanism of CNS involvement and demyelination caused by autoimmune encephalitis in COVID‐19 cases, a condition that results from extreme self‐response and antigen‐driven immune responses [74]. SARS-CoV-2 virus can elicit autoimmune responses through several mechanisms, including molecular mimicry between viral proteins and host self-antigens, epitope spreading due to T‐cell-mediated damage by the virus, bystander activation of T cells by the action of virus-induced inflammatory cytokines, the presentation of cryptic antigens, poly-clonal B cell activation, and viral superantigens [75, 76]. The studies indicated the presence of high-level IgA and IgG antibodies (antineuronal antibodies) in the CSF and serum of patients, demonstrating that these antibodies can reach the brain [77, 78].

Schematic representation of mechanisms that SARS-CoV-2 infection may result in demyelination. One crucial way is the decreased number of CD4 + T cells, CD8 + T cells, B cells, and natural killer (NK) cells. Furthermore, enormous cytokine release creates a pro-inflammatory condition that some of the produced cytokines lead to the death of neurons and oligodendrocytes, increase glial activation and promote the expression of TLR in neural cells. These signals are capable of attracting macrophages, NK, mast cells, and, etc., which eventually may release reactive oxygen species (ROS) and reactive nitrogen species (RNS). Another mechanism is spontaneous or provoked autoimmune reactions and the generation of auto-antibodies against SARS-CoV-2 virus

Schematic representation of toll-like receptors (TLRs) activation and induction of cytokine storm. Two necessary proteins to activate these receptors are the cluster of differentiation 14 (CD14) and myeloid differential protein-2 (MD-2), triggering 2 internal signaling pathways, including the MyD88-dependent and the MyD88-independent pathways, which is known as TRIF-dependent signaling pathway. TLRs trigger the internal signaling pathways to activate some transcription factors responsible for the generation of pro-inflammatory cytokines, like nuclear factor-kβ (NF- kβ), as well as interferon regulatory factors (IRF) that mediate the type I interferon (IFNs)-dependent antiviral response
Thus, in patients with COVID‐19, antibodies that are produced against SARS‐CoV‐2 attack antigens presented in human endothelial or neural cells (through the interrupted BBB), leading to cerebral edema and autoimmune encephalitis [25]. Given the evidence, SARS-CoV-2 infection may take part in proceeding MS and demyelination via several mechanisms, including reduction in the number of CD4 + T cells, CD8 + T cells, B cells, and NK cells, enormous cytokine release, creating a pro-inflammatory condition, and provoked autoimmune reactions (Fig. 1) [11].
Conclusion
This narrative review briefly described the mechanisms of CNS affection, altering the host immunological function, and induction of demyelination by SARS-CoV-2 infection, and summarized the evidence of the myelin sheath demolition induced by this viral infection. In summary, SARS-CoV-2 can enter the host cells in the brain and exert the neurologic symptoms via the binding to ACE2 and integrins presented on existing cells. Given the evidence, SARS-CoV-2 infection is able to cause MS and demyelination via several mechanisms. One important mechanism is the decreased number of CD4 + T cells, CD8 + T cells, B cells, and NK cells. Another mechanism is provoked autoimmune reactions, a condition which results from excessive self‐response and antigen‐driven immune responses. Thus, in patients with COVID‐19, antibodies that are produced against SARS‐CoV‐2 attack antigens in human endothelial cells presented in cerebral vessels or neurons, leading to cerebral edema and autoimmune encephalitis. The most prominent type of mechanism is massive cytokines release by activating TLRs, which creates a pro-inflammatory environment. Some of the released cytokines induce neuronal and oligodendrocyte death and demyelination. They also could enhance glial activation and promote TLR expression in neural cells, intensifying the cytokine storm and inflammatory response.
References
Su S, Wong G, Shi W, Liu J, Lai ACK, Zhou J, Liu W, Bi Y, Gao GF (2016) Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol 24(6):490–502
Schoeman D, Fielding BC (2019) Coronavirus envelope protein: current knowledge. Virol J 16(1):1–22
Chatterjee S (2020) Understanding the nature of variations in structural sequences coding for coronavirus spike, envelope, membrane and nucleocapsid proteins of SARS-CoV-2. Envelope, Membrane and Nucleocapsid Proteins of SARS-CoV-2
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y (2020) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. The Lancet 395(10223):507–513
Xiaohua C, Binhong Z, Yueming Q, Yurou C, Jie X, Yong F, Feng L (2020) Detectable serum SARS-CoV-2 viral load (RNAaemia) is closely associated with drastically elevated interleukin 6 (IL-6) level in critically ill COVID-19 patients. medRxiv
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, Chang J, Hong C, Zhou Y, Wang D (2020) Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol 77(6):683–690
Xiang P, Xu XM, Gao LL, Wang HZ, Xiong HF, Li RH (2020) First case of 2019 novel coronavirus disease with encephalitis. ChinaXiv 202003:00015
Kim J-E, Heo J-H, Kim H-o, Song S-h, Park S-S, Park T-H, Ahn J-Y, Kim M-K, Choi J-P (2017) Neurological complications during treatment of middle east respiratory syndrome. J Clin Neurol 13(3):227–233
Mahammedi A, Saba L, Vagal A, Leali M, Rossi A, Gaskill M, Sengupta S, Zhang B, Carriero A, Bachir S (2020) Imaging of neurologic disease in hospitalized patients with COVID-19: an italian multicenter retrospective observational study. Radiology 297(2):E270–E273
Mao L, Wang M, Chen S, He Q, Chang J, Hong C, Zhou Y, Wang D, Miao X, Hu Y (2020) Neurological manifestations of hospitalized patients with COVID-19 in Wuhan, China: a retrospective case series study
Schirinzi T, Landi D, Liguori C (2020) COVID-19: dealing with a potential risk factor for chronic neurological disorders. J Neurol:1–8
Pithadia AB, Kakadia N (2010) Guillain-Barré syndrome (GBS). Pharmacol Rep 62(2):220–232
Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, Zou W, Zhan J, Wang S, Xie Z (2005) Multiple organ infection and the pathogenesis of SARS. J Exp Med 202(3):415–424
Zhang QL, Ding YQ, Hou JL, He L, Huang ZX, Wang HJ, Cai JJ, Zhang JH, Zhang WL, Geng J (2003) Detection of severe acute respiratory syndrome (SARS)-associated coronavirus RNA in autopsy tissues with in situ hybridization. Di 1 jun yi da xue xue bao= Acad J First Med Coll PLA 23 (11):1125–1127
Montalvan V, Lee J, Bueso T, De Toledo J, Rivas K (2020) Neurological manifestations of COVID-19 and other coronavirus infections: a systematic review. Clin Neurol Neurosurg 194:105921
Nikoletseas MM (2020) Neural and non-neural transport in COVID-19. MICHAEL NIKOLETSEAS,
Mao X-Y, Jin W-L (2020) The COVID-19 pandemic: consideration for brain infection. Neuroscience
Wan Y, Shang J, Graham R, Baric RS, Li F (2020) Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol 94(7)
Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM (2011) Classical renin-angiotensin system in kidney physiology. Compr Physiol 4(3):1201–1228
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell
Parsamanesh N, Pezeshgi A, Hemmati M, Jameshorani M, Saboory E (2020) Neurological manifestations of coronavirus infections: role of angiotensin-converting enzyme 2 in COVID-19. Int J Neurosci 1–11
Doo FX, Kassim G, Lefton DR, Patterson S, Pham H, Belani P (2020) Rare presentations of COVID-19: PRES-like leukoencephalopathy and carotid thrombosis. Clin Imaging 69:94–101
Doobay MF, Talman LS, Obr TD, Tian X, Davisson RL, Lazartigues E (2007) Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol-Regul Integr Compar Physiol 292(1):R373–R381
Xia H, Lazartigues E (2008) Angiotensin-converting enzyme 2 in the brain: properties and future directions. J Neurochem 107(6):1482–1494
Al‐Sarraj S, Troakes C, Hanley B, Osborn M, Richardson MP, Hotopf M, Bullmore E, Everall IP (2020) Invited Review: The spectrum of neuropathology in COVID‐19. Neuropathol Appl Neurobiol
Baig AM (2020) Neurological manifestations in COVID-19 caused by SARS-CoV-2. CNS Neurosci Ther 26(5):499
Baig AM, Khaleeq A, Ali U, Syeda H (2020) Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host–virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci 11(7):995–998
Iadecola C, Anrather J, Kamel H (2020) Effects of COVID-19 on the nervous system. Cell
Davies J, Randeva HS, Chatha K, Hall M, Spandidos DA, Karteris E, Kyrou I (2020) Neuropilin-1 as a new potential SARS-CoV-2 infection mediator implicated in the neurologic features and central nervous system involvement of COVID-19. Mol Med Rep 22(5):4221–4226
Ulrich H, Pillat MM (2020) CD147 as a target for COVID-19 treatment: suggested effects of azithromycin and stem cell engagement. Stem Cell Rev Rep 16(3):434–440
Istifli ES, Tepe AŞ, Sarikürkcü C, Tepe B (2020) Interaction of certain monoterpenoid hydrocarbons with the receptor binding domain of 2019 novel coronavirus (2019-nCoV), transmembrane serine protease 2 (TMPRSS2), cathepsin B, and cathepsin L (CatB/L) and their pharmacokinetic properties. Turk J Biol 44(3):242
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181(2):271–280
Uversky VN, Elrashdy F, Aljadawi A, Ali SM, Khan RH, Redwan EM (2021) Severe acute respiratory syndrome coronavirus 2 infection reaches the human nervous system: how? J Neurosci Res 99(3):750–777
Qiao J, Li W, Bao J, Peng Q, Wen D, Wang J, Sun B (2020) The expression of SARS-CoV-2 receptor ACE2 and CD147, and protease TMPRSS2 in human and mouse brain cells and mouse brain tissues. Biochem Biophys Res Commun 533(4):867–871
Shang J, Wan Y, Luo C, Ye G, Geng Q, Auerbach A, Li F (2020) Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci 117(21):11727–11734
Zheng C, Kar I, Chen CK, Sau C, Woodson S, Serra A, Abboud H (2020) Multiple sclerosis disease-modifying therapy and the COVID-19 pandemic: implications on the risk of infection and future vaccination. CNS Drugs 34(9):879–896
Zanin L, Saraceno G, Panciani PP, Renisi G, Signorini L, Migliorati K, Fontanella MM (2020) SARS-CoV-2 can induce brain and spine demyelinating lesions. Acta Neurochirurgica:1–4
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, Collaboration HLHAS (2020) COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet (London, England) 395(10229):1033
Zoghi A, Ramezani M, Roozbeh M, Darazam IA, Sahraian MA (2020) A case of possible atypical demyelinating event of the central nervous system following COVID-19. Multiple Sclerosis Relat Disord 44:102324
Brun G, Hak J-F, Coze S, Kaphan E, Carvelli J, Girard N, Stellmann J-P (2020) COVID-19—White matter and globus pallidum lesions: Demyelination or small-vessel vasculitis? Neurol-Neuroimmunol Neuroinflamm 7(4):e777
Agarwal A, Pinho M, Raj K, Frank FY, Bathla G, Achilleos M, Oneill T, Still M, Maldjian J (2020) Neurological emergencies associated with COVID-19: stroke and beyond. Emerg Radiol 27(6):747–754
Karapanayiotides T, Geka E, Prassopoulos P, Koutroulou I, Kollaras P, Kiourtzieva E, Pourzitaki C, Veroniki F, Sintila S-A, Astreinidis A (2020) Concentric demyelination pattern in COVID-19-associated acute haemorrhagic leukoencephalitis: a lurking catastrophe? Brain
Khandelwal K, Puranik M, Gupta V, Khandelwal G, Dave PK, Hirve M (2021) COVID-19 associated acute demyelination masquerading as stroke: a case report. Egypt J Radiol Nucl Med 52(1):1–4
Bracaglia M, Naldi I, Govoni A, Ventura DB, De Massis P (2020) Acute inflammatory demyelinating polyneuritis in association with an asymptomatic infection by SARS-CoV-2. J Neurol 267(11):3166–3168
Wong CK, Lam CWK, Wu AKL, Ip WK, Lee NLS, Chan IHS, Lit LCW, Hui DSC, Chan MHM, Chung SSC (2004) Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol 136(1):95–103
Kurtzke JF (1993) Epidemiologic evidence for multiple sclerosis as an infection. Clin Microbiol Rev 6(4):382–427
Gerna G, Percivalle E, Sarasini A, Campanini G, Piralla A, Rovida F, Genini E, Marchi A, Baldanti F (2007) Human respiratory coronavirus HKU1 versus other coronavirus infections in Italian hospitalised patients. J Clin Virol 38(3):244–250
Lu L, Zhang H, Zhan M, Jiang J, Yin H, Dauphars DJ, Li S-Y, Li Y, He Y-W (2020) Preventing mortality in COVID-19 patients: which cytokine to target in a raging storm? Front Cell Dev Biol 8:677
Du N, Chen H, Zhang Q, Che L, Lou L, Li X, Zhang K, Bao W (2020) A case series describing the epidemiology and clinical characteristics of COVID-19 infection in Jilin Province. Virulence 11(1):482–485
Long C, Xu H, Shen Q, Zhang X, Fan B, Wang C, Zeng B, Li Z, Li X, Li H (2020) Diagnosis of the Coronavirus disease (COVID-19): rRT-PCR or CT? Eur J Radiol 126:108961
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet 395(10223):497–506
Guan W-j, Ni Z-y, Hu Y, Liang W-h, Ou C-q, He J-x, Liu L, Shan H, Lei C-l, Hui DSC (2020) Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382(18):1708–1720
Feng X, Li S, Sun Q, Zhu J, Chen B, Xiong M, Cao G (2020) Immune-inflammatory parameters in COVID-19 cases: a systematic review and meta-analysis. Front Med 7:301
Cao X (2020) COVID-19: immunopathology and its implications for therapy. Nat Rev Immunol 20(5):269–270
Cheng C, Xiaorong Z, Zhenyu J, Weifeng H (2020) Research progress on the mechanism of cytokine storm induced by new coronavirus pneumonia and related immunotherapy. Chin J Burns 36
Ameri A, Rahnama N, Bozorgmehr R, Mokhtari M, Farahbakhsh M, Nabavi M, Shoaei SD, Izadi H, Kashi ASY, Dehbaneh HS (2020) Low-dose whole-lung irradiation for COVID-19 pneumonia: short course results. Int Radiat Oncol Biology Phys 108(5):1134–1139
Anwar MM (2021) Immunotherapies and COVID-19 related Neurological manifestations: A Comprehensive review article. J Immunoassay Immunochem 1–16
Henry BM, Vikse J, Benoit S, Favaloro EJ, Lippi G (2020) Hyperinflammation and derangement of renin-angiotensin-aldosterone system in COVID-19: a novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin Chim Acta 507:167–173
Mohammadi S, Moosaie F, Aarabi MH (2020) Understanding the immunologic characteristics of neurologic manifestations of SARS-CoV-2 and potential immunological mechanisms. Mol Neurobiol 57(12):5263–5275
Ellul MA, Benjamin L, Singh B, Lant S, Michael BD, Easton A, Kneen R, Defres S, Sejvar J, Solomon T (2020) Neurological associations of COVID-19. Lancet Neurol
Biswas I, Khan GA (2020) Coagulation disorders in COVID-19: role of toll-like receptors. J Inflamm Res 13:823
Safaei S, Karimi-Googheri M (2020) Letter to the Editor: Toll-like receptor antagonists as a potential therapeutic strategy against cytokine storm in COVID-19-Infected Patients. Viral Immunology
Hasan U, Chaffois C, Gaillard C, Saulnier V, Merck E, Tancredi S, Guiet C, Brière F, Vlach J, Lebecque S (2005) Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J Immunol 174(5):2942–2950
Mukherjee S, Karmakar S, Babu SPS (2016) TLR2 and TLR4 mediated host immune responses in major infectious diseases: a review. Br J Infect Dis 20(2):193–204
Duffy L, O’Reilly SC (2016) Toll-like receptors in the pathogenesis of autoimmune diseases: recent and emerging translational developments. ImmunoTargets Therap 5:69
Kawasaki K, Akashi S, Shimazu R, Yoshida T, Miyake K, Nishijima M (2000) Mouse toll-like receptor 4· MD-2 complex mediates lipopolysaccharide-mimetic signal transduction by Taxol. J Biol Chem 275(4):2251–2254
Kawai T, Akira S (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34(5):637–650
Soy M, Keser G, Atagündüz P, Tabak F, Atagündüz I, Kayhan S (2020) Cytokine storm in COVID-19: pathogenesis and overview of anti-inflammatory agents used in treatment. Clin Rheumatol 39:2085–2094
Khanmohammadi S, Rezaei N (2021) Role of Toll‐like receptors in the pathogenesis of COVID‐19. J Med Virol
Vargas G, Geraldo LHM, Salomão N, Paes MV, Lima FRS, Gomes FCA (2020) Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and glial cells: Insights and perspectives. Brain, Behav Immun Health 7:100127
Li L, Acioglu C, Heary RF, Elkabes S (2020) Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav Immun
Choudhury A, Mukherjee S (2020) In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol 92(10):2105–2113
Brandão SCS, Ramos JdOX, Dompieri LT, Godoi ETAM, Figueiredo JL, Sarinho ESC, Chelvanambi S, Aikawa M (2020) Is Toll-like receptor 4 involved in the severity of COVID-19 pathology in patients with cardiometabolic comorbidities? Cytokine & Growth Factor Reviews
Liu Y, Sawalha AH, Lu Q (2021) COVID-19 and autoimmune diseases. Curr Opin Rheumatol 33(2):155
Salle V (2021) Coronavirus-induced autoimmunity. Clin Immunol 108694
Hussein HM, Rahal EA (2019) The role of viral infections in the development of autoimmune diseases. Crit Rev Microbiol 45(4):394–412
Franke C, Ferse C, Kreye J, Reincke SM, Sanchez-Sendin E, Rocco A, Steinbrenner M, Angermair S, Treskatsch S, Zickler D (2021) High frequency of cerebrospinal fluid autoantibodies in COVID-19 patients with neurological symptoms. Brain Behav Immun 93:415–419
Grimaldi S, Lagarde S, Harlé J-R, Boucraut J, Guedj E (2020) Autoimmune encephalitis concomitant with SARS-CoV-2 infection: insight from 18F-FDG PET imaging and neuronal autoantibodies. J Nucl Med 61(12):1726–1729
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Ethical approval
The manuscript is not be submitted to more than one journal for simultaneous consideration. The submitted work is original and have not been published elsewhere in any form or language (partially or in full). A single study is not be split up into several parts to increase the quantity of submissions and is not submitted to various journals or to one journal over time (i.e. ‘salami-slicing/publishing’). Concurrent or secondary publication is sometimes justifiable, provided certain conditions are met. Examples include: translations or a manuscript that is intended for a different group of readers. Results are presented clearly, honestly, and without fabrication, falsification or inappropriate data manipulation (including image-based manipulation). Authors adhered to discipline-specific rules for acquiring, selecting and processing data. No data, text, or theories by others are presented as if they were the author’s own (‘plagiarism’). Proper acknowledgements to other works must be given (this includes material that is closely copied (near verbatim), summarized and/or paraphrased), quotation marks (to indicate words taken from another source) are used for verbatim copying of material, and permissions secured for material that is copyrighted.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shabani, Z. Demyelination as a result of an immune response in patients with COVID-19. Acta Neurol Belg 121, 859–866 (2021). https://doi.org/10.1007/s13760-021-01691-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13760-021-01691-5