
Abstract
New insights into molecular genetics and pathomechanisms in epidermolysis bullosa (EB), methodological and technological advances in molecular biology as well as designated funding initiatives and facilitated approval procedures for orphan drugs have boosted translational research perspectives for this devastating disease. This is echoed by the increasing number of clinical trials assessing innovative molecular therapies in the field of EB. Despite remarkable progress, gene-corrective modalities, aimed at sustained or permanent restoration of functional protein expression, still await broad clinical availability. This also reflects the methodological and technological shortcomings of current strategies, including the translatability of certain methodologies beyond preclinical models as well as the safe, specific, efficient, feasible, sustained and cost-effective delivery of therapeutic/corrective information to target cells. This review gives an updated overview on status, prospects, challenges and limitations of current gene-targeted therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Several in vivo and ex vivo molecular therapy approaches targeting the underlying cause of EB have entered clinical trials. Most notably, these involve gene and protein replacement, modulation of RNA splicing, PTC readthrough and cell-based therapies |
Despite encouraging prospects, potentially curative options still await broad clinical availability and need to address methodological limitations in terms of efficacy, delivery, feasibility, sustainability and safety |
Disease heterogeneity and inter-patient differences additionally lead to variable treatment outcomes so that the choice of therapy will depend on multiple patient-centric factors |
Long-term effectiveness relies on genetic (i.e., DNA) correction in epidermal stem cells. Alternatively, some therapies are suitable for repeated topical application to ensure and maintain a therapeutic effect. In severe subtypes, systemic therapies may be warranted |
Symptom-relieving approaches have palliative potential to reduce disease burden and may serve as a complementary strategy to gene-targeted therapies to improve QoL of affected individuals |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.14665125.
Background
Epidermolysis bullosa (EB) defines a subset of rare, inherited skin fragility disorders that feature characteristic mucocutaneous blistering of variable severity upon mechanical trauma [1]. To date, mutations in 16 different genes have been identified as the underlying cause of EB. These genes predominantly encode proteins critically involved in maintaining the structural and functional integrity of the epidermis and dermo-epidermal basement membrane zone (BMZ) of the skin and mucous membranes. Mutations resulting in diminished or loss of function of these proteins compromise not only the stability and resilience of the tissue, but in some cases also key cellular processes that impact the tissue’s ability to repair itself and maintain its barrier function [2]. The genetic heterogeneity of the disease is reflected in the high phenotypic variability observed, which can range from mild localized blistering to severe and generalized erosions that are accompanied by several secondary complications. Moreover, expression of disease-associated genes is not limited to the skin but can be ubiquitous in other epithelialized tissues (gastrointestinal, respiratory and urogenital tract) or mesenchymal organs (smooth and skeletal muscle). This renders the more severe subtypes of EB a systemic disease, associated with primary multi-organ involvement and accompanying secondary extracutaneous complications and significant morbidity and mortality (Fig. 1) [3, 4].

Clinical scope of epidermolysis bullosa (EB) subtypes. a Partly hemorrhagic blisters predominantly restricted to mechanically exposed acral sites in localized EB simplex. b Generalized skin involvement with characteristically grouped (“herpetiform”, c) blisters in severe EB simplex. d Widespread erosive skin denudation with crusting and inflammation in severe junctional EB. e Generalized blistering and chronic wounding with crusting and subsequent atrophic scarring in severe recessive dystrophic EB (RDEB). Oral manifestations in severe RDEB include (f) microstomia, enamel defects and excessive caries. g On mechanically exposed skin areas such as hands and feet, chronic acral scarring commonly leads to pseudosyndactily, nail loss and mitten deformity in severe RDEB. h Sites of repetitive tissue damage, chronic wounding and regeneration predispose to the development of aggressive cutaneous squamous cell carcinoma in severe RDEB. i EB naevi are large, eruptive irregularly pigmented and highly dynamic melanocytic lesions that typically arise in sites of previous bullae or erosions. Although clinical, dermatoscopic and histological features may be suggestive of melanoma, their course is usually benign. j Nail dystrophy, k milia formation and atrophic scarring are clinical hallmarks of dominant dystrophic EB
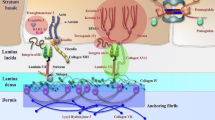
Epidermolysis bullosa is classified into four major types, based on the level of blister formation within the BMZ, which is defined by the tissue localization of the affected protein. EB simplex (EBS), characterized by intraepidermal blistering, is most frequently associated with mutations in KRT5, KRT14 and PLEC1. Blistering within the lamina lucida defines the junctional subtype (JEB), predominantly caused by mutations in LAMB3 and COL17A1. Dystrophic EB (DEB) presents with dermal (i.e., sub-lamina densa) blistering and is caused by alterations in COL7A1. Finally, Kindler EB is typified by mixed levels of tissue cleavage due to mutations in KIND1 (a.k.a. FERMT1). Our increased understanding of the molecular pathology of EB allows further diagnostic stratification to include phenotype (distribution and severity of tissue involvement), mode of inheritance, ultrastructural level of cleavage, associated morphological and clinical findings, involved proteins and genes as well as specific mutations. Ultimately, this has led to the identification of 30 distinct subtypes of the disease so far. The recently updated EB classification further differentiates EB and “EB-related skin fragility disorders.” In the latter, blisters are lacking or are only a minor part of the phenotypic presentation due to a very superficial skin cleavage. This group comprises peeling skin disorders, erosive disorders, hyperkeratotic disorders, connective tissue disorders with skin fragility as well as rare syndromic variants with primary manifestations in other organ systems [2].
Apart from the considerable genetic heterogeneity, other factors at the epigenetic, biochemical and microenvironmental level also contribute to the phenotypic variability and severity of EB [5,6,7,8,9]. In particular, repeated trauma-induced skin barrier disruption results not only in the activation of inflammatory pathways, but eventually in their chronification [10]. While in milder EB variants such inflammatory responses may be mainly restricted to the microenvironment of lesional skin, in patients with severe subtypes and generalized involvement, inflammatory markers could be detected in the blood, signifying systemic involvement [11]. Of note, such injury-driven inflammatory signatures, including elevated levels of IL-1β, IL-2, IL-5, IL-6, IL-12, TGF-β, IFN-γ, TNF-α, TNF-β and T-helper 17 cytokines, significantly contribute to disease-associated pathogenic traits, such as impaired wound healing, tissue remodeling with excessive fibrosis and scarring, burdensome symptoms of itch and pain, and the development of life-threatening skin cancer (Fig. 2) [11,12,13,14,15,16,17].

Pathogenic traits in epidermolysis bullosa. Mutations in genes encoding components essential for the structural and functional integrity of the epidermis and dermo-epidermal junction underlie the four main types of epidermolysis bullosa (EB), featuring skin blistering within the epidermis (EB simplex), the lamina lucida (junctional EB), the upper dermis (DEB) or at mixed levels (Kindler EB). Letters in boxes next to the protein names indicate which subtype of EB results in case of non-functional or lacking protein (S EBS, J JEB, D DEB, K Kindler EB)
Epidermolysis bullosa is currently incurable. Current therapies focus primarily on (preventive) skin and wound care, management of extracutaneous complications and major symptoms such as itch and pain, avoidance of wound infection, nutritional and psychosocial support, and occupational therapy [18,19,20,21]. These primarily symptom-oriented and supportive approaches, however, do not sufficiently address the medical needs of a critical portion of patients, particularly those suffering from severe EB subtypes.
In light of the promising translational prospects, this review focuses on the feasibility and challenges of potentially curative gene- and cell-based therapies that are currently under clinical investigation. The predominantly monogenic disease etiology renders EB an ideal candidate for gene therapy approaches. The goal of these strategies is the sustained or permanent restoration of functional protein expression in EB, through addition, replacement, modification or correction at the DNA, RNA, protein, or cellular level [22]. While these molecular approaches are already under evaluation in clinical trials, the methodological challenges involved, as well as the genotypic heterogeneity of EB, pose major hurdles that make case-specific, tailored approaches mandatory [21, 23].
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Ex Vivo Gene Replacement Therapy
Briefly, ex vivo autologous gene replacement therapy involves isolating patient cells, genetically correcting them in vitro by the introduction of wild-type cDNA copies of the affected gene, expanding the corrected cells into epidermal sheets and grafting these back onto chronic wounds. This form of therapy is mainly investigated in severe EB types (JEB, RDEB), which typically display an autosomal recessive inheritance. In this setting, replacement of a single copy of the functional gene is sufficient to rescue the phenotype. This is in contrast to autosomal dominant traits of inheritance seen in some EBS types, wherein the mutated protein exerts a dominant negative effect over the normal protein. For such variants, different gene therapy approaches are required [24].
The recent observation that continuous renewal and repair of the human epidermis rely on a limited number of epidermal stem cells, known as holoclones, has greatly boosted confidence in the feasibility of gene replacement therapy. The holoclone is the only clonal type that possesses full structural and functional long-term regenerative potential and generates both meroclones (in an intermediate state of differentiation) and differentiated paraclones, which exhibit properties of keratinocyte-forming transit-amplifying progenitors [25, 26].
Current gene delivery techniques are incapable of specifically targeting holoclones in vivo. As such, ensuring efficient targeting of these epidermal stem cells first requires their isolation prior to introduction of the replacement gene ex vivo [23]. This strategy was first implemented in 2006, in a JEB patient harboring mutations in the LAMB3 gene. Autologous epidermal stem cells isolated from skin biopsies were transduced with a retroviral vector expressing full-length, wild-type LAMB3 cDNA. Epidermal skin grafts generated from the molecularly corrected cells were subsequently transplanted onto chronic wounds on the upper leg [27]. The authors reported that most transit-amplifying progenitors were lost within the first few months following grafting. However, long-term restoration of laminin-332 expression and regeneration of morphologically normal, fully differentiated and mechanically stable transgenic skin were observed, along with the preservation of a sustained number of transgenic holoclones [26]. Importantly, in the 15-year follow-up period since this first gene therapy trial in EB, the transgenic skin has been maintained, with no adverse effects observed including tumor development, a potential risk associated with this strategy.
The same approach has been applied twice more in the context of LAMB3-mediated JEB [28, 29].
Most recently, it was applied to the successful replacement of 80% of the body surface area in a 7-year-old boy with intermediate JEB complicated by extensive denudations due to a severe bacterial skin infection. Successful targeting of epidermal stem cells was accompanied by progressive expression of LAMB3-transgenic holoclones and sustained production of functional laminin-332 [29].
Spurred by the favorable outcomes observed following LAMB3 gene replacement therapy, similar trials (NCT03490331, NCT02984085) were launched to evaluate safety and efficacy of autologous epidermal skin grafts expressing COL7A1 and COL17A1 transgenes in recessive DEB (RDEB) and JEB, respectively. Ex vivo gene replacement therapy for RDEB poses several challenges. One is the significantly greater size of the COL7A1 transcript. However, beyond that, LAMB3-associated JEB is linked to a stem cell depletion phenotype due to dysregulated YAP/TAZ mechano-signaling downstream of laminin-332. Proper signaling is restored upon reintroduction of the wild-type LAMB3 cDNA, thereby imparting a replicative advantage to corrected clones. But this is not the case in RDEB, where the YAP/TAZ pathway is unaffected and transduced clones do not gain a replicative advantage. Consequently, the success of this approach has proven far less prominent in RDEB [30]. In a clinical trial involving seven patients treated with autologous epidermal sheets generated from cells retrovirally transduced to express wild-type full-length COL7A1 cDNA, improved wound healing and correct BMZ localization of type VII collagen were observed in most patients, with no serious adverse effects reported. However, transgene expression decreased over time, and persistent wild-type protein expression was detected in only two patients after 2 years [31]. A follow-up phase III trial (NCT04227106) with improvement of wound healing and reduction of pain as primary endpoints was launched in 2020.
Recent evidence suggests that type VII collagen production from both keratinocytes and fibroblasts is a prerequisite for optimal assembly of structurally normal anchoring fibrils, an observation that potentially reflects the influence of microenvironment, extracellular matrix and paracrine signaling on cell engraftment and tissue maturation [32]. Taking this aspect of type VII collagen biology into account, the GENEGRAFT project, including an ongoing phase I/II study (EBGraft, NCT04186650), is based on ex vivo transduction of full-length COL7A1 cDNA in both autologous keratinocytes and fibroblasts, using a self-inactivating (SIN) retroviral vector to circumvent insertional mutagenesis [33, 34]. Corrected cells are then applied to wounds of RDEB patients with residual type VII collagen expression [35]. As of April 2021, one patient has been treated in this study.
Ex vivo gene replacement strategies have advanced the furthest in clinical trials and have enabled insight into patient perspective and experience. The therapy comes with certain drawbacks, including the invasiveness of the approach, which can require multiple biopsies to ensure successful (stem) cell isolation, and the harsh debridement needed for wound bed preparation, which has been shown to increase engraftment success [22, 23, 36]. This places significant burden on this vulnerable patient population. Additionally, several issues, many also shared with other gene therapy approaches (see “Outlook”) will need to be addressed before this therapy can be applied routinely.
In Vivo Gene Replacement Therapy for DEB
Given the interventional burden that accompanies ex vivo approaches, in vivo cDNA replacement strategies via topical delivery to chronic lesional skin has been envisioned, circumventing the need for hospitalization, anesthesia and invasive surgery.
Currently, two approaches are under evaluation. One utilizes a modified, non-integrating, replication-incompetent human herpes simplex virus type 1 (HSV-1) vector formulated into a topical gel to introduce COL7A1 into keratinocytes and fibroblasts in DEB wounds. In a phase II study (NCT03536143), the safety and wound healing efficacy of this therapeutic agent, known as Beremagene Geperpavec gel (B-VEC, formerly known as KB103), was evaluated in four DEB patients. The modified virus leverages both the large packaging capacity (approximately 150 kb) and mucocutaneous tropism of HSV-1 [37]. Data from the phase I/II pilot trial showed complete healing of 90% of wounds within an average time of 17.4 days with B-VEC treatment compared to placebo. Lesions remained closed after an average of 113 days. Imaging analyses additionally revealed production of functional collagen type VII and anchoring fibrils [38]. Notably, the episomal non-integrating nature of this vector increases safety but makes repeated, long-term application necessary [23, 39]. As such, a phase III trial (NCT04491604) is underway to assess the impact of weekly application of B-VEC to DEB wounds in 30 patients.
A second in vivo topical gene delivery therapeutic under investigation for DEB, named AP103, makes use of a highly branched polymer (β-amino ester)-based vector to deliver full-length COL7A1 to both keratinocytes and fibroblasts. In preclinical murine models that had been grafted with human RDEB skin, AP103 therapy resulted in type VII collagen expression and assembly of functional anchoring fibrils for up to 2 weeks following a single application [40, 41]. Potential advantages of this approach include a more stable delivery, reduced risk of inducing immune responses, minimum toxicity, low costs and easy manufacturing. As with B-VEC, the introduced transgene is not integrated, and reapplication is needed. Clinical trials with AP103 are currently in preparation [42, 43].
Ex Vivo Gene Editing
Gene editing, based on programmable nucleases like the CRISPR/Cas9 system, zinc finger nucleases or transcription activator-like effector nucleases (TALEN), aims to permanently correct the genetic defect at the DNA level and is applicable to both autosomal recessive and dominant diseases [44]. In EB, these technologies are evaluated at the preclinical level, where precise gene correction has been achieved in primary skin cells as well as in induced pluripotent stem cells (iPSCs) [45,46,47,48,49,50]. In essence, these technologies induce targeted single- or double-strand breaks in the DNA, followed by activation of the endogenous repair machinery. Gene correction can occur either via non-homologous end joining (NHEJ) or homology direct repair (HDR) utilizing an exogenous donor template. Error-prone NHEJ results in the introduction of small insertions or deletions at the site of the double-strand break, which can be leveraged in several ways. Depending on the context of the mutation, NHEJ can be used to induce disruption of a dominant mutant allele, skipping of a mutant-bearing exon, or reframing of a frameshift mutation. In contrast, HDR is used to achieve traceless repair and full restoration of the wild-type genetic sequence but can suffer from low efficiencies [51]. Low efficiency of HDR repair in epidermal stem cells can be circumvented by performing the editing in differentiated somatic cells that are subsequently reprogrammed into iPSCs through expression of a combination of key transcription factors. These can then be differentiated into skin cells. Alternatively, iPSCs are first generated and then corrected by gene editing [52, 53]. The feasibility of using such gene-corrected iPSCs that are differentiated into keratinocytes and/or fibroblasts to generate transplantable skin equivalents has been demonstrated in preclinical murine models [54,55,56].
A major concern with the use of these gene-editing technologies is their unpredictable off-target effect. Despite careful guide RNA selection, CRISPR/Cas9-editing molecules can bind to genomic sites that are similar but not identical to the intended target site (off-target loci), albeit at low frequency, and cause unintended gene modifications that can potentially drive tumorigenesis [57]. Why some off-target loci are cleaved while others not is not fully understood. Strategies to limit off-target cleavage in the application of CRISPR/Cas9 to EB have included the use of Cas9 nickases in a double-nicking approach and employed ribonucleoprotein (recombinant Cas9-gRNA complexes) rather than plasmids to restrict the amount of time the gene-editing molecules are active in the cells [47, 58].
Despite their great potential, and the continuing and rapid improvement of CRISPR-based technologies, their application has been largely limited to the ex vivo setting. However in 2020, in vivo CRISPR/Cas9 gene-editing therapies were initiated against a congenital retinal degenerative disease (EDIT-101) and hereditary transthyretin amyloidosis (NTLA-2001) (http://www.clinicaltrials.gov). The latter utilizes a two-part genome-editing system (guide RNA specific to the disease-causing gene and messenger RNA encoding the Cas9 nuclease) delivered via lipid nanoparticles and is administered intravenously. This platform presents exciting prospects for a systemic in vivo delivery of gene-editing molecules in RDEB, which frequently present with multi-organ involvement.
Modulation of mRNA Biosynthesis
Targeting the genetic defect at the RNA level induces only a transient effect, thereby requiring long-term treatment. This limitation, on the other hand, potentially improves safety by lowering the risk of mutagenesis. There are several technologies capable of modulating mature mRNA biosynthesis that are being exploited to restore functional protein. Most promising among these is antisense oligonucleotide (AON)-mediated skipping of the mutation-bearing exon during pre-mRNA splicing. In this context, AONs are short 18–22-nt single-stranded DNAs or RNAs that are specifically designed to bind a complementary sequence in the target pre-mRNA that contains critical splicing elements. Thereby, AONs are utilized to mask the mutated exon from the splicing machinery, thereby excluding it from the mature mRNA [59]. This strategy works best on short, in-frame exons encoding non-essential domains, whose deletion from the protein is unlikely to result in major structural or functional changes. In this respect, type VII collagen encodes several such exons, and AON-mediated exon skipping has been successfully applied to exclude mutant-bearing exons 13, 70, 73, 80 and 105, leading to the expression of truncated but functional protein capable of forming anchoring fibrils both in vitro and in vivo and improving dermo-epidermal cohesion [59,60,61,62]. Topical administration of QR-313, an AON that targets exon 73 and is capable of inducing its exclusion from mature mRNA, is currently being evaluated in a phase I–II trial (NCT03605069) in DEB. Preliminary data, however, indicate rather modest efficacy in terms of exon skipping rate, enhanced protein re-expression and reconstitution of anchoring fibrils, which possibly reflects a low tissue uptake at the study dosage [63]. Of note, this highly targeted approach is restricted to a small subset of patients who bear the affected exon [64]. Furthermore, AON-mediated exon skipping is better suited to recessive phenotypes wherein partial protein recovery imparts a greater clinical benefit compared to dominant negative phenotypes [65].
Short interfering RNA (siRNA) capable of specifically inhibiting expression of a mutant mRNA without silencing the wild-type allele is an especially promising strategy to target dominant-negative mutations [66, 67]. This approach relies on the design of highly specific siRNAs capable of single nucleotide sequence discrimination, which binds the mutant mRNA, targeting it for degradation. Application of this strategy has been shown to increase the ratio of functional proteins with an in vitro effectivity of 58% and up to 80% in dominant DEB (DDEB) and EBS, respectively [68,69,70]. Like gene-editing and AON-mediated approaches that are sequence context-dependent, this represents a highly individualized treatment modality.
In contrast, spliceosome-mediated RNA trans-splicing (SMaRT) exploits the endogenous splicing machinery to replace mutated sequences of an endogenous pre-mRNA transcript with wild-type sequences supplied by an exogenous engineered RNA molecule [71]. As large regions of the pre-mRNA can be exchanged, SMaRT aims to be more broadly applicable to many patients. Preclinical studies have shown successful correction of mutations in PLEC1, KRT14, COL7A1 and COL17A1 in vitro and in vivo [72,73,74,75,76]. However, to advance the technology further, it will be pivotal to improve trans-splicing efficiency, minimize the risk of off-target effects and develop a feasible delivery method through the skin barrier [77].
Additionally, recent reports indicate that modulation of regulatory splicing elements could be exploited to favorably alter mutation-based splicing patterns. Manipulation of crucial interactions of the spliceosome, e.g., by drug compounds or small molecules, may thus hold therapeutic potential via augmentation of (functional) full-length protein variants from splice mutations [78, 79].
Replacement of the Gene Product/Missing Protein
Currently, strategies that involve introducing a wild-type recombinant form of the missing or faulty structural protein are only being evaluated for RDEB. Early studies demonstrated that intradermal or intravenous injection of recombinant human type VII collagen (rC7) could be stably incorporated into anchoring fibrils at the BMZ in mice. Despite its large size of 900 kDa, when administered intravenously, rC7 homed to sites of lesional or wounded skin in mice grafted with RDEB skin, restoring anchoring fibril formation and epidermal-dermal adherence [80,81,82]. Notably, rC7 was not detected in any other organs of treated mice. These observations form the basis of a multicenter phase I/II trial (NCT03752905) investigating the safety and efficacy of intravenous infusions of rC7 (PTR-01) in RDEB patients. Interim results from nine patients revealed that serial infusions of up to 1 mg/kg led to a dose-dependent increase of type VII collagen levels in the skin of all patients. Treatment was generally well tolerated, with no serious adverse reactions reported [83]. However, protein size may contribute to the limited skin homing potential as well as accessibility to other extracutaneous tissues. A phase II open-label study with doses of 3 mg/kg of PTR-01 is currently ongoing (NCT04599881).
(Gene Modified) Cell Therapy
Often referred to as a “natural gene therapy,” revertant mosaicism describes a phenomenon wherein the inherited mutation is rescued by a second somatic or postzygotic mutation, yielding a functional protein. Clinically, this manifests as ‘mosaic’ healthy, fairly well-demarcated skin patches that never blister. They usually arise during childhood and stop expanding before adulthood [84]. Various genetic mechanisms (e.g., back mutation, mitotic recombination, second mutation during cell division) have been implicated in this phenomenon [85,86,87], the occurrence of which has been reported in all types of EB and particularly in intermediate JEB [88]. Importantly, such mosaic skin can be leveraged as a source of naturally corrected cells to treat lesional skin. In a LAMB3-mutated JEB patient, treatment of seven chronic ulcers (total area of 7 cm2) with 73 punch biopsy specimens from an area of revertant skin induced healing of both donor and acceptor sites with revertant epidermis. The treated sites exhibited protein expression similar to wild-type skin up to 18 months after transplantation [89]. For treatment of larger areas, however, in vitro expansion of revertant cells is first required.
Recently, treatment of chronic wounds with cultured epidermal autografts (CEA) containing revertant cells displayed an encouraging mean wound epithelialization rate of 81.6% (95% CI 26.6–100.0%) in two out of three RDEB patients as measured 4 weeks post-treatment. Clinical improvement was noted by the authors up to 76 weeks post-treatment. Treatment outcome appeared to correlate with the proportion of revertant haplotypes within the grafts, with the best outcome observed in the subject receiving the CEA with the highest percentage of revertant haplotypes as measured before treatment (95% versus 5.7% in the patient with the worst response) [90]. Presumably, the sustainability of the phenotypic reversion depends on the percentage of revertant stem cells in the graft [91]. Given the invasive nature of the transplantation procedure, molecular subtyping to assess the proportion of revertant stem cells in the CEA can be performed prior to grafting to predict treatment outcome. Using revertant patient cells for generation of iPSCs that are then differentiated into functional (healthy) keratinocytes constitutes a further promising combination of tools, as the risk of immune rejection of “naturally” corrected cells is low [54].
While revertant mosaicism was initially reported solely in epidermal keratinocytes, it was recently observed in dermal fibroblasts in a patient with RDEB, demonstrating that corrected fibroblasts, which also synthesize and secrete type VII collagen albeit to a lesser extent than basal keratinocytes, are fully capable of sustaining healthy skin architecture [92]. This implies that these cells, like keratinocytes, are suitable targets for gene therapy in DEB [93], particularly as an incremental rise to 30–35% of normal wild-type skin levels is believed to be sufficient to achieve substantial improvement in skin stability [94]. Notably, technical handling of fibroblasts is advantageous as they are easier to culture and are more resistant to growth arrest and differentiation [95].
While local injection of allogeneic fibroblasts into RDEB wounds or perilesional skin did not provide solid evidence for improved wound healing [96,97,98,99,100,101], the use of gene-modified autologous fibroblasts demonstrated superior efficacy compared to control in separate phase I and I/II studies (NCT02493816, NCT02810951) [102,103,104]. In both studies, autologous fibroblasts were corrected using (SIN) lentiviral vectors to express functional type VII collagen and injected intradermally into target wounds and intact skin. Neither study reported serious adverse reactions or distinct autoimmune reactions within the observation period of up to 12 months. According to preliminary communications, applications across and in the margins of targeted chronic wounds revealed positive wound-healing trends, with signs of type VII collagen expression in at least 50% of injected sites and restoration of anchoring fibrils within the BMZ up to a year post-administration. In previous studies, however, intradermal injections were described as painful. Moreover, the limited diffusion radius may impede extracutaneous effects. Nevertheless, the preliminary data are encouraging regarding applicability and efficiency of this treatment modality as a maintenance therapy. A follow-up phase III trial (NCT04213261) is currently enrolling patients.
Finally, curative as well as immunomodulatory potential of gene-corrected-iPSC-derived mesenchymal stem cells is currently investigated at a preclinical stage. Regimens allowing for systemic delivery via infusions may additionally enable addressing the extracutaneous manifestations of EB [100, 105].
Premature Termination Codon (PTC) Readthrough Therapies
Nonsense mutations occurring on both alleles are among the most detrimental, as they tend to result in near complete loss of full-length protein expressed from the affected gene. In EB, the most severe variants of the disease are caused by such mutations. Aminoglycoside antibiotics such as gentamicin were shown to induce readthrough of premature termination codons (PTCs) in several monogenic diseases, including EB. PTC readthrough drugs act by inducing a conformational change at the decoding site, leading to reduced translational fidelity and incorporation of near-cognate tRNAs at the stop site (PTC). Thereby, nonsense-mediated decay (NMD) is prevented and production of full-length protein is increased [106,107,108]. Efficacy of this approach depends on the stop codon type, flanking nucleotide sequences and, consequently, the impact of the introduced amino acid on protein folding, stability and post-translational processing.
Topical gentamicin, a commercially available, low-priced drug, led to a molecular correction and protein re-expression in several clinical trials involving RDEB and JEB patients with COL7A1, COL17A1 and LAMB3 PTC mutations [109,110,111]. Clinical improvements lasted up to 3 months post intervention. Recently, daily application of 0.3% gentamicin-containing ophthalmic ointment restored laminin-332 expression in conjunctival cells of a JEB patient within a week, with symptomatic improvement of intermittent corneal erosions lasting up to 5 weeks. The applied concentration proved to be safe and effective for improving corneal epithelial integrity in JEB patients [112].
Systemic application of gentamicin with the intention to treat extracutaneous symptoms, however, harbors the risk of considerable side effects such as ototoxicity and nephrotoxicity. Nevertheless, previous administration of intravenous gentamicin at doses of 7.5 mg/kg/day for 3 weeks was well tolerated in patients with severe JEB. Beneficial impact on skin fragility was noted in four out of five patients, although the drug failed to prevent premature death. Early demise due to failure to thrive, airway obstruction and sepsis within the first 2 years of life is prototypic for this severe EB subtype [113]. Prospective studies to assess the safety and efficacy of intravenous gentamicin are ongoing (NCT03526159, NCT03392909, NCT04140786). Preliminary results of a pilot study with daily administration of intravenous gentamicin for 14 days involving four patients with RDEB and three with JEB revealed synthesis of new full-length, functional collagen fibrils in the skin for up to 3 months after treatment. This was associated with improved wound healing, reduced disease severity and higher quality of life scores. Two RDEB patients further received gentamicin infusions twice weekly for 3 months. Safety assessments were inconspicuous in all these patients, and no autoantibodies to newly introduced type VII collagen or laminin-332 were detected in either skin or blood [114].
Additionally, molecules that act synergistically with aminoglycosides on translation, e.g., by inhibition of translation termination factors or nonsense-mediated decay (NMD) of PTC-bearing mRNAs, may enhance the efficacy of PTC readthrough. The potential of this approach was recently demonstrated in JEB-derived cells with COL17A1 nonsense mutations by combining gentamicin with drugs targeting the translation termination factor eRF3 in vitro [115]. In this context, the anti-inflammatory drug amlexanox has been associated with both NMD inhibition and PTC readthrough [116, 117], and its application in nonsense mutated RDEB patient cells in vitro led to the generation of full-length type VII collagen [116, 117]. Furthermore, its effectiveness at lower concentrations suggests a more favorable toxicity profile compared to aminoglycosides.
A novel option in the readthrough arena is the targeting of the translating ribosome. Previous studies identified ribosomal protein L35 as a translational modulator of LAMB3 PTC mRNA. Specifically, variant or specialized ribosomes with reduced levels of rpL35 were able to promote increased production levels of full-length protein from a LAMB3-PTC-bearing mRNA in vitro. Importantly, the rpL35-deficient sub-population of specialized ribosomes left both translation of a reporter luciferase carrying a different PTC and bulk mRNA translation largely unaltered [118]. Subsequent molecular docking studies identified two FDA-approved drugs, atazanavir and artesunate, as candidate small-molecule binders of rpL35. Molecular interaction studies predicted several binding clusters for both compounds scattered along the rpL35 structure, while NMR titration studies and docking calculations identified an N-terminal domain of rpL35, where the non-identical binding sites for atazanavir and artesunate overlap. This rpl35 domain is accessible when rpL35 is integrated in its natural ribosomal environment, suggesting that treatment with artesunate and atazanavir may target the translating ribosome [119]. In initial studies using artesunate in a suitable cellular model to rescue the LAMB3-PTC defect, persistent increase in production of full-length LAMB3 protein expression could be demonstrated (Breitenbach-Koller et al., unpublished data). Given its favorable side effect profile, artesunate may be well suited for use in newborns with LAMB3-negative severe JEB, particularly to help them survive the critical first 2 years of life.
Complementary Strategies of Symptom-Relieving and Disease-Modifying Therapies
Epidermolysis bullosa patients suffer from a high clinical, psychological, social and economic disease burden [120]. As there are currently no curative treatments for EB, symptom-relieving or disease-modifying therapies constitute a promising complementary palliative approach to improve quality of life (QoL) in affected individuals. These strategies are aimed at the modulation of dysregulated pathways involved in wound healing, inflammation, fibrosis, tissue remodeling, oncogenesis or targeting of burdensome symptoms such as itch or pain. They exploit either innovative agents from the bench or repurposed, thus readily available and well-characterized drugs approved or tested, e.g., for other immunologically mediated diseases with barrier disruption such as atopic dermatitis (Table 1) [6, 11,12,13,14, 43, 121]. While immunomodulatory agents have the potential to address complications and prevent or reduce associated symptoms in EB, their action is generally less precise compared to mutation-specific approaches of molecular therapies. Inflammatory signatures may be dynamic and are modified by various factors, including the distinct molecular aberration, anatomic site/microenvironment, microbial burden and pre-treatment. Moreover, inter- and intra-individual variability questions the prospect of a prototypic inflammatory (cytokine) profile. Therapy approaches based on this may thus lack reproducibility and be compromised by pathway redundancies or bypassing effects. Finally, systemic administration of immunomodulators may adversely affect host defense while extensive cutaneous, mucosal, organ involvement increases infection and tumor risk. In this respect, the extent to which individual immune profiling and targeting is necessary or implementable needs to be determined to translate these palliative approaches into an effective, safe, feasible and tolerable therapeutic rationale.
Outlook
The range of therapeutic strategies to address the underlying cause of EB has expanded dramatically, boosted by methodological and technological advances as well as our increased understanding of the molecular pathomechanisms of the disease. This is reflected in the number of clinical trials underway. The current challenge is bringing these into standard clinical practice. To various extents, gene-targeted therapies face common issues of efficacy, safety, feasibility, tolerability and cost-effectiveness [36]. One critical safety issue, particularly in patients who completely lack expression of the affected protein, is the risk of triggering adverse immune responses against the introduced functional protein, at least part of which could be considered “foreign” in the patient. Hence, clinical trials to date only include patients with residual expression of the affected protein or who demonstrate absence of pre-existing pathogenic antibodies against the intended protein product of the therapy. Research into strategies to modulate immune tolerance mechanisms are warranted to be able to extend these therapeutic options to all patients.
With no approved curative options, current management plans for EB patients comprise various complementary or synergistic combinations of different therapeutic strategies to target distinct symptoms such as fibrosis, itch and skin cancer, including strategies to control systemic inflammation, as well as measures to enhance microbial surveillance. While most strategies discussed here offer an opportunity for a cure, the genetic and phenotypic heterogeneity associated with EB, compounded additionally by inter-individual differences, underscores that there will not be one single strategy that suits all patients. Genetic subtype and clinical phenotype (disease severity, systemic manifestation) can direct treatment course, but will also need to be balanced against therapeutic modality (local, systemic) and feasibility (therapeutic burden, costs) to stratify patients into options tailored to their specific needs. Achieving this, however, requires gathering more data, a monumental task in rare disease populations wherein intrinsically low patient numbers jeopardize meeting sample size requirements and compromise statistical power, while increasing trial duration and costs [122]. The development of new statistical methods for meaningful measurement of clinical outcomes in small populations will aid in designing and conducting clinical trials to prove efficacy and safety of innovative treatment modalities in EB, thus helping to bring these life-changing therapies closer to patients.
References
Fine JD. Inherited epidermolysis bullosa. Orphanet J Rare Dis. 2010;5:12.
Has C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183(4):614–27.
Fine J-D, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part I. Epithelial associated tissues. J Am Acad Dermatol. 2009;61(3):367–84.
Fine J-D, Mellerio JE. Extracutaneous manifestations and complications of inherited epidermolysis bullosa: part II. Other organs. J Am Acad Dermatol. 2009;61(3):387–404.
Turczynski S, et al. Marked intrafamilial phenotypic heterogeneity in dystrophic epidermolysis bullosa caused by inheritance of a mild dominant glycine substitution and a novel deep intronic recessive COL 7A1 mutation. Br J Dermatol. 2016;174(5):1122–5.
Odorisio T, et al. Monozygotic twins discordant for recessive dystrophic epidermolysis bullosa phenotype highlight the role of TGF-β signalling in modifying disease severity. Hum Mol Genet. 2014;23(15):3907–22.
Schumann H, et al. Phenotypic spectrum of epidermolysis bullosa associated with α6β4 integrin mutations. Br J Dermatol. 2013;169(1):115–24.
Kiritsi D, et al. Molecular mechanisms of phenotypic variability in junctional epidermolysis bullosa. J Med Genet. 2011;48(7):450–7.
McGrath JA, et al. Moderation of phenotypic severity in dystrophic and junctional forms of epidermolysis bullosa through in-frame skipping of exons containing non-sense or frameshift mutations. J Investig Dermatol. 1999;113(3):314–21.
Cianfarani F, et al. Pathomechanisms of altered wound healing in recessive dystrophic epidermolysis bullosa. Am J Pathol. 2017;187(7):1445–53.
Annicchiarico G, et al. Proinflammatory cytokines and antiskin autoantibodies in patients with inherited epidermolysis bullosa. Medicine. 2015;94(42):e1528.
Wally V, et al. The pathogenetic role of IL-1β in severe epidermolysis bullosa simplex. J Investig Dermatol. 2013;133(7):1901–3.
Castela E, et al. Epidermolysis bullosa simplex generalized severe induces a T helper 17 response and is improved by apremilast treatment. Br J Dermatol. 2019;180(2):357–64.
Esposito S, et al. Autoimmunity and cytokine imbalance in inherited epidermolysis bullosa. Int J Mol Sci. 2016;17(10):1625.
Twaroski K, et al. Role of transforming growth factor-beta1 in recessive dystrophic epidermolysis bullosa squamous cell carcinoma. Exp Dermatol. 2021;30:664–75.
Ng YZ, et al. Fibroblast-derived dermal matrix drives development of aggressive cutaneous squamous cell carcinoma in patients with recessive dystrophic epidermolysis bullosa. Cancer Res. 2012;72(14):3522–34.
Nyström A, et al. Losartan ameliorates dystrophic epidermolysis bullosa and uncovers new disease mechanisms. EMBO Mol Med. 2015;7(9):1211–28.
Pope E, et al. A consensus approach to wound care in epidermolysis bullosa. J Am Acad Dermatol. 2012;67(5):904–17.
Goldschneider KR, et al. Pain care for patients with epidermolysis bullosa: best care practice guidelines. BMC Med. 2014;12:178.
Chan JM, et al. Occupational therapy for epidermolysis bullosa: clinical practice guidelines. Orphanet J Rare Dis. 2019;14(1):129.
Piñón Hofbauer J, et al. Therapy development for epidermolysis bullosa [online first]. IntechOpen. 2021. https://www.intechopen.com/online-first/therapy-development-for-epidermolysis-bullosa. Accessed 5 April 2021
Marinkovich MP, Tang JY. Gene therapy for epidermolysis bullosa. J Investig Dermatol. 2019;139(6):1221–6.
De Rosa L, et al. Toward combined cell and gene therapy for genodermatoses. Cold Spring Harb Perspect Biol. 2020;12(5):a035667.
Koller U, Bauer JW. Gene replacement therapies for genodermatoses: a status quo. Front Genet. 2021;12:515.
Barrandon Y, Green H. Three clonal types of keratinocyte with different capacities for multiplication. Proc Natl Acad Sci USA. 1987;84(8):2302–6.
De Rosa L, et al. Long-term stability and safety of transgenic cultured epidermal stem cells in gene therapy of junctional epidermolysis bullosa. Stem Cell Rep. 2014;2(1):1–8.
Mavilio F, et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med. 2006;12(12):1397–402.
Bauer JW, et al. Closure of a large chronic wound through transplantation of gene-corrected epidermal stem cells. J Investig Dermatol. 2017;137(3):778–81.
Hirsch T, et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature. 2017;551(7680):327–32.
De Rosa L, et al. Laminin 332-dependent YAP dysregulation depletes epidermal stem cells in junctional epidermolysis bullosa. Cell Rep. 2019;27(7):2036-2049.e6.
Eichstadt S, et al. Phase 1/2a clinical trial of gene-corrected autologous cell therapy for recessive dystrophic epidermolysis bullosa. JCI Insight. 2019;4(19):e130554.
Supp DM, et al. Collagen VII expression is required in both keratinocytes and fibroblasts for anchoring fibril formation in bilayer engineered skin substitutes. Cell Transplant. 2019;28(9–10):1242–56.
Yu SF, et al. Self-inactivating retroviral vectors designed for transfer of whole genes into mammalian cells. Proc Natl Acad Sci USA. 1986;83(10):3194–8.
Zufferey R, et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72(12):9873–80.
Gaucher S, Lwin SM. EBGene trial: patient preselection outcomes for the European GENEGRAFT ex vivo phase I/II gene therapy trial for recessive dystrophic epidermolysis bullosa. Br J Dermatol. 2020;182(3):794–7.
Prodinger C, et al. Epidermolysis bullosa: advances in research and treatment. Exp Dermatol. 2019;28(10):1176–89.
Marinkovich MP, et al. 305 In vivo correction of recessive dystrophic epidermolysis bullosa (RDEB) by direct cutaneous COL7A1 gene replacement: results of a phase 1–2 trial. In: Society for Investigative Dermatology (SID) 2020 Meeting Abstract Supplement. J Investig Dermatol. 2020;140(7):S37.
Krystal biotech announces final update from phase 1/2 clinical trial of KB103 (“bercolagene telserpavec,” “B-VEC”). 2019/10/29, https://ir.krystalbio.com/news-releases/news-release-details/krystal-biotech-announces-final-update-phase-12-clinical-trial. Accessed 5 Mar 2021.
Marconi P, et al. HSV as a vector in vaccine development and gene therapy. Hum Vaccine. 2008;4(2):91–105.
Cutlar L, et al. A non-viral gene therapy for treatment of recessive dystrophic epidermolysis bullosa. Exp Dermatol. 2016;25(10):818–20.
Zeng M, et al. Efficient and robust highly branched poly(β-amino ester)/minicircle COL7A1 polymeric nanoparticles for gene delivery to recessive dystrophic epidermolysis bullosa keratinocytes. ACS Appl Mater Interfaces. 2019;11(34):30661–72.
Sumeray M. OP34: non-viral gene therapy for epidermolysis bullosa. In: EB2020 1st world congress on epidermolysis bullosa, January 19–23, 2020, London, UK. Acta Derm Venereol. 2020;100(220):13.
Prodinger C, Bauer JW, Laimer M. Translational perspectives to treat epidermolysis bullosa-where do we stand? Exp Dermatol. 2020;29(11):1112–22.
Gupta SK, Shukla P. Gene editing for cell engineering: trends and applications. Crit Rev Biotechnol. 2017;37(5):672–84.
Hainzl S, et al. COL7A1 editing via CRISPR/Cas9 in recessive dystrophic epidermolysis bullosa. Mol Ther. 2017;25(11):2573–84.
Benati D, et al. CRISPR/Cas9-mediated in situ correction of LAMB3 gene in keratinocytes derived from a junctional epidermolysis bullosa patient. Mol Ther. 2018;26(11):2592–603.
Kocher T, et al. Cut and paste: efficient homology-directed repair of a dominant negative KRT14 mutation via CRISPR/Cas9 nickases. Mol Ther. 2017;25(11):2585–98.
Bonafont J, et al. Correction of recessive dystrophic epidermolysis bullosa by homology-directed repair-mediated genome editing. Mol Ther. 2021;29(6):2008–18.
Aushev M, et al. Traceless targeting and isolation of gene-edited immortalized keratinocytes from epidermolysis bullosa simplex patients. Mol Ther Methods Clin Dev. 2017;6:112–23.
Osborn MJ, et al. TALEN-based gene correction for epidermolysis bullosa. Mol Ther. 2013;21(6):1151–9.
March OP, Kocher T. Context-dependent strategies for enhanced genome editing of genodermatoses. Cells. 2020;9(1):112.
Webber BR, et al. CRISPR/Cas9-based genetic correction for recessive dystrophic epidermolysis bullosa. NPJ Regen Med. 2016;1:16014.
Itoh M, et al. Footprint-free gene mutation correction in induced pluripotent stem cell (iPSC) derived from recessive dystrophic epidermolysis bullosa (RDEB) using the CRISPR/Cas9 and piggyBac transposon system. J Dermatol Sci. 2020;98(3):163–72.
Umegaki-Arao N, et al. Induced pluripotent stem cells from human revertant keratinocytes for the treatment of epidermolysis bullosa. Sci Transl Med. 2014;6(264):264ra164.
Sebastiano V, et al. Human COL7A1-corrected induced pluripotent stem cells for the treatment of recessive dystrophic epidermolysis bullosa. Sci Transl Med. 2014;6(264):264ra163.
Jacków J, et al. CRISPR/Cas9-based targeted genome editing for correction of recessive dystrophic epidermolysis bullosa using iPS cells. Proc Natl Acad Sci USA. 2019;116(52):26846–52.
Thomas M, Burgio G. Collateral damage and CRISPR genome editing. PLoS Genet. 2019;15(3):e1007994.
Kocher T, et al. Improved double-nicking strategies for COL7A1-editing by homologous recombination. Mol Ther Nucleic Acids. 2019;18:496–507.
Goto M, et al. Targeted skipping of a single exon harboring a premature termination codon mutation: implications and potential for gene correction therapy for selective dystrophic epidermolysis bullosa patients. J Investig Dermatol. 2006;126(12):2614–20.
Turczynski S, et al. Targeted exon skipping restores type VII collagen expression and anchoring fibril formation in an in vivo RDEB model. J Investig Dermatol. 2016;136(12):2387–95.
Bremer J, et al. Antisense oligonucleotide-mediated exon skipping as a systemic therapeutic approach for recessive dystrophic epidermolysis bullosa. Mol Ther Nucleic Acids. 2016;5(10):e379.
Bornert O, et al. Analysis of the functional consequences of targeted exon deletion in COL7A1 reveals prospects for dystrophic epidermolysis bullosa therapy. Mol Ther. 2016;24(7):1302–11.
Marinkovich MP. 306 Topical QR-313, an antisense oligonucleotide, in the treatment of dystrophic epidermolysis bullosa. In: Society for Investigative Dermatology (SID) 2020 Meeting Abstract Supplement. J Investig Dermatol. 2020;140(7):S37.
Bornert O, et al. QR-313, an antisense oligonucleotide, shows therapeutic efficacy for treatment of dominant and recessive dystrophic epidermolysis bullosa: a preclinical study. J Investig Dermatol. 2020;141:883–93.
Bremer J, et al. Natural exon skipping sets the stage for exon skipping as therapy for dystrophic epidermolysis bullosa. Mol Ther Nucleic Acids. 2019;18:465–75.
Cao T, et al. An inducible mouse model for epidermolysis bullosa simplex: implications for gene therapy. J Cell Biol. 2001;152(3):651–6.
Fritsch A, et al. Dominant-negative effects of COL7A1 mutations can be rescued by controlled overexpression of normal collagen VII. J Biol Chem. 2009;284(44):30248–56.
Pendaries V, et al. siRNA-mediated allele-specific inhibition of mutant type VII collagen in dominant dystrophic epidermolysis bullosa. J Investig Dermatol. 2012;132(6):1741–3.
Morgan CP, et al. A mutation-independent therapeutic strategy for dominant dystrophic epidermolysis bullosa. J Investig Dermatol. 2013;133(12):2793–6.
Atkinson SD, et al. Development of allele-specific therapeutic siRNA for keratin 5 mutations in epidermolysis bullosa simplex. J Investig Dermatol. 2011;131(10):2079–86.
Bauer JW, et al. RNA trans-splicing for genodermatoses. Methods Mol Biol. 2013;961:441–55.
Dallinger G, et al. Collagen 17A1 gene correction using spliceosome mediated RNA trans-splicing (SMaRTtrade mark) technology. J Investig Dermatol. 2000;115(2):332.
Wally V, et al. 5’ trans-splicing repair of the PLEC1 gene. J Investig Dermatol. 2008;128(3):568–74.
Wally V, et al. K14 mRNA reprogramming for dominant epidermolysis bullosa simplex. Hum Mol Genet. 2010;19(23):4715–25.
Murauer EM, et al. Functional correction of type VII collagen expression in dystrophic epidermolysis bullosa. J Investig Dermatol. 2011;131(1):74–83.
Peking P, et al. A gene gun-mediated nonviral RNA trans-splicing strategy for Col7a1 repair. Mol Ther Nucleic Acids. 2016;5:e287.
Hong EM, Ingemarsdotter CK, Lever AML. Therapeutic applications of trans-splicing. Br Med Bull. 2020;136(1):4–20.
Mittwollen R, et al. Aberrant splicing as potential modifier of the phenotype of junctional epidermolysis bullosa. J Eur Acad Dermatol Venereol. 2020;34(9):2127–34.
Murrell DF, Uitto J. Splicing of the cake can affect the severity of epidermolysis bullosa. J Eur Acad Dermatol Venereol. 2020;34(9):1879–80.
Woodley DT, et al. Injection of recombinant human type VII collagen restores collagen function in dystrophic epidermolysis bullosa. Nat Med. 2004;10(7):693–5.
Woodley DT, et al. Intravenously injected recombinant human type VII collagen homes to skin wounds and restores skin integrity of dystrophic epidermolysis bullosa. J Investig Dermatol. 2013;133(7):1910–3.
Remington J, et al. Injection of recombinant human type VII collagen corrects the disease phenotype in a murine model of dystrophic epidermolysis bullosa. Mol Ther. 2009;17(1):26–33.
Bruckner AL, et al. Interim update from a Phase 1/2 trial examining the safety and tolerability of PTR-01, a collagen 7 protein replacement therapy, in patients with recessive dystrophic epidermolysis bullosa. Poster presented at: Society of Pediatric Dermatology Virtual Meeting July 2020. https://phoenixtissuerepair.com/wp-content/uploads/SPD_Poster_FINAL-.pdf. Accessed 5 May 2021
van den Akker PC, et al. A “late-but-fitter revertant cell” explains the high frequency of revertant mosaicism in epidermolysis bullosa. PLoS ONE. 2018;13(2):e0192994.
Pasmooij AM, et al. Revertant mosaicism in junctional epidermolysis bullosa due to multiple correcting second-site mutations in LAMB3. J Clin Investig. 2007;117(5):1240–8.
Suzuki S, et al. Revertant mosaicism in ichthyosis with confetti caused by a frameshift mutation in KRT1. J Invest Dermatol. 2016;136(10):2093–5.
Almaani N, et al. Revertant mosaicism in recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2010;130(7):1937–40.
Pasmooij AM, et al. Natural gene therapy may occur in all patients with generalized non-Herlitz junctional epidermolysis bullosa with COL17A1 mutations. J Investig Dermatol. 2012;132(5):1374–83.
Gostyński A, Pasmooij AM, Jonkman MF. Successful therapeutic transplantation of revertant skin in epidermolysis bullosa. J Am Acad Dermatol. 2014;70(1):98–101.
Matsumura W, et al. Cultured epidermal autografts from clinically revertant skin as a potential wound treatment for recessive dystrophic epidermolysis bullosa. J Investig Dermatol. 2019;139(10):2115-2124.e11.
Gostynski A, et al. Adhesive stripping to remove epidermis in junctional epidermolysis bullosa for revertant cell therapy. Br J Dermatol. 2009;161(2):444–7.
Twaroski K, et al. Revertant mosaic fibroblasts in recessive dystrophic epidermolysis bullosa. Br J Dermatol. 2019;181(6):1247–53.
Woodley DT, et al. Normal and gene-corrected dystrophic epidermolysis bullosa fibroblasts alone can produce type VII collagen at the basement membrane zone. J Investig Dermatol. 2003;121(5):1021–8.
Fritsch A, et al. A hypomorphic mouse model of dystrophic epidermolysis bullosa reveals mechanisms of disease and response to fibroblast therapy. J Clin Investig. 2008;118(5):1669–79.
Ortiz-Urda S, et al. Injection of genetically engineered fibroblasts corrects regenerated human epidermolysis bullosa skin tissue. J Clin Investig. 2003;111(2):251–5.
Wong T, et al. Potential of fibroblast cell therapy for recessive dystrophic epidermolysis bullosa. J Investig Dermatol. 2008;128(9):2179–89.
Nagy N, et al. HB-EGF induces COL7A1 expression in keratinocytes and fibroblasts: possible mechanism underlying allogeneic fibroblast therapy in recessive dystrophic epidermolysis bullosa. J Investig Dermatol. 2011;131(8):1771–4.
Petrof G, et al. Fibroblast cell therapy enhances initial healing in recessive dystrophic epidermolysis bullosa wounds: results of a randomized, vehicle-controlled trial. Br J Dermatol. 2013;169(5):1025–33.
Venugopal SS, et al. A phase II randomized vehicle-controlled trial of intradermal allogeneic fibroblasts for recessive dystrophic epidermolysis bullosa. J Am Acad Dermatol. 2013;69(6):898-908.e7.
Conget P, et al. Replenishment of type VII collagen and re-epithelialization of chronically ulcerated skin after intradermal administration of allogeneic mesenchymal stromal cells in two patients with recessive dystrophic epidermolysis bullosa. Cytotherapy. 2010;12(3):429–31.
Petrof G, et al. Potential of systemic allogeneic mesenchymal stromal cell therapy for children with recessive dystrophic epidermolysis bullosa. J Investig Dermatol. 2015;135(9):2319–21.
Lwin SM, et al. Safety and early efficacy outcomes for lentiviral fibroblast gene therapy in recessive dystrophic epidermolysis bullosa. JCI Insight. 2019;4(11):e126243.
Mauro VP, Chappell SA. A critical analysis of codon optimization in human therapeutics. Trends Mol Med. 2014;20(11):604–13.
Marinkovich MP. P123: a phase 1/2 study of genetically corrected, collagen VII expressing autologous human dermal fibroblasts injected into the skin of patients with recessive dystrophic epidermolysis bullosa (RDEB). In: EB2020 1st world congress on epidermolysis bullosa, January 19–23, 2020, London, UK. Acta Derm Venereol. 2020;100(220):75.
Roop D. OP26: state of the art IPSCs and alternative methods for delivering cells derived from gene-edited IPSCs. In: EB2020 1st world congress on epidermolysis bullosa, January 19–23, 2020, London, UK. Acta Derm Venereol. 2020;100(220):11.
Cogan J, et al. Aminoglycosides restore full-length type VII collagen by overcoming premature termination codons: therapeutic implications for dystrophic epidermolysis bullosa. Mol Ther. 2014;22(10):1741–52.
Barton-Davis ER, et al. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Investig. 1999;104(4):375–81.
Linde L, Kerem B. Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008;24(11):552–63.
Woodley DT, et al. Gentamicin induces functional type VII collagen in recessive dystrophic epidermolysis bullosa patients. J Clin Investig. 2017;127(8):3028–38.
Kwong A, et al. Gentamicin induces laminin 332 and improves wound healing in junctional epidermolysis bullosa patients with nonsense mutations. Mol Ther. 2020;28(5):1327–38.
Li Y, et al. Gentamicin induces COL17A1 nonsense mutation readthrough in junctional epidermolysis bullosa. J Dermatol. 2020;47(3):e82–3.
Hung JH, et al. Topical gentamicin ointment induces LAMB3 nonsense mutation readthrough and improves corneal erosions in a patient with junctional epidermolysis bullosa. Clin Exp Ophthalmol. 2021;49:309–12.
Hammersen J, et al. Attenuation of severe generalized junctional epidermolysis bullosa by systemic treatment with gentamicin. Dermatology. 2019;235(4):315–22.
Hao M, et al. Intravenous gentamicin therapy for epidermolysis bullosa. In: AAD 2020 virtual meeting late-breaking research. 2020. https://aad.wistia.com/medias/lh2wwwc7pc. Accessed 1 May 2021.
Baradaran-Heravi A, et al. Effect of small molecule eRF3 degraders on premature termination codon readthrough. Nucleic Acids Res. 2021;49:3692–708.
Atanasova VS, et al. Amlexanox enhances premature termination codon read-through in COL7A1 and expression of full length type VII collagen: potential therapy for recessive dystrophic epidermolysis bullosa. J Investig Dermatol. 2017;137(9):1842–9.
Gonzalez-Hilarion S, et al. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J Rare Dis. 2012;7(1):58.
Bauer JW, et al. Specialized yeast ribosomes: a customized tool for selective mRNA translation. PLoS ONE. 2013;8(7):e67609.
Rathner A, et al. Drug development for target ribosomal protein rpL35/uL29 for repair of LAMB3R635X in rare skin disease epidermolysis bullosa. Skin Pharmacol Physiol. 2021;34:1–16.
Tang JY, et al. A systematic literature review of the disease burden in patients with recessive dystrophic epidermolysis bullosa. Orphanet J Rare Dis. 2021;16(1):175.
Has C, South A, Uitto J. Molecular therapeutics in development for epidermolysis bullosa: update 2020. Mol Diagn Ther. 2020;24(3):299–309.
Prodinger C, et al. Profiling trial burden and patients’ attitudes to improve clinical research in epidermolysis bullosa. Orphanet J Rare Dis. 2020;15(1):182.
Ebens CL, McGrath JA. Bone marrow transplant with post-transplant cyclophosphamide for recessive dystrophic epidermolysis bullosa expands the related donor pool and permits tolerance of nonhaematopoietic cellular grafts. Br J Dermatol. 2019;181(6):1238–46.
El-Darouti M, et al. Treatment of dystrophic epidermolysis bullosa with bone marrow non-hematopoeitic stem cells: a randomized controlled trial. Dermatol Ther. 2016;29(2):96–100.
Rashidghamat E, et al. Phase I/II open-label trial of intravenous allogeneic mesenchymal stromal cell therapy in adults with recessive dystrophic epidermolysis bullosa. J Am Acad Dermatol. 2020;83(2):447–54.
Riedl J, et al. ABCB5+ dermal mesenchymal stromal cells with favorable skin homing and local immunomodulation for recessive dystrophic epidermolysis bullosa treatment. Stem Cells. 2021. https://doi.org/10.1002/stem.3356.
Lee SE, et al. Intravenous allogeneic umbilical cord blood-derived mesenchymal stem cell therapy in recessive dystrophic epidermolysis bullosa patients. JCI Insight. 2021;6(2):e143606.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Authorship Contributions
TW, CP, JPH and ML wrote the manuscript. AH, JWB and HBK provided critical feedback and contributed to manuscript review.
Authorship
All authors approved the final version. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Martin Laimer (Grant/Research/Clinical Trial Support: Bristol-Myers Squibb, Onconova Therapeutics, Amryt Pharma, Rheacell, Holostem Terapie Avanzate, Castle Creek Pharmaceuticals), Johann W Bauer (Grant/Research/Clinical Trial Support: Onconova Therapeutics, Amryt Pharma, Rheacell, Holostem Terapie Avanzate, Castle Creek Pharmaceuticals; Shareholder Diaderm Ges.m.b.H: commercialisation of diacerein creme), Hannelore Breitenbach-Koller (Grant/ Research/Translational research support: WISS 2015 P_147200_30, Land Salzburg; Grant/ Translational Research Support: Hans-Groeber Stiftung PFA149050_01; KBHB Consult GmBH). JPH is supported by DEBRA Austria. Tobais Welponer, Christine Prodinger and Arno Hintersteininger have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Welponer, T., Prodinger, C., Pinon-Hofbauer, J. et al. Clinical Perspectives of Gene-Targeted Therapies for Epidermolysis Bullosa. Dermatol Ther (Heidelb) 11, 1175–1197 (2021). https://doi.org/10.1007/s13555-021-00561-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-021-00561-5