
Abstract
The analysis of mitochondrial DNA polymorphism has been applied in multiple organisms to obtain information about species biology, ecology, population dynamics, and evolution. In this manuscript, the complete sequencing and characterization of the mitochondrial genome (mtGenome) of Tetrapedia diversipes are reported and discussed from comparative and evolutionary perspectives among all mtGenomes available for bees so far. The T. diversipes mtGenome is 15,358 bp long and exhibits the typical set of genes and an A+T-rich region of 588 bp. The overall base composition is biased towards A/T (84.3%), with 42.6% A, 41.7% T, 9.8% C, and 5.9% G nucleotides. The obtained data also comprise the mitochondrial DNA methylation and single-nucleotide polymorphic sites of one T. diversipes population. Transcription follows the “tRNA punctuation” model, with at least three primary polycistronic transcripts that are posteriorly processed. Additionally, higher expression rates of the 16S gene suggest the existence of an exclusive transcription site in this region, and the differential expression of the 12S gene between larvae and adults reveals different isoforms for this gene. The sequence order of protein-coding and rRNA genes is conserved across different bee lineages, and differences are restricted to tRNA gene positions. The present results characterize numerous understudied aspects of bee mtGenomes, and a major evolutionary review of this molecule within the group is provided. Therefore, this work is a valuable resource for studying mitochondrial molecular biology and evolution in bees.
Similar content being viewed by others
1 Introduction
Since the completion of the first mitochondrial genome (mtGenome) from humans in 1981 (Anderson et al. 1981), thousands of these molecules have been sequenced. At present, according to the NCBI database, the genome of this organelle is the most sequenced eukaryotic genome, and approximately 80 complete or nearly complete mtGenomes of bees have been made publicly available. In contrast to this trend, other characteristics of mitochondrial DNA (mtDNA) molecules have rarely been described (Smith 2015; Tian and Smith 2016). Therefore, data on other mitochondrial characteristics, such as mitochondrial transcription (Stewart and Beckenbach 2009; Margam et al. 2011) and methylation (Iacobazzi et al. 2013; Mawlood et al. 2016), are missing for most organisms.
Nevertheless, a great number of studies rely on the analysis of mtDNA sequence polymorphisms to obtain information on species biology, ecology, population dynamics, and evolution (Avise et al. 1987; Beheregaray 2008; Hickerson et al. 2010). In Hymenoptera, mtGenomes have been especially useful for evolutionary and phylogenetic analyses because rearrangements in this molecule are frequent in this group (Mao et al. 2015).
The vast majority of bee species are solitary (Michener 2007), and although they are top pollinators of crops and wild plants, solitary bees are extremely underrepresented in genetic studies (Neumann and Seidelmann 2006). Tetrapedia diversipes Klug (1810) is a solitary oil-collecting bee of the tribe Tetrapediini (Michener 2007) distributed from Costa Rica to Argentina (Moure 2012) that nests in pre-existing holes in wood, including trap nests (Aguiar and Martins 2002; Alves-dos-Santos 2003; Gazola and Garófalo 2009). Given its wide distribution and nesting aggregation behavior, this bee species is easy to manage and sample, which makes T. diversipes a useful Neotropical solitary bee model.
In the present manuscript, the sequencing and characterization of the T. diversipes mtGenome is described and discussed from an evolutionary perspective. The obtained data comprise the complete sequencing and annotation (including the A+T region) results for the molecule along with its transcriptomic and DNA methylation profiles and the identification of single-nucleotide polymorphism sites (SNPs) from one population. In addition to the identified genomic characteristics, inferences about the evolutionary dynamics of the mtDNA in bees were made based on a major comparative review with other bee mtGenome sequences available at GenBank.
2 Materials and methods
2.1 Sampling and DNA extraction
All samples were obtained from the same trap nest aggregation in São Paulo, Brazil (23° 33′ S), thus representing one population of T. diversipes. To increase the sequencing coverage and avoid numts (nuclear copies of mitochondrial origin), some DNA extracts were enriched for mtGenome sequences using the protocol described by Françoso et al. (2015). Library preparation and sample sequencing on both Illumina® and Sanger platforms were performed by Macrogen (South Korea). For DNA methylation analyses, whole-bisulfite sequencing was conducted on the Illumina® NextSeq 500 platform at the University of Georgia following the protocol described in (Urich et al. 2015).
2.2 The complete mitochondrial genome sequence, assembly and annotation
To guarantee assembly quality and sequence completeness, different strategies were combined to obtain the entire mtGenome of T. diversipes:
-
1.
Mitochondrial enrichment followed by NGS—the first partial assembly. Under this approach, Illumina® sequencing of 4,000,000 single reads (approximate size of 100 bp), representing more than 2,000-fold coverage, was performed in DNA extracts enriched for mtDNA molecules from one female bee. Library preparation was performed according to the instructions of the manufacturer as adapted by Monica Carlsen (personal communication). The quality of the Illumina reads was evaluated using the FastQC v0.11.2 program (Andrews 2010). This dataset was then independently assembled in two ways. [1A] First, Geneious Pro v5.6.3 software (Kearse et al. 2012) and the raw reads were used. Reference assemblies (based on the mtGenomes of Apis mellifera [NC_001566] and Bombus ignitus [NC_010967]) and denovo assembly were performed. Contigs congruent in all three assemblies were then extended, realigning all reads to them iteratively. After each iteration, the extended sequence was manually curated. [1B] The second assembly strategy relied on the MITObim v1.8 program (Hahn et al. 2013). Under this approach, reads were initially cleaned by removing the first two nucleotides with the FASTX v0.0.14 toolkit (Gordon 2009), and low-quality bases (phred score below 20) and small reads (less than 20 nucleotides) were removed with Seqyclean v1.9.10 (Zhbannikov et al. 2017). To maintain a maximum coverage of 50-fold, as indicated for the MITObim pipeline, the cleaned reads were digitally normalized before assembly following the protocol of Brown et al. (2012). Paired reads were interleaved using khmer v2.0 (Crusoe et al. 2015). The final MITObim assembly was conducted with a combination of three different approaches using denovo and reference-based extensions, referred to here named ASB0, ASB1, and ASB2. For the de novo assembly (ASB0), a fragment of the Cytochrome C Oxidase I (CO1) gene from T. diversipes was used as the seed. In the reference assembly (ASB1), the complete mitochondrial genome from B. ignitus was used as a reference. For the ASB2 assembly, the contigs resulting from ASB1 were used as extension seeds for a new denovo assembly. Posteriorly, ASB0, ASB1, and ASB2 were aligned using Geneious to obtain a manually curated consensus sequence combining all three assemblies. This consensus sequence was used as a reference in a new assembly with MITObim, which returned the final 1B assembly. Finally, assemblies 1A and 1B were compared to generate a consensus sequence.
-
2.
NGS of nonenriched DNA—the second partial assembly. Independent sequencing of the total DNA from one T. diversipes male was performed using the TruSeq DNA PCR-Free kit for paired-end library preparation and the Illumina® HiSeq 2500 platform, generating 293,600,062 paired reads. Based on the first partial genome as a reference, these new sequencing data were assembled under a reference-guided approach with Geneious v9.1.6. All parameters used were the defaults, and the Bowtie2 aligner was set to “fast accurate read mapper and end to end alignment”. Due to the presence of paired-end reads, the coverage of the aligned reads from this new dataset was more homogeneous than that generated previously, allowing the extension and completion of some missing regions compared with the first partial mtGenome.
-
3.
Sanger sequencing—mtGenome final assembly. PCR extension followed by Sanger sequencing was performed to solve low-support regions (i.e., regions with low coverage) and ambiguities between assemblies and to recover missing regions. Several primers were designed using Primer3 (Rozen and Skaletsky 2000) (Table S1; Figure S1) and tested using all the possible combinations for the L and H strands. The PCR conditions followed Françoso and Arias (2013), with the annealing temperatures ranging from 38 to 56 °C. The amplification of the A+T-rich region failed in direct sequencing attempts; thus, this region was cloned into the pGEM plasmid vector (PROMEGA), which was then used to transform competent Escherichia coli DH5-α cells prior to sequencing. Genome annotation was performed using the MITOS web server (Bernt et al. 2013), which employs a specialized algorithm that uses similarities and structure-based searches to improve mitochondrial genome annotation.
2.3 Transcript assembly
The RNA-Seq data for T. diversipes adults and larvae were obtained from Araujo et al. (2018). Mitochondrial transcripts from both developmental stages were assembled using the reference and denovo assembly methods. The reference assembly of the transcripts was generated using the HISAT2 v2.0.5 (Kim et al. 2015) and StringTie v1.2.2 (Pertea et al. 2015) programs based on the final mtGenome. In the denovo assembly, mitochondrial transcripts were identified through a blastn search (minimum e-value 1e-5) from the complete transcriptome of T. diversipes (Araujo et al. 2018) against the complete mtGenome. Redundant transcripts, i.e., transcripts overlapping the same mitochondrial region in the denovo assembly, were manually curated so that only the largest transcript was retained.
2.4 SNP identification
Transcriptomic data were also used to identify SNPs in the mitochondrial genome of T. diversipes. Therefore, the SNP information represents the mitochondrial diversity of 36 individuals from the studied population (Araujo et al. 2018). The cleaned read alignments (bam files) used previously for transcriptome reference assembly were analyzed via the variant-calling pipeline combining the SAMtools mpileup (v0.1.19) and BCFtools view (v0.1.19) utilities (Li et al. 2009). Only SNPs with a minimum quality of 30 and 30-fold coverage were selected. SNPs were manually curated through alignment checking of the reads with the IGV tool (Robinson et al. 2011).
2.5 DNA methylation analyses
DNA methylation data were obtained from the whole-body DNA extract of one founder female. Reads obtained from bisulfite sequencing were cleaned using the Trim Galore v0.4.3 (Krueger 2012) wrapper script, with default parameters. The alignment of the reads to the mitochondrial genome and methylation calling were executed following the Bismark v0.17.0 pipeline (Krueger and Andrews 2011). Alignment quality was evaluated using Qualimap v2.2 (García-Alcalde et al. 2012).
2.6 mtGenome comparisons
Twenty-three mtGenomes, comprising the genomes of bees from thirteen genera and six families, were compared. The wasp Philanthus triangulum (Apoidea), and Squilla mantis, which represents the ancestral pancrustacean mitochondrial genome organization (Cook et al. 2005), were used as external groups. All genomes were aligned using the MUSCLE algorithm (Edgar 2004) implemented in Geneious 9.1.6, and the rearrangements were visually compared. The length and non-ambiguous base composition were also obtained with Geneious. Only one complete mtGenome per genus of the Anthophila lineage was used in the analyses, except when differences in arrangement were observed. When more than one mtGenome was available per genus, we selected the most complete molecule (i.e., the molecule containing the greatest number of protein-coding genes, PCGs) including the A+T-rich region and the longest sequence assembled as the representative. Partial genomes were also used when no others were available to represent a genus, but only mtGenomes verified at GenBank were employed.
3 Results
3.1 Genomic characterization
3.1.1 mtGenome sequence
The complete mtGenome of T. diversipes was 15,358 bp in length and exhibited the typical set of genes (Table I), including 13 PCGs, 22 transfer RNAs (tRNA), two ribosomal RNAs (rRNA) and the A+T-rich noncoding region (GenBank accession number: MN732885). The overall base composition was biased towards A/T (84.3%), with 42.6% A, 41.7% T, 9.8% C, and 5.9% G. The PCGs CO1, CO2, and CytB presented the lowest A/T content among the other PCGs (Table S2), and, in general, a lower A/T content is correlated with higher average coverage of short reads (Figures S2 and S3).
3.1.2 Transcription analyses
From the adult RNA-Seq analysis, 6,408,822 paired reads were aligned to the mtGenome (mean coverage of 37,323-fold, 167,141 s.d.), and from the larval data, 9,495,025 paired reads were aligned (mean coverage of 55,313-fold, 261,975 s.d.) [alignments available at NCBI—BioProject ID: PRJNA590962]. The assembly of the transcripts resulted in three contigs for both developmental stages when the reference genome was used and six contigs for larvae and ten for adults via the denovo assembly method. The reduction in coverage was more pronounced in regions encoding tRNAs, affecting transcript continuity, especially when the de novo assembly method is used (Figure 1). In the same figure, it is possible to note the alignment of few reads in a small portion of the A+T-rich region (between 164 and 362 bp and 432 and 502 bp).

. Transcription of the mitochondrial genome of Tetrapedia diversipes. From inner to outer circles: 1, representation of genomic positions; 2, genome annotation. Gray: protein-coding genes; orange: tRNA; purple: rRNA, and red: A+T-rich region. 3, GC content graph, where the outer peaks represent guanine or cytosine nucleotide bases; 4 and 7, in pink: mitochondrial transcripts obtained using the reference assembly method (4 from larvae and 7 from adult data); 5 and 8, in dark purple: mitochondrial transcripts obtained using the denovo assembly method (5 from larvae and 8 from adult data); 6 and 9: expression coverage of RNA-Seq sequencing, in which blue areas represent coverage greater than 50-fold. In circle 6, larval sample coverage is represented, and the red areas are regions with coverage < 50; in circle 9, adult sample coverage is shown, and orange areas represent coverage < 50.
Coverage analyses of the transcripts also indicated a bias in the expression of ribosomal RNAs. In both developmental stages, the second-highest-coverage region of the mtGenome was obtained for the CO1 gene (with approximately 250,000-fold maximum coverage). Nevertheless, the coverage in the 16S region reached values six-fold greater than those in adults (≅ 1,450,000-fold maximum coverage) and up to nine-fold greater in larvae (≅ 2,200,000-fold maximum coverage) (Figure 2). This enormous increase in coverage was only observed for this gene; therefore, it was not skewed by a richer GC content and did not include the 12S ribosomal gene region (200-fold maximum coverage in adults and 51-fold in larvae). Additionally, the comparison of the 12S sequences from adults and larvae revealed a sudden decrease in coverage in the 5′ portion of 12S in larvae (Figure S4).

. Bulk RNA-Seq coverage and cytosine methylation (mC) across the mtGenome. From top to bottom: the first panel shows the bulk distribution of RNA-Seq reads; the second panel shows the GC percentage for comparison; the third panel shows the percentage of methylation estimated for all cytosines from a female founder sample; and in the last panel, PCG positions are indicated along the mtGenome for reference. In the methylation panel, the red dashed line marks the mean mC level in the whole mtGenome. Below the panels, the order of the main mitochondrial genes verified for T. diversipes is presented.
3.1.3 SNPs
Eighty-one SNPs were identified in the studied population (Table S3). Most of the SNPs (64.2%) were in protein-coding regions, particularly at the third codon position, which led to synonymous mutations in most cases. Only 18.5% of the SNPs resulted in nonsynonymous substitutions. The ND4 gene presented the highest number of nonsynonymous SNPs (five) and the greatest number of SNPs (twelve). However, CO3 was the PCG with the highest SNP ratio (Figure S5). The number of SNPs observed in coding regions was correlated with the region size (Pearson coefficient = 0.74) (Figure S5).
3.1.4 DNA methylation
Bisulfite sequencing coverage across the mtDNA was uniform, with a mean coverage of 1,566-fold (279 s.d.). Altogether, 161,789 reads with a mean length of 148 bp were aligned to the mitochondrial genome [alignments available at NCBI—BioProjectID: PRJNA590962]. The mean overall methylation level of the genome was 0.96%, with 0.6% methylated cytosines in the CpG context, 0.7% in the CHG context and 1.0% in the CHH context. Among all the cytosines identified as methylated, 96.6% occurred at a non-CpG site in the genome. In Figure 2, where the methylation profile of all cytosines is indicated, it can be noted that that C-methylation generally showed the opposite pattern to the GC content across the genome. Indeed, GC content and C-methylation were negatively correlated in the T. diversipes mtGenome (Pearson coefficient = −0.54).
3.1.5 Genomic comparisons across multiple bee species
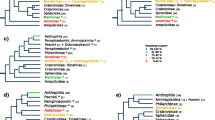
The general profiles of the analyzed genomes are provided in Table S4. Most of the 23 mtGenomes analyzed presented the typical gene content of 13 PCGs, two rRNAs, 22 tRNAs, and one extra copy of each of tRNALeu and tRNASer. Apis koschevnikovi nevertheless presented a duplication of tRNAMet rather than tRNASer. Among the 21 bee genomes, Melipona bicolor presented the highest A/T ratio (87%), and bees from the Andrenidae family exhibited the lowest (78.6% and 79.4%). The longest mtGenome was from Bombus consobrinus, which consisted of 17,966 bp (Table S4). The order and orientation of the PCGs and rRNAs were conserved in all species. Differences were restricted to changes in tRNA positions, possibly due to local inversions, translocations, and shuffling of adjacent tRNA clusters (Table I, Figure 3).

. Putative synapomorphies in bee mitochondrial genomes. The wasp Philanthus triangulum and Squilla mantis, which represents the ancestral pancrustacean mitochondrial genome organization, were used as external groups.
Most of the events were translocations, especially from tRNA cluster 6 (ND3-ND5 junction) to cluster 1 (A+T-rich region-ND2 junction), and many of the variations represented putative synapomorphies at the family or genus level (Figure 3). Compared with the ancestral pancrustacean genome, represented here by Squilla mantis, T. diversipes differed in cluster 6 (including a shuffling of tRNAMet, shuffling and inversion of tRNAArg, inversion of tRNAGlu, and translocation of tRNAAla from cluster 6 to cluster 1), in cluster 2 (shuffling of tRNATrp), and in cluster 4 (shuffling between tRNALys and tRNAAsp) (Figure 3).
4 Discussion
4.1 Genomic characterization
Due to the repetitive nature and high A/T content of the mitochondrial genomes of bees, the completion of the T. diversipes mtGenome was challenging, even with the use of high-throughput sequencing. Therefore, multiple approaches for sequencing and data analyses were necessary. Although Illumina sequencing using enriched mtDNA and total DNA generated a very high average coverage, this coverage was extremely heterogeneous and was correlated with the GC content across the genome (Figures S2 and S3). In general, regions with a lower A/T content, such as the CO1, CO2, and CytB genes, were well represented, while other areas, such as the genes encoding tRNAs, ND2, and 12S and the A+T-rich region, presented lower coverage and a reduced mapping quality (Table S2; Figure S2). This methodological bias was most likely induced by problems in the alignment of short reads into repetitive regions, and it greatly impacted the assembly of these areas. Consequently, the effective completion of the A+T-rich region was only possible by traditional methods of amplification with cloning and Sanger sequencing.
In addition to the complete sequence of the mtGenome, we also report the polymorphic sites of the studied population in this molecule (Table S3). These data might be useful for population genetic, phylogenetic and conservation studies (revised by Smith 2015) and are especially relevant considering all of the efforts recently applied to develop conservation strategies for native bees (Dicks et al. 2016; Potts et al. 2016), due to their importance as pollinators of native and commercial plants (Garibaldi et al. 2014, 2016). In general, the number of SNPs was correlated with gene size (Figure S5), but a higher ratio of SNPs could be observed in the CO3 and ND4 genes, suggesting that these genes are good candidates for phylogenetic and taxonomic studies.
Analyses of the mitochondrial transcriptome led to some insightful observations about mtDNA expression dynamics in this species. These results suggest that mitochondrial genome transcription in T. diversipes follows the “tRNA punctuation” model (Ojala et al. 1981), with the formation of at least three primary polycistronic transcripts that are posteriorly processed at tRNA positions, leading to the potential formation of 13 mitochondrial mRNAs. This is supported by the reduction in the coverage of the mitochondrial transcriptome in tRNA regions and the reconstruction of three distinct transcripts after mitochondrial transcriptome assembly. This processing mechanism of mitochondrial mRNA through tRNA punctuation is apparently conserved in many organisms (Taanman 1999; Stewart and Beckenbach 2009), including bees (Crozier and Crozier 1993). However, mRNA transcription and processing in each species is variable, and the number of primary and processed transcripts may differ (Taanman 1999; Stewart and Beckenbach 2009; Neira-Oviedo et al. 2011; Tian and Smith 2016).
As can be observed in Figure 2, the coverage of the RNA-Seq data across the mtDNA showed a considerably higher expression rate of 16S. Higher expression of this gene has also been observed in other insects such as Drosophila (Torres et al. 2009), mosquitoes (Neira-Oviedo et al. 2011), and one bee (Araujo and Arias 2019). In mammals, increased expression of ribosomal genes has been associated with the existence of an exclusive transcription site for the 16S and the 12S genes (Taanman 1999). In T. diversipes mtDNA, the existence of this differential transcription site is also supported by the increase in the coverage of 16S; however, the region of higher sequence coverage does not comprise the smaller ribosomal gene. Reduced transcription coverage of 12S decoupled from 16S has also been reported in Drosophila (Torres et al. 2009) and M. bicolor (Araujo and Arias 2019) and might be driven by a methodological bias caused by the reduced polyA tail of the 12S mRNA, rather than by differences in transcription initiation itself (Stewart and Beckenbach 2009). Considering that the library preparation method used in the present study for RNA-Seq sequencing relied on the polyA structure to select mRNAs, it is possible that the employed methodology was not appropriate to efficiently capture 12S transcripts (Neira-Oviedo et al. 2011). Therefore, although some evidence suggests the existence of an alternative transcription site for 16S in T. diversipes and other insects, it is still unclear if 12S is also transcribed.
Another intriguing result from transcription data was the great decrease in coverage observed in the 12S 5′ region in larvae compared with that in adults (Figure S4). This is unlikely to be a result of methodological bias because the two samples were prepared using the same methods; thus, this difference suggests that T. diversipes presents at least two distinct isoforms of this mitochondrial gene, one of which is transcribed in the larval stage and the other in the adult stage. Evidence of multiple isoforms of the 12S has been reported previously in the bee M. bicolor, in which the RNASeq coverage in 12S conflicted with the complete annotation of this gene in the 5′ region (Araujo and Arias 2019) and for the stink bug Erthesina fullo on the basis of long read sequencing (Gao et al. 2016).
Low levels of gene expression in the control region of mtDNA have been reported in mammals, where they are associated with the production of an initiation primer or a long non-coding RNA that functions in the control of both the transcription and replication of the mitochondrial genome (Taanman 1999; Gao et al. 2018). Accordingly, it is possible that this small portion of the A+T-rich region in the RNA-Seq read alignment (between 164 and 362 pb and 432 and 502 pb), refers to a similar region functionally relevant for the initiation of transcription and/or replication of the mtGenome in T. diversipes (and possibly other bees, Araujo and Arias 2019). However, as discussed previously, the A+T-rich region presents low complexity and is very repetitive; consequently, the alignment of short reads to this region cannot be trusted without further evidences.
The first studies on DNA methylation in mitochondria were performed in the 1970s (Nass 1973), but it was not until 2011, after the association of the DNMT1, DNMT3A, and DNMT3B methyltransferases with the mitochondria (as reviewed in Iacobazzi et al. 2013), that the importance of DNA methylation in mtDNA dynamics began to receive more attention. Evidence suggests that, similar to methylation of nuclear DNA, changes in mtDNA methylation are driven by environmental elements (Iacobazzi et al. 2013). Differential DNA methylation in mitochondria has been associated with aging (Mawlood et al. 2016), diseases (Infantino et al. 2011; Iacobazzi et al. 2013) and metabolic processes that play a role in oxidative stress (as discussed in van der Wijst and Rots 2015). Specifically, for T. diversipes foundresses, changes in mitochondrial gene expression are related to differences between individuals from different reproductive generations (Araujo et al. 2018). Therefore, it would be interesting to determine whether the changes in the pattern of mtDNA methylation documented here are associated with these changes and other mechanisms of expression control in mitochondria.
4.2 Genomic comparisons across multiple bee species
Although mitochondrial genomes have been described as highly conserved (Wolstenholme 1992), the order in which genes are arranged is more variable than initially predicted, especially for tRNA genes. For example, cluster 6 (ND3-ND5 junction) is a region of frequent rearrangements in Hymenoptera that are rarely described in other groups of Insecta (Dowton et al. 2003). Interestingly, tRNA translocations are not reciprocal in this region; i.e., this cluster tends to lose genes instead of gaining them from other clusters in bee mtGenomes (Dowton et al. 2003).
On the basis of the comparison of Apis, Melipona, and Bombus, Dowton et al. (2009) suggested that the translocation of tRNAAla to cluster 1 is an Apidae synapomorphy. In the present work, this hypothesis was corroborated by the analyses of the genus Tetrapedia and Nomada and extended to the family Megachilidae. It was also suggested that the shuffling between tRNAAsp and tRNALys in cluster 4 (CO2-ATP8 junction) would erroneously phylogenetically group Bombus and Apis because this shuffling was not present in Melipona (Silvestre et al. 2008). However, in Figure 3, it is possible to see that this event is actually distributed among all bees. Thus, the translocation of tRNALys is instead a synapomorphy in Melipona.
Although frequent tRNA rearrangements are common, some genomic positions are highly conserved. For example, tRNAPhe in cluster 6 (ND3-ND5 junction) and tRNAPro in cluster 8 (ND4L-ND6 junction) exhibit the same position and orientation in all bees. It has been suggested that the tRNAPhe position might be under selective constraint because this gene is located at a site where transcription polarity changes; therefore, it could be a putative signal for endonucleolytic cleavage during the maturation of the primary polycistronic transcript (Ojala et al. 1981; Dowton et al. 2003). The same reasoning can be used to explain the position of tRNAPro in cluster 8 (Table I; Figure 1), since both genes are phylogenetically conserved and located at sites of polarity changes. Additionally, these tRNAs are positioned adjacent to cleavage sites of the polycistronic transcripts assembled for T. diversipes (Figure 1), reinforcing the hypothesis of their functional role as maturation signals.
5 Conclusions
Here, we provide a valuable dataset for the mitochondrial genome of T. diversipes, including its complete sequence and annotation, transcription patterns in two life stages, methylated sites in females during nest foundation and population genomic diversity determined through SNP identification. Additionally, we combined the present sequencing data with database sequences to understand the molecular mechanisms underlying mitochondrial genomic evolution in bees through a comparative review of all available bee mtGenomes. The results highlight the importance of tRNA rearrangement events in the evolution of this molecule in bees and the existence of DNA methylation in the T. diversipes mitochondrial genome in a predominant non-CG context, and they show some intricate mechanisms involved in gene expression regulation. The reported analyses and datasets may be used to address important evolutionary questions not only concerning T. diversipes but also for other bee species, especially regarding the underrepresented group of solitary bees.
References
Aguiar AJC, Martins CF (2002) Abelhas e vespas solitárias em ninhos-armadilha na Reserva Biológica Guaribas (Mamanguape, Paraíba, Brasil). Rev Bras Zool 19:101–116. https://doi.org/10.1590/S0101-81752002000500005
Alves-dos-Santos I (2003) Trap-nesting bees and wasps on the university campus in São Paulo, Southeastern Brazil (Hymenoptera: Aculeata). J Kansas Entomol Soc 76:328–334
Anderson S, Bankier AT, Barrell BG, et al (1981) Sequence and organization of the human mitochondrial genome. Nature 290:457–465. https://doi.org/10.1038/290457a0
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Araujo NS, Arias MC (2019) Mitochondrial genome characterization of Melipona bicolor: Insights from the control region and gene expression data. Gene 705:55–59. https://doi.org/10.1016/j.gene.2019.04.042
Araujo NS, Santos PKF, Arias MC (2018) RNA-Seq reveals that mitochondrial genes and long non-coding RNAs may play important roles in the bivoltine generations of the non-social Neotropical bee Tetrapedia diversipes. Apidologie 49:3–12. https://doi.org/10.1007/s13592-017-0542-2
Avise JC, Arnold J, Ball RM, et al (1987) Intraspecific phylogeography: the mitochondrial DNA bridge between population genetics and systematics. Annu Rev Ecol Syst 18:489–522. https://doi.org/10.1146/annurev.es.18.110187.002421
Beheregaray LB (2008) Twenty years of phylogeography: the state of the field and the challenges for the Southern Hemisphere. Mol Ecol 17:3754–3774. https://doi.org/10.1111/j.1365-294X.2008.03857.x
Bernt M, Donath A, Jühling F, et al (2013) MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol 69:313–319. https://doi.org/10.1016/j.ympev.2012.08.023
Brown CT, Howe A, Zhang Q, et al (2012) A Reference-free algorithm for computational normalization of shotgun sequencing data. arXiv 1203.4802 [q-bio.GN]
Cook CE, Yue Q, Akam M (2005) Mitochondrial genomes suggest that hexapods and crustaceans are mutually paraphyletic. Proc R Soc B Biol Sci 272:1295–1304. https://doi.org/10.1098/rspb.2004.3042
Crozier RH, Crozier YC (1993) The mitochondrial genome of the honeybee Apis mellifera: complete sequence and genome organization. Genetics 133:97–117
Crusoe MR, Alameldin HF, Awad S, et al (2015) The khmer software package: enabling efficient nucleotide sequence analysis. F1000Research. 4:900. 1–10. https://doi.org/10.12688/f1000research.6924.1
Dicks L V., Viana B, Bommarco R, et al (2016) Ten policies for pollinators. Science (80) 354:975–976. https://doi.org/10.1126/science.aai9226
Dowton M, Cameron SL, Dowavic JI, et al (2009) Characterization of 67 mitochondrial tRNA gene rearrangements in the hymenoptera suggests that mitochondrial tRNA gene position is selectively neutral. Mol Biol Evol 26:1607–1617. https://doi.org/10.1093/molbev/msp072
Dowton M, Castro LR, Campbell SL, et al (2003) Frequent mitochondrial gene rearrangements at the hymenopteran nad3-nad5 junction. J Mol Evol 56:517–526. https://doi.org/10.1007/s00239-002-2420-3
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. https://doi.org/10.1093/nar/gkh340
Françoso E, Arias MC (2013) Cytochrome c oxidase I primers for corbiculate bees: DNA barcode and mini-barcode. Mol Ecol Resour 13:844–850. https://doi.org/10.1111/1755-0998.12135
Françoso E, Gomes F, Arias MC (2015) A protocol for isolating insect mitochondrial genomes: a case study of NUMT in Melipona flavolineata (Hymenoptera: Apidae). Mitochondrial DNA 27:1–4. https://doi.org/10.3109/19401736.2015.1028049
Gao S, Ren Y, Sun Y, et al (2016) PacBio full-length transcriptome profiling of insect mitochondrial gene expression. RNA Biol 13:820–825. https://doi.org/10.1080/15476286.2016.1197481
Gao S, Tian X, Chang H, et al (2018) Two novel lncRNAs discovered in human mitochondrial DNA using PacBio full-length transcriptome data. Mitochondrion 38:41–47. https://doi.org/10.1016/j.mito.2017.08.002
García-Alcalde F, Okonechnikov K, Carbonell J, et al (2012) Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 28:2678–2679. https://doi.org/10.1093/bioinformatics/bts503
Garibaldi LA, Carvalheiro LG, Leonhardt SD, et al (2014) From research to action: enhancing crop yield through wild pollinators. Front Ecol Environ 12:439–447. https://doi.org/10.1890/130330
Garibaldi LA, Carvalheiro LG, Vaissiere BE, et al (2016) Mutually beneficial pollinator diversity and crop yield outcomes in small and large farms. Science 351:388–391. https://doi.org/10.1126/science.aac7287
Gazola AL, Garófalo CA (2009) Trap-nesting bees (Hymenoptera: Apoidea) in forest fragments of the State of São Paulo, Brazil. Genet Mol Res 8:607–622. https://doi.org/10.4238/vol8-2kerr016
Gordon A (2009) FASTX-Toolkit. http://hannonlab.cshl.edu/fastx_toolkit/
Hahn C, Bachmann L, Chevreux B (2013) Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads - A baiting and iterative mapping approach. Nucleic Acids Res 41:e129. https://doi.org/10.1093/nar/gkt371
Hickerson MJ, Carstens BC, Cavender-Bares J, et al (2010) Phylogeography’s past, present, and future: 10 years after Avise, 2000. Mol Phylogenet Evol 54:291–301. https://doi.org/10.1016/j.ympev.2009.09.016
Iacobazzi V, Castegna A, Infantino V, Andria G (2013) Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol Genet Metab 110:25–34. https://doi.org/10.1016/j.ymgme.2013.07.012
Infantino V, Castegna A, Iacobazzi F, et al (2011) Impairment of methyl cycle affects mitochondrial methyl availability and glutathione level in Down’s syndrome. Mol Genet Metab 102:378–382. https://doi.org/10.1016/j.ymgme.2010.11.166
Kearse M, Moir R, Wilson A, et al (2012) Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12:357–360. https://doi.org/10.1038/nmeth.3317
Krueger F (2012) Trim Galore. http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
Krueger F, Andrews SR (2011) Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27:1571–1572. https://doi.org/10.1093/bioinformatics/btr167
Li H, Handsaker B, Wysoker A, et al (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Mao M, Gibson T, Dowton M (2015) Higher-level phylogeny of the Hymenoptera inferred from mitochondrial genomes. Mol Phylogenet Evol 84:34–43. https://doi.org/10.1016/j.ympev.2014.12.009
Margam VM, Coates BS, Hellmich RL, et al (2011) Mitochondrial genome sequence and expression profiling for the legume pod borer Maruca vitrata (Lepidoptera: Crambidae). PLoS One 6:e16444. https://doi.org/10.1371/journal.pone.0016444
Mawlood SK, Dennany L, Watson N, et al (2016) Quantification of global mitochondrial DNA methylation levels and inverse correlation with age at two CpG sites. Aging (Albany NY) 8:636–641. https://doi.org/10.18632/aging.100892
Michener CD (2007) The bees of the world, 2nd edn. The Johns Hopkins University Press, Baltimore
Moure JS (2012) Tetrapediini Michener & Moure, 1957. In: Moure JS, Urban D, Melo GAR (Orgs). Catalogue of Bees (Hymenoptera, Apoidea) in the Neotropical Region - online version. http://www.moure.cria.org.br/catalogue. Accessed 8 Nov 2013
Nass MMK (1973) Differential methylation of mitochondrial and nuclear DNA in cultured mouse, hamster and virus-transformed hamster cells In vivo and in vitro methylation. J Mol Biol 80:155–175. https://doi.org/10.1016/0022-2836(73)90239-8
Neira-Oviedo M, Tsyganov-Bodounov A, Lycett GJ, et al (2011) The RNA-Seq approach to studying the expression of mosquito mitochondrial genes. Insect Mol Biol 20:141–152. https://doi.org/10.1111/j.1365-2583.2010.01053.x
Neumann K, Seidelmann K (2006) Original article Microsatellites for the inference of population structures in the Red Mason bee Osmia rufa (Hymenoptera , Megachilidae). Apidologie 37:75–83. https://doi.org/10.1051/apido
Ojala D, Montoya J, Attardi G (1981) tRNA punctuation model of RNA processing in human mitochondria. Nature 290:470–474. https://doi.org/10.1038/290470a0
Pertea M, Pertea GM, Antonescu CM, et al (2015) StringTie enables improved reconstruction of a transcriptome from RNA-Seq reads. Nat Biotechnol 33:290–295. https://doi.org/10.1038/nbt.3122
Potts SG, Imperatriz-Fonseca V, Ngo HT, et al (2016) Safeguarding pollinators and their values to human well-being. Nature 540:220–229. https://doi.org/10.1038/nature20588
Robinson JT, Thorvaldsdóttir H, Winckler W, et al (2011) Integrative genomics viewer. Nat Biotechnol 29:24–26. https://doi.org/10.1038/nbt.1754
Rozen S, Skaletsky H (2000) Primer3 on the WWW for General Users and for Biologist Programmers. In: Bioinformatics Methods and Protocols. Humana Press, New Jersey, pp 365–386
Silvestre D, Downton M, Arias MC (2008) The mitochondrial genome of the stingless bee Melipona bicolor (Hymenoptera, Apidae, Meliponini): Sequence, gene organization and a unique tRNA translocation event conserved across the tribe Meliponini. Genet Mol Biol 31:451–460. https://doi.org/10.1590/S1415-47572008000300010
Smith DR (2015) The past, present and future of mitochondrial genomics: have we sequenced enough mtDNAs? Brief Funct Genomics 15:47–54. https://doi.org/10.1093/bfgp/elv027
Stewart JB, Beckenbach AT (2009) Characterization of mature mitochondrial transcripts in Drosophila, and the implications for the tRNA punctuation model in arthropods. Gene 445:49–57. https://doi.org/10.1016/j.gene.2009.06.006
Taanman J-W (1999) The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta Bioenerg 1410:103–123. https://doi.org/10.1016/S0005-2728(98)00161-3
Tian Y, Smith DR (2016) Recovering complete mitochondrial genome sequences from RNA-Seq: A case study of Polytomella non-photosynthetic green algae. Mol Phylogenet Evol 98:57–62. https://doi.org/10.1016/j.ympev.2016.01.017
Torres TT, Dolezal M, Schlötterer C, Ottenwälder B (2009) Expression profiling of Drosophila mitochondrial genes via deep mRNA sequencing. Nucleic Acids Res 37:7509–7518. https://doi.org/10.1093/nar/gkp856
Urich MA, Nery JR, Lister R, et al (2015) MethylC-seq library preparation for base-resolution whole-genome bisulfite sequencing. Nat Protoc 10:475–483. https://doi.org/10.1038/nprot.2014.114
van der Wijst MGP, Rots MG (2015) Mitochondrial epigenetics: an overlooked layer of regulation? Trends Genet 31:353–356. https://doi.org/10.1016/j.tig.2015.03.009
Wolstenholme DR (1992) Animal mitochondrial DNA: structure and evolution. Int Rev Cytol 141:173–216. https://doi.org/10.1016/S0074-7696(08)62066-5
Zhbannikov IY, Hunter SS, Foster JA, Settles ML (2017) SeqyClean: a pipeline for high-throughput sequence data preprocessing. In: Proceedings of the 8th ACM International Conference on Bioinformatics, Computational Biology,and Health Informatics - ACM-BCB ‘17. ACM Press, New York, pp 407–416
Acknowledgments
The authors would like to thank Dr. Lucia Lohmann and Dr. Monica Carlsen for the discipline with which the first mtGenome was generated; Dr. Isabel Alves dos Santos and Dr. Guaraci Duran Cordeiro for their support in specimen sampling; Susy Coelho for laboratory maintenance; Dr. Bob Schmitz (University of Georgia) for the whole-bisulfite sequencing and library preparation of the methylation data; and Dr. Denis Jacob Machado for useful advice about assembly and annotation methods for mitochondrial genomes.
Funding
Financial support was provided by the Fundação de Amparo à Pesquisa do Estado de São Paulo (Proc. 10/50597-5, 12/18531-0 and 13/12530-4; scholarship to EF 2009/07124-1, 2010/20548-2, 2013/03961-1 and 201425023-6), CAPES—Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil (Finance Code 001 and scholarship to PKFS), CNPq—Conselho Nacional de Desenvolvimento Científico e Tecnológico (research sponsorship to MCA, Process number 306932/2016-4), and the Research Center on Biodiversity and Computing (BioComp) of the Universidade São Paulo (USP), supported by the USP Provost’s Office for Research.
Author information
Authors and Affiliations
Contributions
EF: work idealization, genome assembly; NSA: genome assembly, transcription, methylation, and SNP analyses; ARZ: assistance in data analysis, comparative analyses /rearrangements; PCR: PCR amplification and cloning, assistance in data analyses; PKFS: genome assembly, comparative analyses /rearrangements; and MCA: project advising. All authors contributed to the preparation of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Responsible editor: Marina Meixner
Perspectives d’évolution de l’ADNmt des abeilles à partir d’analyses “mito-OMIQUES” d’une espèce solitaire.
organisation du génome mt / méthylation mitochondriale / transcription mitochondriale / abeilles collectrices d’huile / Tetrapedia diversipes.
Evolutionäre Perspektiven zur mtDNA von Bienen aus der Analyse von “mito-OMICS” Daten einer solitären Art.
Organisation des mt Genoms / mitochondriale Methylierung / mitochondriale Transkription / Ölsammelnde Bienen / Tetrapedia diversipes.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1.
(DOCX 905 kb)
Rights and permissions
About this article

Cite this article
Françoso, E., de Souza Araujo, N., Ricardo, P.C. et al. Evolutionary perspectives on bee mtDNA from mito-OMICS analyses of a solitary species. Apidologie 51, 531–544 (2020). https://doi.org/10.1007/s13592-020-00740-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-020-00740-x