
Abstract
Lysosomes are acidic, membrane-bound organelles that serve as the primary catabolic compartment of the cell. They are crucial to a variety of cellular processes from nutrient storage to autophagy. Given the diversity of lysosomal functions, it is unsurprising that lysosomes are also emerging as important players in aging. Lysosomal dysfunction is implicated in several aging-related neurodegenerative diseases including Alzheimer’s, Parkinson’s, amyotrophic lateral sclerosis/frontotemporal dementia, and Huntington’s. Although the precise role of lysosomes in the aging brain is not well-elucidated, some insight into their function has been gained from our understanding of the pathophysiology of age-dependent neurodegenerative diseases. Therapeutic strategies targeting lysosomes and autophagic machinery have already been tested in several of these diseases with promising results, suggesting that improving lysosomal function could be similarly beneficial in preserving function in the aging brain.
Similar content being viewed by others

Introduction
Lysosomes are dynamic organelles that function as a major catabolic center in the cell. They participate in cellular homeostasis by serving as the terminal degradation compartment in autophagy, modulating nutrient status and interacting with other organelles [1]. There is growing evidence for the role of lysosomes in aging. Some studies suggest that processes such as lysosomal acidification and autophagy decrease with age, potentially altering nutrient storage and clearance of damaged proteins and organelles [2]. The importance of lysosomes in the aging brain is also supported by the many neurodegenerative diseases presenting with lysosomal dysfunction [3]. Thus, improved knowledge of the pathophysiology of these diseases as well as identification of therapeutic targets will strengthen our understanding of and ability to preserve lysosomal function in the aging brain.
Lysosomal Functions Related to Aging
Lysosomes participate in several cellular functions including nutrient processing, autophagy, and inter-organelle contacts. There is emerging evidence that these functions may be perturbed in age-related disease processes, including those affecting the central nervous system, as well as in physiological aging.
Nutrient Storage and Sensing
Lysosomes play a critical role in nutrient homeostasis, serving as both centers for nutrient processing via a host of degradative enzymes and sites for storage of metabolites including amino acids, lipids, and ions. The transport of metabolites into the lysosome depends on transmembrane transporters and channels, whose activity is coupled to a proton gradient established by the highly conserved H+-ATPase or V-ATPase [4, 5]. V-ATPase pumps protons into the vacuolar lumen, and the subsequent extrusion of these protons drives ions and metabolites inward [6, 7].
Amino Acids
Lysosomes sense nutrient availability through the activities of V-ATPase and the conserved serine–threonine kinase, mTORC1, and both are sensitive to changes in amino acid levels [2, 8]. The activity of the V-ATPase is regulated through association–dissociation of its many subcomplexes [9]. In states of low amino acids, the V-ATPase becomes fully associated, generating a proton gradient to promote the influx of substrates into the lysosome [10]. The nutrient sensing function of the V-ATPase is additionally linked to the mTORC1 pathway via the V-ATPase, Rag-GTPases, and Ragulator complex. During states of nutrient abundance, mTORC1 associates with lysosomes to promote cellular growth and limit autophagy [11, 12]. The relationship between mTORC1 and lysosomal amino acid sensing is further evidenced by the lysosomal arginine transporter, SLC38A9, which communicates amino acid status to mTORC1 [13, 14]. This transporter mediates efflux of nonpolar amino acids from the lysosome to be utilized in processes such as protein synthesis [15]. A recent lysosomal metabolomics study identified both V-ATPase and mTORC1 as important mediators of amino acid efflux from the lysosome [16].
Evidence highlighting the importance of the V-ATPase and the mTORC1 pathways in aging is also growing. Acidification by V-ATPase or overexpression of V-ATPase components promoted long-term mitochondrial stability [17] and extended lifespan in yeast, respectively [18]. Similarly, mTORC1 inhibition was shown to increase lifespan in a range of organisms including yeast, flies, and mice [2].
Calcium
Lysosomes, along with the endoplasmic reticulum (ER), serve as main cellular reservoirs for Ca2+, an ion implicated in several signaling pathways and organelle homeostasis [19]. Although the mammalian lysosomal Ca2+ importer is unknown, various efflux channels have been characterized including transient receptor potential channel mucolipin-1 (TRPML1) and two-pore channels 1 and 2 (TPC1/2) [20]. Importantly, Ca2+ level has been correlated with lifespan as elevation of either intracellular or extracellular Ca2+ shortens replicative lifespan in yeast []. Moreover, loss of lysosomal function results in a rapid increase in cellular Ca2+ in yeast [22], and activation of the TRPML1 channel facilitates Ca2+-induced Ca2+ release from the ER, further increasing intracellular Ca2+ [23, 24]. TRPML1 has also been linked to V-ATPase function as blockage of V-ATPase or knockdown of its V0a1 subunits results in an increase in TRPML1-mediated Ca2+ efflux from lysosomes, suggesting a close interplay of the 2 proteins in Ca2+ homeostasis [25]. Regulation of cellular Ca2+ also has important implications for overall cell survival as Ca2+ overload and subsequent accumulation in the mitochondria can induce mitochondrial permeability transition pore opening, promoting the release of pro-apoptotic factors [26].
In the central nervous system, Ca2+ plays an important role in modulating electrical activity and neurotransmitter release [27]. Aged neurons show a variety of alterations in Ca2+ homeostasis including diminished efflux from the plasma membrane [28], decreased mitochondrial buffering capacity [29], and enhanced release from the ER [30, 31]. In the aging brain, this results in a net increase in cellular Ca2+, which perturbs neuronal excitability and long-term processes such as synaptic plasticity [27]. Although the function of lysosome-dependent Ca2+ homeostasis in the aging brain remains poorly understood, the ability of lysosomes to regulate cellular Ca2+ in non-neuronal cells suggests the possibility of a similar role for lysosomes in neurons.
Iron
Another key ion regulated by lysosomes is Fe2+ which enters the cell via the endolysosomal system [32, 33]. Tight regulation of cellular Fe2+ is crucial as it is an important cofactor for enzymes in the electron transport chain [34]; however, high levels may promote the formation of toxic reactive oxygen species (ROS) [35]. Of note, Fe2+ levels have been reported to be increased with age, thereby promoting mitochondrial protein aggregation and decreased lifespan in Caenorhabditis elegans [36].
Fe2+ is also important for central nervous system function via its involvement in myelin and neurotransmitter synthesis [33]. Fe2+ levels in the brain increase with age due to increased blood–brain barrier permeability, alterations in Fe2+ homeostasis, and neuroinflammation [37]. Moreover, changes to brain Fe2+ distribution in areas like the substantia nigra and basal ganglia occur with age [38]. In non-nervous tissues, free Fe2+ remains low via binding to ferritin and transferrin, whereas in catecholaminergic neurons, free Fe2+ is regulated through the formation of neuromelanin–iron complexes, which have been found to increase with age [39, 40]. A connection between lysosomes, Fe2+, and neuromelanin was recently highlighted with the characterization of neuromelanin-containing organelles as specialized autolysosomes that accumulate undegraded proteins and lipids in substantia nigra neurons [41]. The importance of iron homeostasis in the brain is also supported by a group of inherited disorders called neurodegeneration with brain iron accumulation (NBIA). Several NBIA genes are related to lysosomal function [42], and it was recently reported that iron overload in NBIA mutant cells resulted in both lysosomal and mitochondrial dysfunction [43].
Lipids and Glycoproteins
The role of lysosomes in glycoprotein and lipid homeostasis is evidenced by the plethora of lysosomal storage disorders (LSDs) that result from complete loss or decreased activity of specific lysosomal hydrolases. In LSDs, undigested lipids, glycoproteins, and mucopolysaccharides accumulate in the lysosome, leading to cellular toxicity and compromised survival [44]. A comprehensive description of the LSDs can be found in previously published reviews [45, 46]. Importantly, lysosomal dysfunction due to storage of accumulated substrates in LSDs results in decreased lysosomal degradation and various pathologies in the CNS that resemble adult-onset neurodegenerative diseases—for example, Parkinson’s disease (PD)-like Lewy body accumulation in Gaucher disease and Alzheimer’s disease (AD)-like plaques and tangles in Niemann–Pick disease type C [46]. This suggests that alterations in lysosomal degradative capacity may contribute to age-related neuropathologies.
Autophagy
Autophagy is a critical physiological process that maintains intracellular homeostasis by degrading and recycling cytoplasmic material, including damaged organelles, long-lived proteins, and protein aggregates, via delivery to the lysosome. Though the process was previously thought to occur nonselectively, studies have shown that autophagy can be stimulated under stress conditions and by cellular insults. Additionally, dysfunctional organelles like the mitochondria may also undergo degradation by the lysosome via selective autophagy.
Mechanism of Autophagy
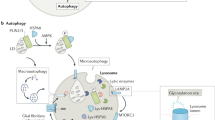
Macroautophagy, hereafter referred to as autophagy, is 1 of 3 modes of autophagy alongside microautophagy and chaperone-mediated autophagy. It is the primary mode for degrading large parts of cytoplasm through sequestration inside double-membrane vesicles, termed autophagosomes, which then fuse with lysosomes [47]. Several highly conserved AuTophaGy-related (Atg) genes have been identified as directly involved in autophagosome formation, maturation, and fusion with the lysosome.
Autophagosome formation is initiated by the phagophore, an open double-membrane structure that forms around proteins and organelles in the cytoplasm [47]. The membrane is derived from proximal structures including the ER, endosomes, mitochondria, Golgi, and plasma membrane [48, 49]. Unc-51-like kinase 1 (ULK1) is a critical protein in phagophore induction through its interaction with autophagy proteins including mAtg13 and the scaffolding protein FIP200 [50]. Other players in autophagosome formation include a transient interaction with mAtg9 [51], which may provide additional membrane sources to the growing phagophore.
In humans, the class III phosphatidylinositol 3-kinase, vesicular protein sorting 34 (hVps34), and its complex 1 binding partners—Beclin 1, p150, and Atg14L—are also necessary for phagophore formation [52,53,54,55]. hVps34 complex 1 interactions stimulate hVps34 activity, promoting phosphatidylinositol to phosphatidylinositol 3-phosphate conversion, which facilitates recruitment of other Atgs to the forming autophagosome [52, 55]. hVps34 also has a set of complex 2 interactors, in which the conserved hVps34–Beclin 1 complex backbone interacts with ultraviolet irradiation resistant-associated gene (UVRAG) [56, 57]. UVRAG association with complex 2 modulates several functions including membrane bending, negative and positive regulation of autophagy, and promotion of autophagosome fusion with endosomes/lysosomes [58].
Elongation of the phagophore membrane involves 2 ubiquitin-dependent conjugation systems. In the first, Atg7 activates Atg12, which is transferred to Atg10 to stimulate Atg12–Atg5 covalent linkage [59,60,61]. The Atg12–Atg5 complex then interacts with Atg16L to form the Atg16L complex, which drives phagophore elongation [60, 61]. The second system involves the microtubule-associated protein light chain 3 (LC3) [62]. Upon autophagy induction, LC3 is cleaved to cytosolic LC3-I which is then conjugated to phosphatidylethanolamine by Atg7 and Atg3 to generate lipidated LC3-II [59, 60]. LC3-II associates with both sides of the phagophore membrane to mediate cargo selection through its recruitment of the autophagy receptor, p62, and components involved in autophagosome–lysosome fusion [63,64,65,66,67]. The final step involves fusion of the outer membrane of the autophagosome with the lysosome. Various SNAREs and tethering/adaptor proteins on late endosomes/lysosomes and autophagosomes have been identified and have been discussed in previous reviews [68].
Regulation of Autophagy
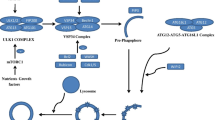
Studies have revealed a close interplay between lysosomes and mTORC1 in the regulation of autophagy. Under a nutrient-rich environment, mTORC1 is recruited to the lysosomal membrane via a complex consisting of the membrane-tethered Ragulator complex and Rag GTPases [12, 69], where it is activated by Ras homolog enriched in brain (Rheb) [70]. Activation of mTORC1 along with suppression of its inhibition by tuberous sclerosis tumor suppressor complex (TSC1–TSC2) in nutrient-replete conditions inhibits autophagy [71]. Autophagy initiation and lysosome biogenesis are also controlled at a transcriptional level through the transcription factor EB (TFEB). Nutrient deprivation induces nuclear translocation of TFEB to enhance expression of autophagosome-related genes [72, 73]. However, under basal conditions, activated mTORC1 phosphorylates TFEB, resulting in cytoplasmic retention of the transcription factor and inhibition of autophagy [74, 75].
mTORC1-independent mechanisms of autophagy induction have also been identified, including inhibition of inositol-1,4,5-trisphosphate (IP3) levels and attenuation of Ca2+-related pathways [76, 77]. IP3 is released from the plasma membrane upon phospholipase C processing of phosphatidylinositol 4,5-bisphosphate (PIP2) [78]. High levels of IP3 can inhibit cellular clearance [79, 80] likely by stimulating Ca2+ release through IP3 receptors and disrupting Ca2+ gradients needed for autophagosome–lysosome fusion. Elevated cytosolic Ca2+ also regulates autophagy via activation of the Ca2+-dependent cysteine protease, calpain. A negative correlation likely exists between calpain activity and autophagy initiation as calpain inhibition has been shown to promote autophagosome formation [77, 81, 82]. In addition, calpain activity could increase IP3 levels via G-protein signaling downstream of GPCRs [77]. Lastly, inhibition of TFEB through phosphorylation by the serine–threonine kinase, Akt, can block autophagy independent of mTORC1 [83]. Conversely, TFEB dephosphorylation by the Ca2+-dependent phosphatase, calcineurin, enables nuclear translocation, thereby inducing autophagy [84]. Notably, it was also shown that calcineurin is activated by local Ca2+ release from lysosomes [84].
Mitophagy
Mitophagy is a selective form of autophagy in which damaged mitochondria are eliminated, and serves as a quality control mechanism to preserve cellular homeostasis [85,86,87]. The best characterized pathway for mitophagy involves the Parkinson’s disease-linked proteins, PTEN-induced kinase 1 (PINK1) and Parkin [85,86,87]. When mitochondria are damaged, PINK1 stabilizes on the outer mitochondrial membrane to promote Parkin translocation and E3 ubiquitin ligase activity, leading to the incorporation of ubiquitinated cargo into autophagosomes [88]. In addition, PINK1 and Parkin are involved in a mechanism of mitochondrial clearance that delivers mitochondrial-derived vesicles (MDVs) enriched in oxidized proteins to lysosomes for degradation [89].
Additional pathways of mitophagy have also been identified. Transmembrane receptor-mediated mitophagy, in which LC3-interacting regions mediate recognition of damaged mitochondria, have been described for several outer mitochondrial membrane proteins including Nix [90] and FUN14 Domain containing 1 [91, 92], which were observed to be induced to drive mitophagy in response to hypoxia. Moreover, cardiolipin-mediated mitophagy, where exposure of the inner mitochondrial membrane phospholipid, cardiolipin, on the mitochondrial surface allows its interaction with LC3, has been shown to activate mitophagy independent of PINK1/Parkin [93]. Finally, other stimuli, such as iron chelators [94] and increased mitochondrial fission [95, 96], also induce mitophagy in a PINK1/Parkin-independent manner.
Although several mechanisms of mitophagy, such as the PINK1/Parkin and cardiolipin-mediated pathways, have been observed in neurons, whether additional neuronal mitophagy pathways exist and how different pathways interact and are regulated is still incompletely understood. Nevertheless, the elimination of damaged mitochondria is critical for neurons, which cannot alleviate cellular stress by division and require functional mitochondria for energy and Ca2+ homeostasis. The importance of mitophagy in neurons is also demonstrated by the various mutations in mitophagy-related genes that are causal or linked to neurodegenerative diseases such as PD and amyotrophic lateral sclerosis–frontotemporal dementia (ALS–FTD) [97] as well as by evidence suggesting that decreased lysosomal acidification impairs mitophagy in cellular models of AD [98]. Thus, understanding how mitophagy is dysregulated in neurodegeneration may further elucidate how lysosomal function in neurons is altered in age-related pathologies.
Interactions with Mitochondria
The regulation of nutrient homeostasis by lysosomes is intimately associated with mitochondrial function, and the modulation of mitochondrial function by lysosomes has various connections to aging. For example, vacuolar acidity is decreased with age in yeast, resulting in reduced lifespan and impaired mitochondrial function [17]. Similarly, lifespan and mitochondrial function are compromised in cells lacking certain subunits of the V-ATPase, leading to altered mitochondrial morphology, protein import, and potential [17, 99, 100].
Recent studies have elucidated the physical interaction between lysosomes and mitochondria. A tethering complex, vCLAMP, was identified between the yeast vacuole and mitochondria and was hypothesized to regulate lipid and metabolite transport between the 2 compartments [101, 102]. Importantly, mitochondria–lysosome contact sites were recently identified in mammalian cells and were found to be regulated by GTP hydrolysis of the lysosomal GTPase, Rab7 [103]. These contact sites allow for bidirectional regulation of mitochondrial and lysosomal dynamics, whereby 1) mitochondria regulate lysosomal dynamics by modulating the GTP-binding status of Rab7 via the mitochondrial-localized Rab7-GAP (TBC1D15), and conversely 2) lysosomes regulate mitochondrial dynamics by marking the majority of mitochondrial fission sites [103]. Mitochondria–lysosome contacts may have additional functions including the transfer of metabolites such as lipids or ions [103, 104].
Lysosomes may also affect mitochondrial function indirectly through the modulation of amino acid homeostasis. High levels of cytosolic basic amino acids have been shown to induce cellular toxicity, impair mitochondrial function, and increase ROS generation [105]. Similarly, increased levels of nonpolar, unbranched amino acids in the cytosol can accumulate in the mitochondria and overload mitochondrial metabolic pathways [106, 107].
A hallmark feature of the aging brain is reduction in mitochondrial function with a concurrent increase in ROS and oxidative damage. The ability of ROS to modulate lysosomal function is mediated in part by the lysosomal TRPML1 channel, which acts as a ROS sensor. When cellular ROS is increased, activated TRPML1 releases Ca2+ into the cytosol, inducing both TFEB nuclear translocation for lysosome biogenesis and autophagy [108]. Because both TRPML1 activation and mitochondrial ROS production have been shown to increase in response to inhibition of V-ATPase, lysosomal de-acidification may serve as an upstream pathophysiological event modulating lysosomal Ca2+ release and mitochondrial function [109]. Further evidence linking ROS to lysosomal dysfunction include studies showing that loss of mitochondrial function in brain tissue following deletion of mitochondrial proteins or inhibition of oxidative phosphorylation is sufficient to impair lysosomal function in a ROS-dependent manner [110]. Although there is still debate as to whether ROS is a cause or consequence of aging, it is likely that it contributes to both; initial increases in ROS due to age further exacerbate aging by enhancing lysosomal dysfunction.
Lysosomal Function in Neurodegenerative Disease
Several studies have revealed the importance of lysosome-related functions, such as autophagy, in neuronal homeostasis. Mouse models with neural cell-specific knockout of Atg5 or Atg7, leading to impaired autophagosome-lysosome clearance, exhibited neurodegeneration with motor deficits and shorter lifespan [111, 112].
Conversely, induction of autophagy has been linked to longevity and resistance to protein aggregation and oxidative stress in neurons [71, 113]. Selective activation of lysosomal activity via TFEB was also found to clear protein aggregates and increase activation of quiescent neural stem cells during aging [114]. Although the role of lysosomes in the aging brain is still being elucidated, our understanding of age-dependent neurodegenerative diseases, many of which implicate lysosome-related genes, has provided insight into the changes in lysosomal function with age (Fig. 1).

Genes implicated in neurodegenerative disease related to autophagic and lysosomal function. Genes involved in several neurodegenerative diseases affect various steps of autophagy as well as have direct impacts on lysosomal function. Bolded correspond to causative genes, unbolded correspond to risk genes. Genes implicated in PD (blue), AD (green), ALS/FTD (red), and HD (purple)
Alzheimer’s Disease
AD is the most common neurodegenerative disease affecting more than 24 million individuals over the age of 65 [115]. The major pathological hallmarks include the accumulation of extracellular amyloid-beta (Aβ) plaques and intracellular tau-induced neurofibrillary tangles (NFTs) [115]. Normally, Aβ is trafficked to the lysosome via the autophagy pathway and degraded by cathepsin D [116]. However, recent studies suggest that release of Aβ into the extracellular space may also be in part regulated through autophagy [117]. Disruptions in autophagy were first linked to AD when autophagosome/lysosome accumulation was observed in dystrophic neurites [118]. Follow-up studies were able to show the presence of Aβ sequestered in the retained autophagosomes [119], thus highlighting the importance of the autophagy pathway in AD progression.
Clusterin, encoded by CLU, is 1 of the top candidate genes for AD development and is linked to autophagy through its involvement in autophagosome biogenesis via interaction with LC3 [120, 121]. In human studies, clusterin expression positively correlates with increasing levels of Aβ40/42 in brain areas normally afflicted in AD [122]. Furthermore, clusterin interacts directly with Aβ and reduces its aggregation, thereby protecting against toxic effects [123]. Although associations between clusterin and tau are still debated, 1 study found that tau overexpression increases clusterin expression [124], which could potentially stimulate autophagy-mediated degradation of tau.
PSEN1, another causal AD gene which encodes presenilin 1, is directly linked to autophagy through an identified role in lysosomal acidification [25, 125, 126]. Presenilin 1 is responsible for regulating gamma-secretase, which processes amyloid precursor protein (APP) into Aβ [127]. Mutations affecting presenilin 1 function may disrupt lysosomal proteolysis and promote accumulation of autophagic cargo within the cell [128, 129]. In support of this, cerebrospinal fluid (CSF) from PSEN1 patients exhibited less Aβ than normal individuals [130], suggesting that Aβ retention may lead to cellular toxicity when autophagy is impaired.
Several risk genes for AD also have links to autophagy. Atg7, which mediates the formation of the Atg12–Atg5 complex during phagophore maturation [131], is involved in incorporation of Aβ into multivesicular bodies (MVBs)/late endosomes [132, 133]. Increased transport of Aβ into MVBs could affect downstream fusion with autophagosomes. Recent studies suggest that release of Aβ into the extracellular space may be in part regulated by autophagy [117]. Inhibition of autophagy through genetic ablation of Atg7 decreases Aβ plaque formation in mouse models [117]. However, complete loss of autophagy also led to the accumulation of Aβ inside neurons [117], which can potentiate neurotoxicity. Atg7 is also linked to the tau degradation as knockout mice exhibited increased phosphorylated tau [134]. Altogether, this suggests that extreme modulation of autophagy may protect against 1 AD phenotype but exacerbate another.
Associations between Bcl-2 expression and memory have been reported in AD patients [135], highlighting an important role for Bcl-2 in AD pathogenesis. Treatment with Aβ decreases Bcl-2 expression and overexpression of Bcl-2 in APP mutant mice was found to be neuroprotective [135, 136]. Bcl-2 overexpression also decreases APP processing, extracellular Aβ deposits, and intracellular NFTs [137]. As stated previously, Beclin 1 is responsible for initiating autophagy and regulating autophagosome formation [138]. Beclin 1 heterozygous knockout mice displayed disruptions in autophagy [139], suggesting that alterations in Beclin 1 levels and function can contribute to AD progression. In agreement with this, presenilin-null cellular models and postmortem analysis of AD brains reported a decrease in Beclin 1 level [140]; however, a recent large-scale quantitative study found no significant changes in Beclin 1 gene expression or protein levels across multiple stages of AD [141].
Cathepsin D, the lysosomal protein involved in Aβ processing [116], is encoded by CTSD which confers risk in AD [142, 143]. Although the mechanism of cathepsin D in AD pathology is not well understood, 1 potential mechanism is through altered functioning of lysosomal proteolysis potentially mediated by presenilin 1 mutations which ultimately result in disrupted lysosomal acidification [25, 125, 126]. Cathepsin D levels were reportedly lower in bone marrow-derived monocytes, fibroblasts, and peripheral blood lymphocytes in AD patients [144,145,146], whereas elevated CTSD gene expression and protein levels were observed in AD brains and neurites [141, 147]. Interestingly, 1 additional study found that cathepsin D levels were increased in postmortem CSF albeit in an inactive form of the protein [148]. This suggests that like Beclin 1, alterations in cathepsin D function can be detrimental in AD pathogenesis when compounded with disease-associated mutations like those found in PSEN1.
Another hallmark of AD is dysfunction in the endosomal–lysosomal system, such as endosomal enlargement, which is 1 of the earliest pathological features of disease [149]. APP, in which mutations cause autosomal dominant, early-onset AD, has been implicated in AD-related endosomal aberrations. It was demonstrated that the APP β C-terminal fragment directly interacts with the adaptor protein containing pleckstrin homology domain, phosphotyrosine binding domain, and leucine zipper motif (APPL1), which also stabilizes active Rab5 on the endosomal membrane [150]. Overactivation of Rab5 by APP β C-terminal fragment mediates downstream events including altered endosomal motility and signaling and may also contribute to AD phenotypes such as defects in long-term potentiation [149, 151,152,153] and cholinergic degeneration. In addition, APP and Rab5 overactivation has been linked to defective lysosomal morphology and proteolysis as well as autophagy in various cellular and mouse models of AD [151, 152, 154].
The E4 allele of apolipoprotein E (ApoE4), a major risk factor for sporadic AD, has similarly been linked to endolysosomal dysfunction including increased endocytosis of APP [155] and impaired endosomal recycling [156, 157]. ApoE4 intermediates have also been shown to disrupt lysosomal membranes in neuronal cultures, leading to leakage of hydrolases and induction of apoptosis, which is further potentiated by Aβ(1-42) [158, 159]. A recent transcriptomics analysis in ApoE4-targeted replacement mice showed significant enrichment of genes involved in endosomal–lysosomal processing, further implicating ApoE4 in endolysosomal function [160].
Parkinson’s Disease
PD is the most common movement disorder with an estimated global prevalence of ~400 out of 100,000 by the age of 65 [161]. The cardinal motor symptoms—bradykinesia, rigidity, and tremor—are primarily attributed to dopaminergic neuron deterioration. In the majority of PD cases, Lewy bodies composed of proteins, lipids, and undegraded organelles including autolysosomes and damaged mitochondria accumulate in several brain regions [162,163,164].
Several autosomal dominant PD genes are directly linked to lysosomal function including SNCA, leucine-rich repeat kinase 2 (LRRK2), and vacuolar protein sorting-associated protein 35 (VPS35). The first PD gene identified, SNCA, encodes for alpha-synuclein (αSyn) [165], a major constituent of Lewy bodies [166]. Phenotypically, multiplication of the SNCA gene leads to earlier onset PD, indicating that increased expression of SNCA promotes disease pathogenesis [167, 168]. αSyn overexpression impedes autophagy by prohibiting Atg9 from initiating phagophore formation [169, 170]. Moreover, high endogenous αSyn in human iPSC-derived dopaminergic neurons reduces lysosomal proteolysis by delaying the delivery of hydrolases such as cathepsin B and glucocerebrosidase (GCase) to the lysosome [171]. SNCA missense mutations, which have an increased propensity to oligomerize, are also linked to familial PD [172, 173]. These aberrant forms of αSyn are resistant to degradation and reduce the efficiency of autophagy [174, 175]. Inhibition of autophagy and lysosomal function increase αSyn release, potentially as a compensatory mechanism [176,177,178,179,180,181,182,183].
Gain-of-function mutations in LRRK2 are the most common cause of familial late-onset PD, and polymorphisms in the locus increase PD risk [184]. LRRK2 can be localized on membranes of synaptic vesicles, late endosomes, and lysosomes [185, 186], and several Rabs involved in vesicle formation, trafficking, and docking/fusion are phosphorylated by LRRK2 [186, 187]. The consequences of pathogenic LRRK2 mutations on synaptic integrity, autophagy, and lysosomal function require further elucidation. Studies have described that patient G2019S LRRK2 fibroblasts exhibited higher lysosomal abundance and protein clearance at baseline [188], and several LRRK2 mutant fibroblasts showed decreased autophagic flux following serum/amino acid deprivation [189]. Moreover, G2019S LRRK2 iPSC-derived dopaminergic neurons were more susceptible to oxidative stress and retained higher loads of autophagic vacuoles and lipid droplets [190, 191].
VPS35, causally linked to autosomal dominant PD, is part of the retromer complex and is important for trafficking of cathepsin D, Lamp2A, and Atg9a [172, 192,193,194], thereby regulating autophagy and endolysosomal processing. Mitochondrial quality control also requires VPS35 for both fusion/fission dynamics and trafficking of MDVs to the lysosome for degradation [89, 195].
Loss-of-function of the cytosolic E3 ubiquitin ligase, Parkin, and the mitochondrial kinase, PINK1, are linked to early-onset autosomal recessive PD [172], and both proteins are also involved in mitochondrial quality control via mitophagy and MDVs [88, 89]. Parkin deficiency has been shown to destabilize Rab7 [196], which is crucial for both mitochondrial fission and autophagosome–lysosome fusion.
DJ-1, a chaperone with antioxidant functions, has been shown to protect dopaminergic neurons against oxidative stress in PD pathogenesis [197, 198]. iPSC-derived dopaminergic neurons from DJ-1 patients show an accelerated pathological cascade beginning with increased oxidative stress which further induces dopamine oxidation [199]. Oxidized dopamine in turn inhibits the lysosomal enzyme glucosylceramidase (GCase) and is paralleled by increased levels of soluble and insoluble αSyn [199].
The juvenile-onset atypical parkinsonism, Kufor–Rakeb syndrome, is caused by autosomal recessive mutations in PARK9, which deplete P5-type ATPase 13A2 (ATP13A2) from endolysosomes [200]. Studies using patient fibroblasts, iPSC-derived neurons, and knockout mice report mitochondrial dysfunction, lysosomal acidification and macroautophagy deficits, as well as lipid deposition preceding αSyn accumulation [201,202,203,204]. Moreover, ATP13A2 is considered to be important for maintaining intracellular ion homeostasis [205,206,207,208], a function which may be critical for autophagy/endolysosomal processing efficiency.
Various PD risk factors further implicate lysosomal dysfunction in PD pathogenesis. Autosomal recessive mutations in GBA1, which encode GCase, increase the risk for PD and cause the LSD, Gaucher disease [209, 210]. Some Gaucher patients present with parkinsonism as well as dopaminergic neurodegeneration and Lewy body pathology [209]. iPSC-derived neurons from GBA1-associated PD patients show reduced lysosomal GCase activity, macroautophagy, and Ca2+ buffering [211, 212]. Reduced GCase activity is linked to elevated glucosylceramide levels, which further promote αSyn accumulation and higher-order assembly within lysosomes [211, 213, 214]. Variants of the acid sphingomyelinase-encoding SMPD1 gene are linked to the LSDs, Niemann–Pick disease types A and B, and may follow a similar mechanism to GBA1 variants [215,216,217]. Furthermore, the PD risk factor SCARB2 encodes the lysosomal integral membrane protein type 2 (LIMP2), which sorts GCase to the lysosome and is important for autophagy, αSyn proteostasis, and dopaminergic viability [218,219,220]. Additional PD risk loci linked to lysosome-related genes (e.g., GALC, CTSB, ATP6V01A) were recently revealed by a large meta-analysis of genome-wide association studies [210]. Collectively, this points to the necessity of improving lysosomal function in PD.
Amyotrophic Lateral Sclerosis and Frontotemporal Dementia
ALS is characterized by the progressive degeneration of motor neurons. Like other neurodegenerative diseases, ALS is a proteinopathy where pathological DNA-binding protein 43 (TDP-43) is found accumulated in the cytosol of affected neurons [221, 222]. Aggregated TDP-43 plays a pathogenic role in both ALS and FTD [221], indicating that both ALS and FTD belong to a spectrum of the same disease. To date, a large number of genes have been linked to familial ALS, but strikingly, many of these genes are directly linked to autophagic function, suggesting that improving autophagy may alleviate ALS-related pathologies.
Superoxide dismutase 1 (SOD1) was 1 of the first enzymes associated with ALS. Inhibition of autophagy led to aggregation of mutant SOD1 [223], indicating that dysfunctional SOD1 is largely degraded by autophagy. The autophagy receptor protein p62, encoded by the ALS-linked gene SQSTM1, is responsible for loading of cargo into the extending phagophore/autophagosome [224,225,226]. Interestingly, p62 has been shown to interact with polyubiquitinated mutant SOD1 [227, 228], which enhances its interaction with lipidated LC3-II on the phagophore membrane [228], suggesting that autophagy is activated in the presence of aggregated mutant SOD1. These interactions lead to the formation of protein inclusions within motor neurons [228]. Autophagy was also reportedly increased in mutant SOD1 mice potentially due to decreased phosphorylation of ULK1 [229], which activates autophagy [230]. In addition, proteins involved in autophagy regulation such as TFEB and Beclin 1 were found upregulated in the spinal cords of mutant SOD1 mice [231].
Several genes associated with ALS also encode RNA-binding proteins that aggregate when mutated. Both TDP-43 and fused in sarcoma (FUS) are found in intracellular aggregates in brain autopsy [232, 233]. In order to maintain homeostasis, autophagy is known to degrade aggregated TDP-43 and FUS [234,235,236]. Not surprisingly, increased expression of TDP-43 and FUS promote aggregation and stall autophagy in ALS models that could be rescued upon autophagy induction [234, 237, 238]. Furthermore, TDP-43 depletion increases translocation of TFEB into the nucleus, thereby stimulating autophagy [239]. However, downregulation of TDP-43 has also been shown to decrease Atg7 expression leading to reduced autophagic flux and autophagosome–lysosome fusion [240]. Thus, studies on the mechanism of action for TDP-43 in relation to autophagy have given both gain and loss of function results.
A subset of ALS-linked genes encode autophagy receptors, such as p62 and optineurin (OPTN) [241], or regulators of such receptors like Tank binding kinase 1 (TBK1) [242, 243]. As autophagy receptors, both p62 and optineurin contain a ubiquitin-associated domain (UBA) and an LC3-interacting region [241]. Generally, the proteasome is responsible for the degradation of ubiquitinated proteins. However, the discovery of autophagic receptors with UBA domains suggests that ubiquitinated cargo may also be targeted to and degraded by lysosomes. Many ALS-associated mutations affecting p62 and optineurin activity fall within the UBA domain [244, 245]. Furthermore, mutations located in the UBA domain may affect the protein’s ability to be phosphorylated by TBK1 [242, 243], suggesting that loss of TBK1 activity can enhance autophagic dysfunction in ALS.
The most commonly mutated gene in ALS, C9orf72 [246], has also been linked to autophagy via its interaction with endocytic proteins, Rab5 and Rab7 [247, 248], which regulate endosomal maturation [249]. Proper maturation and positioning of late endosomes is critical for autophagosome fusion [249]. C9orf72 also interacts with the ULK1 complex directly, and loss of C9orf72 prevents autophagy in an ULK1-mediated manner [250, 251]. As C9orf72 was previously shown to interact with Rab1a, an early autophagic regulator involved in phagophore formation [247, 251], C9orf72 may also regulate the recruitment of ULK1 to the initiation complex. Conversely, loss of C9orf72 leads to inactivation of mTORC1, increased TFEB nuclear translocation, and subsequent activation of autophagy [250, 252]. Therefore, C9orf72 is involved in several conflicting steps of autophagy but ultimately has been implicated in the maintenance of lysosomal function.
FTD is the second most common young-onset dementia and is characterized by atrophy of the frontal and temporal lobes [253, 254]. FTD is divided into subgroups based on the pathological hallmarks that develop with disease progression, including FTD with tau (FTD-tau) or TDP-43 (FTD-TDP)-positive inclusions [253]. The majority of FTD cases are attributed to mutations in microtubule-associated protein tau, progranulin (GRN), and C9orf72 [253, 254]. The remaining familial-associated FTD cases are linked to mutations in valosin-containing protein (VCP), FUS, TDP-43, and charged multivesicular body protein 2B (CHMP2B), which play distinct roles in the endolysosomal pathway [253, 254]. This suggests that disruptions in normal endosome to lysosome maturation may delay the degradation of autophagic cargo or impair the delivery of lysosomal hydrolases. For example, it was previously shown that VCP patients exhibited autophagic defects including an increase in the number of enlarged cathepsin B-positive autophagic vesicles [255]. Moreover, patients with mutations in VCP and CHMP2B were also reported to have decreased autophagic clearance and lysosomal function [255,256,257,258,259].
It was previously posited that FTD and LSDs were related diseases as they both shared similar common pathophysiological phenotypes. However, a concrete link was established with the discovery of a family affected by both neuronal ceroid lipofuscinosis (NCL), a young-onset LSD, and FTD [260]. Linkage analysis revealed independent heterozygous and homozygous mutations in the GRN gene [260], which encodes granulin, thus linking lysosomal dysfunction to GRN mutants and FTD. The proprotein, progranulin, is cleaved into individual granulins in the endolysosomal pathway [261, 262]. Recent work further demonstrated granulin E is responsible for modulating cathepsin D activity [263, 264], suggesting that GRN can directly alter lysosomal function through resident hydrolases. In agreement with this, GRN knockout mice displayed an increase in the levels of TMEM106B, as well as an accumulation of lipofuscin puncta in lysosomes [265], both of which are indicators of lysosomal dysfunction commonly found in NCLs. Furthermore, CTSD knockout mice exhibited increases in progranulin and TMEM106B along with saposin D, which is normally found accumulated in NCL10 patients [265]. Altogether, the genetics of ALS/FTD strongly implicate dysfunctions in both autophagy and endolysosomal systems in disease pathogenesis.
Huntington’s Disease
Huntington’s disease (HD) is an autosomal dominant, trinucleotide repeat disorder characterized by cognitive dysfunction, psychiatric disturbances, and loss of motor control [266]. The brain region most affected in HD is the striatum, an area critical to initiating and controlling movement, although other areas of the brain such as the cortex and cerebellum are also affected as the disease progresses [267]. Although defects in various pathways have been associated with HD, including alterations in Ca2+ handling, vesicle transport, and ER homeostasis [268], recent studies have implicated defective autophagy as a key feature of the disease, highlighting the potential of lysosomes and other autophagy machinery as druggable targets in HD.
The genetic basis of HD is a polyglutamine (polyQ) repeat expansion near the N-terminus of the huntingtin (htt) protein [269]. A repeat expansion of greater than 36Q is causative of disease, and like in other repeat disorders, greater repeat lengths correlate with earlier disease onset [268]. The hallmark feature of HD pathology is the accumulation and aggregation of mutant htt protein (mhtt). These aggregates form both intranuclear and intracytoplasmic neuronal inclusions, although whether these aggregates are pathogenic is still debated [270, 271]. Furthermore, htt can be proteolyzed to smaller fragments, which may also contribute to disease pathogenesis [272,273,274,275].
Although the function of wild-type htt is not fully understood, htt has been shown to interact with autophagy machinery. For instance, human and mouse HD samples exhibit an increase in autophagosomes [276, 277], and mhtt was shown to activate autophagy via sequestration and inactivation of mTORC1 [278]. Despite increasing autophagosome formation, mhtt appears to alter cargo recognition and loading through an unidentified mechanism, ultimately leading to accumulation of damaged organelles and protein aggregates [279]. Additional aberrations in autophagy are linked to effects of mhtt on vesicle trafficking including deficits in autophagosome motility [280] and autophagosome–lysosome fusion [281]. Silencing of either htt or its binding partner, huntingtin-associated protein 1, blocked retrograde transport of autophagosomes along the axonal compartment, whereas depletion of mhtt also resulted in an accumulation of autophagosomes containing undegraded cargo [280]. Alterations in mRNA of autophagy-associated proteins were also observed in the striatum of HD patients. Notably, LC3A and LAMP2 mRNA are increased, consistent with an overall increase in autophagic flux, whereas PINK1 is decreased, suggesting defects in mitophagy [282]. Finally, a polymorphism in Atg7, which is required for LC3 lipidation, is associated with earlier HD onset, thus providing strong genetic evidence for the close interplay between HD and autophagy [283].
Another important feature of htt is its ability to modulate autoclearance through posttranslational modifications. Ubiquitination of htt at K6, K9, and K15 is associated with increased degradation, whereas SUMOylation of these residues leads to decreased clearance [284]. Wild-type htt is further ubiquitinated at the K48 linkage, which is a classical marker of proteosomal degradation, whereas mhtt is ubiquitinated at K63, which is correlated with increased aggregation [275]. The ubiquitination and SUMOylation of htt is regulated through phosphorylation of S13 and S16, which are critical for mediating mhtt toxicity [285]. Acetylation at K444 is also important for regulating htt degradation and has been shown to increase mhtt clearance through enhanced binding to p62 [286].
In addition to the effects of mhtt on the autophagic pathway, recent studies have suggested a direct interaction of mhtt with lysosomes. In mouse striatum, mhtt alters lysosomal positioning via increased perinuclear localization which correlated with increased autophagic flux in response to nutrient deprivation [287]. Furthermore, it was recently shown that mhtt is able to be secreted from neurons through an unconventional endolysosomal pathway, suggesting that interactions of mhtt with the endolysosomal system may also contribute to disease pathogenesis [288].
Targeting Lysosomal Pathways in Neurodegenerative Disease
As discussed, several neurodegenerative diseases present with changes to lysosomal and autophagic function, and these alterations may in turn correlate to physiological aging processes. At present, several therapeutic approaches converge on targeting autophagy and lysosomes in neurodegenerative diseases. Therefore, combating the pathophysiological mechanisms implicated in these diseases will improve our knowledge and assist in the development of lysosome-targeted therapies to reduce disease burden and ultimately preserve lysosomal function in the aging brain.
Targeting Autophagy in Neurodegeneration
The strong association between neurodegenerative diseases and aberrations in autophagy implicates the autophagic pathway as a prime therapeutic target for the treatment of these diseases. Various therapeutic approaches targeting both mTOR-dependent and mTOR-independent pathways of autophagy have now been tested preclinically and clinically (Fig. 2, Table 1).

Pharmacological agents used in preclinical and clinical investigational therapies for neurodegeneration and their known or proposed targets related to autophagy and lysosomes. Agents whose action relates to suppression of mTORC1 activity (1), reduction of cytosolic calcium and IP3 levels as well as enhancement of TFEB activity (2-4) are expected to generally induce cellular autophagy. Targeting lysosomal acidity or glucocerebrosidase activity (5) can specifically induce lysosomal clearance efficiency
mTOR-Dependent Targets
The macrolide allosteric mTOR inhibitor, rapamycin, and its analogue, temsirolimus, have been tested in multiple neurodegenerative disease models. mTOR inhibitors were shown to induce autophagy, prevent dopaminergic loss, and revert motor, cognitive, and affective symptoms in several PD mouse models [289,290,291,292]. Moreover, rapamycin was able to improve pathology and disease phenotypes in AD, ALS, and HD models [237, 278, 333,334,335,336,337]. Other mTOR-dependent stimulators of autophagy, including GTM-1, carbamazepine, and latrepirdine, were shown to decrease Aβ accumulation and APP metabolites in AD cell models and also protect against memory dysfunction in AD patients [310,311,312,313].
mTOR-dependent stimulators have been most extensively tested in PD. The GSK3B inhibitor 6-Bio was neuroprotective in PD mouse model, and reduced αSyn aggregation in vitro [294]. The insulin sensitizer, MSDC-0160, prevented and restored dopaminergic loss and motor deficits in lesioned mice while additionally exerting an anti-inflammatory effect [293]. Similarly, pathways targeted by the anti-diabetic drug, exendin-4/exenatide are implicated in the preservation or restoration of dopaminergic neuron integrity in lesioned PD rodent models [338, 339], yet autophagy-related effects are unclear. Exenatide, now in phase I clinical trials for early stage PD, showed potential for sustainable motor score improvement for PD patients in a previous Phase II study [340]. Lastly, the c-Ab1 tyrosine kinase inhibitor and AMPK activator nilotinib, was shown to induce autophagy, enhance dopaminergic viability, and improve motor function in PD mouse models [341,342,343], yet mTOR involvement is unclear. The compound, which is currently in phase II clinical trials, showed potential to improve symptoms in both late-stage PD and Lewy body dementia patients [344].
mTOR-Independent Targets
Despite the promise of mTOR-dependent activation of autophagy, side effects associated with modulating mTOR have led to the emergence of mTOR-independent activators of autophagy as possible alternatives. Trehalose, a disaccharide, has been proposed to promote lysosomal clearance via inhibition of Akt and prevention of inhibitory TFEB phosphorylation [83]. The compound was shown to enhance clearance of mutant αSyn and Htt aggregates in cell lines, attenuate dopaminergic loss in PD mice, and protect mitochondrially challenged human iPSC-derived dopaminergic neurons [296,297,298, 345]. Moreover, trehalose delayed neurodegeneration while improving pathology and phenotypes in both HD and SOD1 ALS mouse models [323,324,325, 327].
Mood-stabilizing agents, such as lithium and valproate, also function as mTOR-independent autophagy activators through inositol depletion via inhibition of inositol monophosphatase and inositol 3-phosphate synthase, respectively [78]. These agents have been shown to enhance clearance of aggregate-prone htt and protect dopaminergic integrity in PD mouse models [76, 295], alleviate pathology and motor phenotypes in HD models [328, 329], and improve cognitive decline in AD patients with few adverse effects [314]. Resveratrol, another autophagy stimulator, was found to delay symptoms and pathology in SOD1 ALS mice [326], as well as decrease Aβ pathology in AD models in an AMPK-dependent manner [313, 314]. Although 1 study found that AD patients treated with resveratrol show an increase in plasma Aβ40 levels [317], another study demonstrated decreased levels of Aβ42 in the CSF with long-term treatment in patients [318]. Therefore, whether resveratrol is beneficial for AD patients in the long term remains to be determined. Calpain inhibitors such as calpastatin, Gabadur, and Neurodur have also been shown to be beneficial in PD and HD mouse models in an mTOR-independent manner [346, 347].
Other putative mTOR-independent autophagy inducers have been tested in specific disease models. AUTEN-99, which inhibits the myotubularin-related phosphatase MTMR14/Jumpy, a negative regulator of autophagic membrane formation, phenotypically rescues PD-associated outcomes in Drosophila models [299]. Memantine, an NMDA receptor agonist, displays efficacy in patients with moderate to severe AD [319, 320], though the compound’s effects on autophagy remain to be explored. Rilmenidine, an antihypertensive, attenuates HD pathology and accumulation of mhtt in a mouse model via an mTOR-independent mechanism [330]. Lastly, L-type Ca2+ channel antagonist verapamil, ATP-sensitive potassium channel opener minoxidil, and the G-signaling modulator clonidine, have been proposed to promote autophagy via modulation of IP3 levels, intracellular Ca2+, and calpain activity, and have been suggested as potential therapeutic agents in HD and other neurodegenerative diseases [77].
It is not currently known whether induction of autophagy can confer long-term improvements in neuropathology, delay disease progression, or ideally prevent neurodegeneration. Apart from its central role in cellular degradation and recycling, autophagy has emerging roles in DNA repair, inflammation, and protein secretion [348,349,350], suggesting that upstream targeting of this mechanism can have multiple effects which cannot be precisely predicted based on current knowledge. Though autophagy is an attractive target in neurodegeneration, the prolonged and systemic administration of upstream autophagy stimulators comes with the risks. For instance, although rapamycin shows positive outcomes in preclinical studies, higher doses of rapamycin inhibit mTORC2 thereby potentially compromising survival, whereas immunosuppression and other side effects have also been reported [351]. For several other compounds, the mode of action is not thoroughly characterized, and the effect on autophagy may not be specific or direct. In addition, a very recent study in C. elegans demonstrated that increasing autophagy can also be detrimental and compromise lifespan, depending on the status of the mitochondria [352]. This becomes a concern for the systemic induction of autophagy because different cell types or tissues exhibit baseline metabolic differences and could be differentially affected by aging and neurodegeneration. An additional consideration is the timing of intervention as well as the genetic background of patients. Indeed, enhancing autophagy at early disease stages may be effective in delaying the accumulation of disease-related proteins. On the contrary, increasing autophagosome formation at later disease stages or in patients whose lysosomal function is likely already compromised may exacerbate neurodegenerative pathology due to exceeding the cellular capacity for degradation. For these reasons, more targeted approaches could have more efficacious, powerful, and specific outcomes for patients.
Targeting Lysosomes in Neurodegeneration
More direct lysosome-targeted therapies have been proposed and tested in PD, AD, and HD (Table 1). Given the selective vulnerability of substantia nigra neurons in PD, direct gene targeting is a potentially relevant therapeutic strategy. Preclinical models suggest that overexpression of Beclin 1, Lamp2A, and TFEB could induce lysosomal function and confer neuroprotection [290, 353, 354]. Another approach to target lysosomes in PD involves the direct delivery of nanoparticles to enhance lysosomal acidification, which restored lysosomal activity in ATP13A2 and GBA1 PD models [300].
Decreased lysosomal hydrolase activity has been attributed to many neurodegenerative diseases. Thus, patients may also benefit from direct targeting and enhancement of these hydrolases. For example, AAV delivery of GRN to progranulin knockout mice significantly reversed lysosomal dysfunction associated with both FTD and NCL [355]. In addition, viral expression of GBA1 or reduction of GCase substrate levels via pharmacological glucosylceramide synthase inhibition reduced αSyn pathology and rescued dopaminergic integrity and cognitive function in rodents [308, 309, 356]. Venglustat, which inhibits glucosylceramide synthase, is currently in phase II clinical trials for early-stage GBA1-PD patients [307]. Reduction of glucosylceramide levels can also be achieved via small molecule non-inhibitory chaperones. NCGC758, which activates GCase specifically in the lysosomal compartment, preserved physiological αSyn conformation and increased the efficiency of trafficking and maturation of GCase in the endolysosomal compartment in PD iPSC-derived midbrain neurons [214, 305]. Similarly, NCGC607, also a small molecule non-inhibitory chaperone was found to reduce glucosphingolipids and αSyn levels in iPSC-derived dopaminergic neurons from Gaucher patients with parkinsonism [306]. Other compounds, such as the pharmacological chaperones ambroxol and isofagomine, have also been shown to increase GCase levels and activity and preserve αSyn homeostasis in PD/GBA1-PD and also wild-type cell and animal models [301,302,303,304]. Currently, ambroxol is in phase II clinical trials for GBA1-PD and PD with dementia patients [307]. Furthermore, the compound LTI-291, which stimulates GCase activity in the brain, is being assessed in phase II trials in GBA1-PD patients [307]. Unlike enzyme replacement therapies which would require invasive CNS delivery, pharmacological chaperones target specific lysosomal hydrolases, such as GCase, and could be orally administered and cross the blood–brain barrier. Examining the distribution of these compounds in disease-relevant CNS regions is important, to which end both wild-type and lysosomal enzyme-deficient animal models are necessary. Furthermore, GCase activation via pharmacological chaperones such as isofagomine binding to the active site of the enzyme, requires washout in order to achieve maximal lysosomal activity induction [357, 358]. The administration to patients should therefore be optimized so that the chaperone is rapidly removed from the lysosome, allowing the enzyme sufficient time to interact with its endogenous substrate. Future studies focusing on the efficacy of non-inhibitory or allosteric chaperones [305, 306, 359] could provide significant advancements, because the enzyme could interact with the endogenous substrate without the necessity of compound removal.
In AD, a class of interventions is focused on directly improving lysosomal function. Current clinical trials suggest that protein phosphatase 2A agonists, such as metformin, have beneficial effects by inhibiting hyperphosphorylation of tau [321, 322]. As phosphorylated tau constitutes a major component of NFTs in neurons [321, 322], metformin in conjunction with other treatments that reduce Aβ pathology may be useful for improving both pathological hallmarks of AD. Furthermore, TFEB transduction in the hippocampi of AD mouse models reduced AD-associated pathologies by upregulating lysosome biogenesis [360].
Because of growing evidence for htt regulation of autophagy and lysosomal function in HD, direct targeting of htt was proposed as a therapeutic approach in disease. The monogenetic etiology of HD makes it particularly attractive for gene therapies, specifically those targeting mhtt DNA and mRNA. DNA-targeting therapies, such as zinc finger proteins and CRIPSR-Cas9, to transduce cells with functional htt, have been tested in animal models. Zinc finger repressors reduced mhtt expression in the brains of R6/2 mice [331], and CRISPR-Cas9 gene editing ameliorated neurotoxicity in an HD mouse model [332]. Moreover, mRNA-targeting therapies against mhtt, such as antisense oligonucleotides (ASOs), nucleotide-based therapies that bind mRNA to trigger degradation, have already entered clinical trials [361]. The first of these, IONIS-HTTRx, was well-tolerated and resulted in dose-dependent reductions in mhtt levels in the CSF of early HD patients [362] and has now entered phase III clinical trials. The development of therapies directly targeting mhtt is advantageous in that it may not only mitigate lysosomal defects in disease but improve other aspects of disease pathophysiology and symptomology as well.
Overall, lysosomal function is a promising and specific target in neurodegeneration, with the possibility to benefit patients of various genetic backgrounds at early or even later stages of disease. Further elucidation of lysosomal functions and regulation of lysosome-related pathways will be instrumental to shaping the design and specificity of future therapeutic interventions. In this endeavor, a significant step forward has been made by the development of iPSC and CRISPR/Cas9 technologies, which in combination have improved our ability to assess the efficacy of therapies targeting lysosomal function in patient-derived neurons in both sporadic and familial disease. Nevertheless, a major challenge of the future will be the identification of sensitive and specific biomarkers for both diagnosing neurodegenerative diseases early and evaluating patient responses to therapies, which may include assessment of lysosomal efficiency in disease-relevant regions of the CNS.
Conclusion
Lysosomal function in the aging brain is an area that is just beginning to be explored. The lysosome is cardinal to various cellular functions including autophagy, nutrient and ion homeostasis, and inter-organelle interactions. As our understanding of the role of lysosomes in aging continues to take shape, it will be necessary to identify how various lysosomal nutrient storage, sensing, and processing pathways are engaged in brain-specific cell types in order to facilitate the discovery of therapeutic targets. For now, the best insight into lysosomal alterations with age will come from the detailed investigation of pathophysiological mechanisms in age-related neurodegenerative diseases that often present with hallmarks of lysosomal and autophagic dysfunction. Several therapeutic strategies to combat these diseases have already emerged and entered into clinical trials, including those promoting the upregulation of autophagy and those directly aimed at rescuing lysosomal function. The promise of these treatments in neurodegeneration beckons the question of whether these therapies will be similarly beneficial in preserving lysosomal function in the aging brain.
Change history
24 July 2019
This article was corrected to include a revised version of figure 2 overlooked by the Publisher during the production process.
References
Lim CY, Zoncu R. The lysosome as a command-and-control center for cellular metabolism. J Cell Biol 2016;214(6):653–664.
Carmona-Gutierrez D, Hughes AL, Madeo F, Ruckenstuhl C. The crucial impact of lysosomes in aging and longevity. Ageing Res Rev 2016;32:2–12.
Wang C, Telpoukhovskaia MA, Bahr BA, Chen X, Gan L. Endo-lysosomal dysfunction: a converging mechanism in neurodegenerative diseases. Curr Opin Neurobiol 2018;48:52–58.
Sekito T, Fujiki Y, Ohsumi Y, Kakinuma Y. Novel families of vacuolar amino acid transporters. IUBMB Life 2008;60(8):519–525.
Russnak R, Konczal D, McIntire SL. A family of yeast proteins mediating bidirectional vacuolar amino acid transport. J Biol Chem 2001;276(26):23849–23857.
Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 2007;8(11):917–929.
Chapel A, Kieffer-Jaquinod S, Sagne C, Verdon Q, Ivaldi C, Mellal M, et al. An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol Cell Proteomics 2013;12(6):1572–1588.
Colacurcio DJ, Nixon RA. Disorders of lysosomal acidification—the emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res Rev 2016;32:75–88.
Stransky LA, Forgac M. Amino acid availability modulates vacuolar H+-ATPase assembly. J Biol Chem 2015;290(45):27360–27369.
Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med 2012;18(9):524–533.
Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008;320(5882):1496–1501.
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141(2):290–303.
Jung J, Genau HM, Behrends C. Amino acid-dependent mTORC1 regulation by the lysosomal membrane protein SLC38A9. Mol Cell Biol 2015;35(14):2479–2494.
Rebsamen M, Pochini L, Stasyk T, de Araujo ME, Galluccio M, Kandasamy RK, et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature. 2015;519(7544):477–481.
Wyant GA, Abu-Remaileh M, Wolfson RL, Chen WW, Freinkman E, Danai LV, et al. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell. 2017;171(3):642–54.e12.
Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, et al. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017;358(6364):807–813.
Hughes AL, Gottschling DE. An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature. 2012;492(7428):261–265.
Ruckenstuhl C, Netzberger C, Entfellner I, Carmona-Gutierrez D, Kickenweiz T, Stekovic S, et al. Lifespan extension by methionine restriction requires autophagy-dependent vacuolar acidification. PLoS Genet 2014;10(5):e1004347.
Raffaello A, Mammucari C, Gherardi G, Rizzuto R. Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem Sci 2016;41(12):1035–1049.
Xu H, Ren D. Lysosomal physiology. Annu Rev Physiol 2015;77:57–80.
Tsubakiyama R, Mizunuma M, Gengyo A, Yamamoto J, Kume K, Miyakawa T, et al. Implication of Ca2+ in the regulation of replicative life span of budding yeast. J Biol Chem 2011;286(33):28681–28687.
Ohya Y, Umemoto N, Tanida I, Ohta A, Iida H, Anraku Y. Calcium-sensitive cls mutants of Saccharomyces cerevisiae showing a Pet- phenotype are ascribable to defects of vacuolar membrane H(+)-ATPase activity. J Biol Chem 1991;266(21):13971–13977.
Kilpatrick BS, Yates E, Grimm C, Schapira AH, Patel S. Endo-lysosomal TRP mucolipin-1 channels trigger global ER Ca2+ release and Ca2+ influx. J Cell Sci 2016;129(20):3859–3867.
Garrity AG, Wang W, Collier CM, Levey SA, Gao Q, Xu H. The endoplasmic reticulum, not the pH gradient, drives calcium refilling of lysosomes. eLife. 2016;5.
Lee JH, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, et al. Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep 2015;12(9):1430–1444.
Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta 2016;1863(12):2977–2992.
Nikoletopoulou V, Tavernarakis N. Calcium homeostasis in aging neurons. Front Genet 2012;3:200.
Michaelis ML, Bigelow DJ, Schoneich C, Williams TD, Ramonda L, Yin D, et al. Decreased plasma membrane calcium transport activity in aging brain. Life Sci 1996;59(5–6):405–412.
Xiong J, Verkhratsky A, Toescu EC. Changes in mitochondrial status associated with altered Ca2+ homeostasis in aged cerebellar granule neurons in brain slices. J Neurosci 2002;22(24):10761–10771.
Kumar A, Foster TC. Enhanced long-term potentiation during aging is masked by processes involving intracellular calcium stores. J Neurophysiol 2004;91(6):2437–2444.
Gant JC, Sama MM, Landfield PW, Thibault O. Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J Neurosci 2006;26(13):3482–3490.
Sheftel AD, Zhang AS, Brown C, Shirihai OS, Ponka P. Direct interorganellar transfer of iron from endosome to mitochondrion. Blood 2007;110(1):125–132.
Ashraf A, Clark M, So PW. The Aging of Iron Man. Front Aging Neurosci 2018;10:65.
Todkar K, Ilamathi HS, Germain M. Mitochondria and lysosomes: discovering bonds. Front Cell Dev Biol 2017;5:106.
Gammella E, Recalcati S, Cairo G. Dual role of ROS as signal and stress agents: iron tips the balance in favor of toxic effects. Oxidative Med Cell Longev 2016;2016:8629024.
Klang IM, Schilling B, Sorensen DJ, Sahu AK, Kapahi P, Andersen JK, et al. Iron promotes protein insolubility and aging in C. elegans. Aging (Albany NY). 2014;6(11):975–991.
Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol 2014;13(10):1045–1060.
Connor JR, Snyder BS, Arosio P, Loeffler DA, LeWitt P. A quantitative analysis of isoferritins in select regions of aged, parkinsonian, and Alzheimer’s diseased brains. J Neurochem 1995;65(2):717–724.
Zecca L, Bellei C, Costi P, Albertini A, Monzani E, Casella L, et al. New melanic pigments in the human brain that accumulate in aging and block environmental toxic metals. Proc Natl Acad Sci U S A 2008;105(45):17567–17572.
Zecca L, Casella L, Albertini A, Bellei C, Zucca FA, Engelen M, et al. Neuromelanin can protect against iron-mediated oxidative damage in system modeling iron overload of brain aging and Parkinson’s disease. J Neurochem 2008;106(4):1866–1875.
Zucca FA, Vanna R, Cupaioli FA, Bellei C, De Palma A, Di Silvestre D, et al. Neuromelanin organelles are specialized autolysosomes that accumulate undegraded proteins and lipids in aging human brain and are likely involved in Parkinson’s disease. NPJ Parkinsons Dis 2018;4:17.
Schneider SA, Dusek P, Hardy J, Westenberger A, Jankovic J, Bhatia KP. Genetics and pathophysiology of neurodegeneration with brain iron accumulation (NBIA). Curr Neuropharmacol 2013;11(1):59–79.
Seibler P, Burbulla LF, Dulovic M, Zittel S, Heine J, Schmidt T, et al. Iron overload is accompanied by mitochondrial and lysosomal dysfunction in WDR45 mutant cells. Brain 2018;141(10):3052–3064.
Mony VK, Benjamin S, O'Rourke EJ. A lysosome-centered view of nutrient homeostasis. Autophagy 2016;12(4):619–631.
Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med 2015;66:471–486.
Mc Donald JM, Krainc D. Lysosomal proteins as a therapeutic target in neurodegeneration. Annu Rev Med 2017;68:445–458.
Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007;9(10):1102–1109.
Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol 2010;12(9):831–835.
Mari M, Tooze SA, Reggiori F. The puzzling origin of the autophagosomal membrane. F1000 Biol Rep. 2011;3:25.
Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009;284(18):12297–12305.
Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 2012;23(10):1860–1873.
Zeng X, Overmeyer JH, Maltese WA. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci 2006;119(Pt 2):259–270.
Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex—at the crossroads of autophagy and beyond. Trends Cell Biol 2010;20(6):355–362.
Willinger T, Flavell RA. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc Natl Acad Sci U S A 2012;109(22):8670–8675.
Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013;15(7):741–750.
Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 2008;19(12):5360–5372.
Itakura E, Mizushima N. Atg14 and UVRAG: mutually exclusive subunits of mammalian Beclin 1-PI3K complexes. Autophagy 2009;5(4):534–536.
Munson MJ, Allen GF, Toth R, Campbell DG, Lucocq JM, Ganley IG. mTOR activates the VPS34-UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J 2015;34(17):2272–2290.
Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 2007;282(52):37298–37302.
Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 2008;19(5):2092–2100.
Otomo C, Metlagel Z, Takaesu G, Otomo T. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol 2013;20(1):59–66.
Xie Z, Nair U, Klionsky DJ. Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell 2008;19(8):3290–3298.
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000;19(21):5720–5728.
Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004;117(Pt 13):2805–2812.
Sou YS, Tanida I, Komatsu M, Ueno T, Kominami E. Phosphatidylserine in addition to phosphatidylethanolamine is an in vitro target of the mammalian Atg8 modifiers, LC3, GABARAP, and GATE-16. J Biol Chem 2006;281(6):3017–3024.
Noda NN, Kumeta H, Nakatogawa H, Satoo K, Adachi W, Ishii J, et al. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells 2008;13(12):1211–1218.
Rogov V, Dotsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell 2014;53(2):167–178.
Kriegenburg F, Ungermann C, Reggiori F. Coordination of autophagosome-lysosome fusion by Atg8 family members. Curr Biol 2018;28(8):R512-R518.
Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334(6056):678–683.
Lawrence RE, Cho KF, Rappold R, Thrun A, Tofaute M, Kim DJ, et al. A nutrient-induced affinity switch controls mTORC1 activation by its Rag GTPase-Ragulator lysosomal scaffold. Nat Cell Biol 2018;20(9):1052–1063.
Leidal AM, Levine B, Debnath J. Autophagy and the cell biology of age-related disease. Nat Cell Biol 2018;20(12):1338–1348.
Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473–477.
Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011;332(6036):1429–1433.
Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5(228):ra42.
Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci 2016;129(13):2475–2481.
Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005;170(7):1101.
Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, et al. Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat Chem Biol 2008;4(5):295–305.
Ju S, Greenberg ML. 1D-myo-inositol 3-phosphate synthase: conservation, regulation, and putative target of mood stabilizers. Clin Neurosci Res 2004;4(3):181–187.
Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ 2007;14:1029.
Vicencio JM, Ortiz C, Criollo A, Jones AWE, Kepp O, Galluzzi L, et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ 2009;16:1006.
Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006;8:1124.
Russo R, Berliocchi L, Adornetto A, Varano GP, Cavaliere F, Nucci C, et al. Calpain-mediated cleavage of Beclin-1 and autophagy deregulation following retinal ischemic injury in vivo. Cell Death Dis. 2011;2(4):e144-e.
Palmieri M, Pal R, Nelvagal HR, Lotfi P, Stinnett GR, Seymour ML, et al. Corrigendum: mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat Commun 2017;8:15793.
Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 2015;17(3):288–299.
Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005;8(1):3–5.
Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008;183(5):795–803.
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314.
Pickles S, Vigie P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 2018;28(4):R170-R185.
Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J 2014;33(19):2142–2156.
Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res 2014;24(7):787–795.
Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 2012;14(2):177–185.
Lv M, Wang C, Li F, Peng J, Wen B, Gong Q, et al. Structural insights into the recognition of phosphorylated FUNDC1 by LC3B in mitophagy. Protein & Cell 2017;8(1):25–38.
Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 2013;15(10):1197–1205.
Allen GF, Toth R, James J, Ganley IG. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep 2013;14(12):1127–1135.
Dagda RK, Cherra SJ, 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 2009;284(20):13843–13855.
Burman JL, Pickles S, Wang C, Sekine S, Vargas JNS, Zhang Z, et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J Cell Biol 2017;216(10):3231–3247.
Chu CT. Mechanisms of selective autophagy and mitophagy: implications for neurodegenerative diseases. Neurobiol Dis 2019;122:23–34.
Martin-Maestro P, Gargini R, Sproul AA, Garcia E, Anton LC, Noggle S, et al. Mitophagy failure in fibroblasts and iPSC-derived neurons of Alzheimer’s disease-associated presenilin 1 mutation. Front Mol Neurosci. 2017;10:291.
Dimmer KS, Fritz S, Fuchs F, Messerschmitt M, Weinbach N, Neupert W, et al. Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiae. Mol Biol Cell 2002;13(3):847–853.
Merz S, Westermann B. Genome-wide deletion mutant analysis reveals genes required for respiratory growth, mitochondrial genome maintenance and mitochondrial protein synthesis in Saccharomyces cerevisiae. Genome Biol 2009;10(9):R95.
Elbaz-Alon Y, Rosenfeld-Gur E, Shinder V, Futerman AH, Geiger T, Schuldiner M. A dynamic interface between vacuoles and mitochondria in yeast. Dev Cell 2014;30(1):95–102.
Honscher C, Mari M, Auffarth K, Bohnert M, Griffith J, Geerts W, et al. Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev Cell 2014;30(1):86–94.
Wong YC, Ysselstein D, Krainc D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature. 2018;554(7692):382–386.
Wong YC, Kim S, Peng W, Krainc D. Regulation and function of mitochondria-lysosome membrane contact sites in cellular homeostasis. Trends Cell Biol 2019.
Klionsky DJ, Herman PK, Emr SD. The fungal vacuole: composition, function, and biogenesis. Microbiol Rev 1990;54(3):266–292.
Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 2009;9(4):311–326.
Wellen KE, Thompson CB. Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell 2010;40(2):323–332.
Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun 2016;7:12109.
Marino ML, Fais S, Djavaheri-Mergny M, Villa A, Meschini S, Lozupone F, et al. Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. Cell Death Dis 2010;1:e87.
Demers-Lamarche J, Guillebaud G, Tlili M, Todkar K, Belanger N, Grondin M, et al. Loss of mitochondrial function impairs lysosomes. J Biol Chem 2016;291(19):10263–10276.
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885.
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J-i, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006;441:880.
Rubinsztein David C, Mariño G, Kroemer G. Autophagy and aging. Cell 2011;146(5):682–695.
Leeman DS, Hebestreit K, Ruetz T, Webb AE, McKay A, Pollina EA, et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 2018;359(6381):1277–1283.
Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(8).
McDermott JR, Gibson AM. Degradation of Alzheimer’s beta-amyloid protein by human cathepsin D. Neuroreport. 1996;7(13):2163–2166.
Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, et al. Abeta secretion and plaque formation depend on autophagy. Cell Rep 2013;5(1):61–69.
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 2005;64(2):113–122.
Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, et al. Macroautophagy—a novel beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol 2005;171(1):87–98.
Yu JT, Ma XY, Wang YL, Sun L, Tan L, Hu N, et al. Genetic variation in clusterin gene and Alzheimer’s disease risk in Han Chinese. Neurobiol Aging. 2013;34(7):1921 e17–23.
Zhang F, Kumano M, Beraldi E, Fazli L, Du C, Moore S, et al. Clusterin facilitates stressinduced lipidation of LC3 and autophagosome biogenesis to enhance cancer cell survival. Nat Commun 2014;5:5775.
Miners JS, Clarke P, Love S. Clusterin levels are increased in Alzheimer’s disease and influence the regional distribution of Abeta. Brain Pathol 2017;27(3):305–313.
Beeg M, Stravalaci M, Romeo M, Carra AD, Cagnotto A, Rossi A, et al. Clusterin binds to Abeta1-42 oligomers with high affinity and interferes with peptide aggregation by inhibiting primary and secondary nucleation. J Biol Chem 2016;291(13):6958–6966.
Zhou Y, Hayashi I, Wong J, Tugusheva K, Renger JJ, Zerbinatti C. Intracellular clusterin interacts with brain isoforms of the bridging integrator 1 and with the microtubule-associated protein Tau in Alzheimer’s disease. PLoS One 2014;9(7):e103187.
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141(7):1146–1158.
Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, Nixon RA. Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur J Neurosci 2013;37(12):1949–1961.
Murphy MP, Uljon SN, Fraser PE, Fauq A, Lookingbill HA, Findlay KA, et al. Presenilin 1 regulates pharmacologically distinct gamma-secretase activities. Implications for the role of presenilin in gamma-secretase cleavage. J Biol Chem 2000;275(34):26277–26284.
Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, et al. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron 2015;85(5):967–981.
Coffey EE, Beckel JM, Laties AM, Mitchell CH. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 2014;263:111–124.
Portelius E, Andreasson U, Ringman JM, Buerger K, Daborg J, Buchhave P, et al. Distinct cerebrospinal fluid amyloid beta peptide signatures in sporadic and PSEN1 A431E-associated familial Alzheimer’s disease. Mol Neurodegener 2010;5:2.
Nakatogawa H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem 2013;55:39–50.
Nilsson P, Sekiguchi M, Akagi T, Izumi S, Komori T, Hui K, et al. Autophagy-related protein 7 deficiency in amyloid beta (Abeta) precursor protein transgenic mice decreases Abeta in the multivesicular bodies and induces Abeta accumulation in the Golgi. Am J Pathol 2015;185(2):305–313.
Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, et al. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A 2006;103(30):11172–11177.
Inoue K, Rispoli J, Kaphzan H, Klann E, Chen EI, Kim J, et al. Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol Neurodegener 2012;7:48.
Paradis E, Douillard H, Koutroumanis M, Goodyer C, LeBlanc A. Amyloid beta peptide of Alzheimer’s disease downregulates Bcl-2 and upregulates bax expression in human neurons. J Neurosci 1996;16(23):7533–7539.
Karlnoski R, Wilcock D, Dickey C, Ronan V, Gordon MN, Zhang W, et al. Up-regulation of Bcl-2 in APP transgenic mice is associated with neuroprotection. Neurobiol Dis 2007;25(1):179–188.
Rohn TT, Vyas V, Hernandez-Estrada T, Nichol KE, Christie LA, Head E. Lack of pathology in a triple transgenic mouse model of Alzheimer’s disease after overexpression of the anti-apoptotic protein Bcl-2. J Neurosci 2008;28(12):3051–3059.
Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011;18(4):571–580.
Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 2008;118(6):2190–2199.
Neely KM, Green KN, LaFerla FM. Presenilin is necessary for efficient proteolysis through the autophagy-lysosome system in a gamma-secretase-independent manner. J Neurosci 2011;31(8):2781–2791.
Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S, et al. Autophagy flux in CA1 neurons of Alzheimer hippocampus: increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016;12(12):2467–2483.
Albayrak O, Tirniceriu A, Riemenschneider M, Kurz A, Scherag A, Egensperger R. The cathepsin D (224C/T) polymorphism confers an increased risk to develop Alzheimer’s disease in men. J Gerontol A Biol Sci Med Sci 2010;65(3):219–224.
Mariani E, Seripa D, Ingegni T, Nocentini G, Mangialasche F, Ercolani S, et al. Interaction of CTSD and A2M polymorphisms in the risk for Alzheimer’s disease. J Neurol Sci 2006;247(2):187–191.
Urbanelli L, Emiliani C, Massini C, Persichetti E, Orlacchio A, Pelicci G, et al. Cathepsin D expression is decreased in Alzheimer’s disease fibroblasts. Neurobiol Aging 2008;29(1):12–22.
Tian L, Zhang K, Tian ZY, Wang T, Shang DS, Li B, et al. Decreased expression of cathepsin D in monocytes is related to the defective degradation of amyloid-beta in Alzheimer’s disease. J Alzheimers Dis 2014;42(2):511–520.
Straface E, Matarrese P, Gambardella L, Vona R, Sgadari A, Silveri MC, et al. Oxidative imbalance and cathepsin D changes as peripheral blood biomarkers of Alzheimer disease: a pilot study. FEBS Lett 2005;579(13):2759–2766.
Adamec E, Mohan PS, Cataldo AM, Vonsattel JP, Nixon RA. Up-regulation of the lysosomal system in experimental models of neuronal injury: implications for Alzheimer’s disease. Neuroscience 2000;100(3):663–675.
Schwagerl AL, Mohan PS, Cataldo AM, Vonsattel JP, Kowall NW, Nixon RA. Elevated levels of the endosomal-lysosomal proteinase cathepsin D in cerebrospinal fluid in Alzheimer disease. J Neurochem 1995;64(1):443–446.
Nixon RA. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: inseparable partners in a multifactorial disease. FASEB Journal: official publication of the Federation of American Societies for Experimental Biology 2017;31(7):2729–2743.
Kim S, Sato Y, Mohan PS, Peterhoff C, Pensalfini A, Rigoglioso A, et al. Evidence that the rab5 effector APPL1 mediates APP-betaCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease. Mol Psychiatry 2016;21(5):707–716.
Lauritzen I, Pardossi-Piquard R, Bauer C, Brigham E, Abraham JD, Ranaldi S, et al. The beta-secretase-derived C-terminal fragment of betaAPP, C99, but not Abeta, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J Neurosci. 2012;32(46):16243-1655a.
Lauritzen I, Pardossi-Piquard R, Bourgeois A, Pagnotta S, Biferi MG, Barkats M, et al. Intraneuronal aggregation of the beta-CTF fragment of APP (C99) induces Abeta-independent lysosomal-autophagic pathology. Acta Neuropathol 2016;132(2):257–276.
Tamayev R, Matsuda S, Arancio O, D'Adamio L. beta- but not gamma-secretase proteolysis of APP causes synaptic and memory deficits in a mouse model of dementia. EMBO Mol Med 2012;4(3):171–179.
Hung COY, Livesey FJ. Altered gamma-secretase processing of APP disrupts lysosome and autophagosome function in monogenic Alzheimer’s disease. Cell Rep. 2018;25(13):3647–60.e2.
He X, Cooley K, Chung CH, Dashti N, Tang J. Apolipoprotein receptor 2 and X11 alpha/beta mediate apolipoprotein E-induced endocytosis of amyloid-beta precursor protein and beta-secretase, leading to amyloid-beta production. J Neurosci 2007;27(15):4052–4060.
Heeren J, Grewal T, Laatsch A, Becker N, Rinninger F, Rye KA, et al. Impaired recycling of apolipoprotein E4 is associated with intracellular cholesterol accumulation. J Biol Chem 2004;279(53):55483–55492.
Rellin L, Heeren J, Beisiegel U. Recycling of apolipoprotein E is not associated with cholesterol efflux in neuronal cells. Biochim Biophys Acta 2008;1781(5):232–238.
Ji ZS, Miranda RD, Newhouse YM, Weisgraber KH, Huang Y, Mahley RW. Apolipoprotein E4 potentiates amyloid beta peptide-induced lysosomal leakage and apoptosis in neuronal cells. J Biol Chem 2002;277(24):21821–21828.
Ji ZS, Mullendorff K, Cheng IH, Miranda RD, Huang Y, Mahley RW. Reactivity of apolipoprotein E4 and amyloid beta peptide: lysosomal stability and neurodegeneration. J Biol Chem 2006;281(5):2683–2692.
Nuriel T, Peng KY, Ashok A, Dillman AA, Figueroa HY, Apuzzo J, et al. The endosomal-lysosomal pathway is dysregulated by APOE4 expression in vivo. Front Neurosci 2017;11:702.
Dorsey ER, Elbaz A, Nichols E, Abd-Allah F, Abdelalim A, Adsuar JC, et al. Global, regional, and national burden of Parkinson’s disease, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 2018;17(11):939–953.
Anglade P, Vyas S, Hirsch EC, Agid Y. Apoptosis in dopaminergic neurons of the human substantia nigra during normal aging. Histol Histopathol 1997;12(3):603–610.
Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper J, et al. Chaperone-mediated autophagy markers in parkinson disease brains. Arch Neurol 2010;67(12):1464–1472.
Shahmoradian SH, Lewis AJ, Genoud C, Graff-Meyer A, Hench J, Moors T, et al. Lewy pathology in Parkinson’s disease consists of a crowded organellar, membranous medley. bioRxiv. 2018:137976.
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276(5321):2045–2047.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alphasynuclein in Lewy bodies. Nature 1997;388(6645):839–840.
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alphaSynuclein locus triplication causes Parkinson’s disease. Science. 2003;302(5646):841.
Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, et al. Alphasynuclein locus duplication as a cause of familial Parkinson’s disease. Lancet (London, England). 2004;364(9440):1167–9.
Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006;313(5785):324–328.
Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, et al. alpha-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J Cell Biol 2010;190(6):1023–1037.
Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D. alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci U S A 2016;113(7):1931–1936.
Hernandez DG, Reed X, Singleton AB. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J Neurochem 2016;139 Suppl 1:59–74.
Lazaro DF, Rodrigues EF, Langohr R, Shahpasandzadeh H, Ribeiro T, Guerreiro P, et al. Systematic comparison of the effects of alpha-synuclein mutations on its oligomerization and aggregation. PLoS Genet 2014;10(11):e1004741.
Tanik SA, Schultheiss CE, Volpicelli-Daley LA, Brunden KR, Lee VM. Lewy body-like alpha-synuclein aggregates resist degradation and impair macroautophagy. J Biol Chem 2013;288(21):15194–15210.
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004;305(5688):1292–1295.
Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, et al. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis 2011;42(3):360–367.
Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, et al. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 2010;30(20):6838–6851.
Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, et al. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener 2012;7:42.
Ejlerskov P, Rasmussen I, Nielsen TT, Bergstrom AL, Tohyama Y, Jensen PH, et al. Tubulin polymerization-promoting protein (TPPP/p25alpha) promotes unconventional secretion of alphasynuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem 2013;288(24):17313–17335.
Lee H-J, Cho E-D, Lee KW, Kim J-H, Cho S-G, Lee S-J. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp Mol Med 2013;45:e22.
Poehler AM, Xiang W, Spitzer P, May VE, Meixner H, Rockenstein E, et al. Autophagy modulates SNCA/alpha-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014;10(12):2171–2192.
Fussi N, Höllerhage M, Chakroun T, Nykänen N-P, Rösler TW, Koeglsperger T, et al. Exosomal secretion of α-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis 2018;9(7):757.
Minakaki G, Menges S, Kittel A, Emmanouilidou E, Schaeffner I, Barkovits K, et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 2018;14(1):98–119.
Alessi DR, Sammler E. LRRK2 kinase in Parkinson’s disease. Science 2018;360(6384):36.
Alegre-Abarrategui J, Christian H, Lufino MM, Mutihac R, Venda LL, Ansorge O, et al. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet 2009;18(21):4022–4034.
Eguchi T, Kuwahara T, Sakurai M, Komori T, Fujimoto T, Ito G, et al. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc Natl Acad Sci 2018;115(39):E9115.
Steger M, Diez F, Dhekne HS, Lis P, Nirujogi RS, Karayel O, et al. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. eLife 2017;6.
Bravo-San Pedro JM, Niso-Santano M, Gomez-Sanchez R, Pizarro-Estrella E, Aiastui-Pujana A, Gorostidi A, et al. The LRRK2 G2019S mutant exacerbates basal autophagy through activation of the MEK/ERK pathway. Cell Mol Life Sci 2013;70(1):121–136.
Manzoni C, Mamais A, Dihanich S, McGoldrick P, Devine MJ, Zerle J, et al. Pathogenic Parkinson’s disease mutations across the functional domains of LRRK2 alter the autophagic/lysosomal response to starvation. Biochem Biophys Res Commun 2013;441(4):862–866.
Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 2011;8(3):267–280.
Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, Jimenez-Delgado S, Caig C, Mora S, et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med 2012;4(5):380–395.
Follett J, Norwood SJ, Hamilton NA, Mohan M, Kovtun O, Tay S, et al. The Vps35 D620N mutation linked to Parkinson’s disease disrupts the cargo sorting function of retromer. Traffic (Copenhagen, Denmark). 2014;15(2):230–244.
Zavodszky E, Seaman MN, Moreau K, Jimenez-Sanchez M, Breusegem SY, Harbour ME, et al. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat Commun 2014;5:3828.
Tang FL, Erion JR, Tian Y, Liu W, Yin DM, Ye J, et al. VPS35 in dopamine neurons is required for endosome-to-golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for alpha-synuclein degradation and prevention of pathogenesis of Parkinson’s disease. J Neurosci 2015;35(29):10613–10628.
Tang FL, Liu W, Hu JX, Erion JR, Ye J, Mei L, et al. VPS35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep 2015;12(10):1631–1643.
Song P, Trajkovic K, Tsunemi T, Krainc D. Parkin modulates endosomal organization and function of the endo-lysosomal pathway. J Neurosci 2016;36(8):2425–2437.
Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003;299(5604):256.
Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010;468(7324):696–700.
Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017;357(6357):1255–1261.
Lees AJ, Singleton AB. Clinical heterogeneity of ATP13A2 linked disease (Kufor-Rakeb) justifies a PARK designation. Neurology 2007;68(19):1553–1554.
Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, et al. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A 2012;109(24):9611–9616.
Grunewald A, Arns B, Seibler P, Rakovic A, Munchau A, Ramirez A, et al. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol Aging. 2012;33(8):1843.e1–7.
Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci 2012;32(12):4240–4246.
Schultheis PJ, Fleming SM, Clippinger AK, Lewis J, Tsunemi T, Giasson B, et al. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and agedependent sensorimotor deficits. Hum Mol Genet 2013;22(10):2067–2082.
Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet 2009;41(3):308–315.
Kong SM, Chan BK, Park JS, Hill KJ, Aitken JB, Cottle L, et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alpha-Synuclein externalization via exosomes. Hum Mol Genet 2014;23(11):2816–2833.
Tsunemi T, Krainc D. Zn(2)(+) dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum Mol Genet 2014;23(11):2791–2801.
Narayanaswamy N, Chakraborty K, Saminathan A, Zeichner E, Leung K, Devany J, et al. A pH-correctable, DNA-based fluorescent reporter for organellar calcium. Nat Methods 2019;16(1):95–102.
Bultron G, Kacena K, Pearson D, Boxer M, Yang R, Sathe S, et al. The risk of Parkinson’s disease in type 1 Gaucher disease. J Inherit Metab Dis 2010;33(2):167–173.
Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, et al. A metaanalysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 2017;49(10):1511–1516.
Schondorf DC, Aureli M, McAllister FE, Hindley CJ, Mayer F, Schmid B, et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun 2014;5:4028.
Fernandes HJ, Hartfield EM, Christian HC, Emmanoulidou E, Zheng Y, Booth H, et al. ER stress and autophagic perturbations lead to elevated extracellular alpha-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Reports 2016;6(3):342–356.
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146(1):37–52.
Zunke F, Moise AC, Belur NR, Gelyana E, Stojkovska I, Dzaferbegovic H, et al. Reversible conformational conversion of alpha-synuclein into toxic assemblies by glucosylceramide. Neuron. 2018;97(1):92–107.e10.
Gan-Or Z, Ozelius LJ, Bar-Shira A, Saunders-Pullman R, Mirelman A, Kornreich R, et al. The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 2013;80(17):1606–1610.
Dagan E, Schlesinger I, Ayoub M, Mory A, Nassar M, Kurolap A, et al. The contribution of Niemann-Pick SMPD1 mutations to Parkinson disease in Ashkenazi Jews. Parkinsonism Relat Disord 2015;21(9):1067–1071.
Deng S, Deng X, Song Z, Xiu X, Guo Y, Xiao J, et al. Systematic genetic analysis of the SMPD1 gene in Chinese patients with Parkinson’s disease. Mol Neurobiol 2016;53(7):5025–5029.
Hopfner F, Schulte EC, Mollenhauer B, Bereznai B, Knauf F, Lichtner P, et al. The role of SCARB2 as susceptibility factor in Parkinson’s disease. Mov Disord 2013;28(4):538–540.
Blanz J, Groth J, Zachos C, Wehling C, Saftig P, Schwake M. Disease-causing mutations within the lysosomal integral membrane protein type 2 (LIMP-2) reveal the nature of binding to its ligand β-glucocerebrosidase. Hum Mol Genet 2010;19(4):563–572.
Rothaug M, Zunke F, Mazzulli JR, Schweizer M, Altmeppen H, Lullmann-Rauch R, et al. LIMP-2 expression is critical for beta-glucocerebrosidase activity and alpha-synuclein clearance. Proc Natl Acad Sci U S A 2014;111(43):15573–15578.
Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci 2010;30(2):639–649.
van Eersel J, Ke YD, Gladbach A, Bi M, Gotz J, Kril JJ, et al. Cytoplasmic accumulation and aggregation of TDP-43 upon proteasome inhibition in cultured neurons. PLoS One 2011;6(7):e22850.
Kabuta T, Suzuki Y, Wada K. Degradation of amyotrophic lateral sclerosis-linked mutant Cu,Zn-superoxide dismutase proteins by macroautophagy and the proteasome. J Biol Chem 2006;281(41):30524–30533.
Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 2011;68(11):1440–1446.
Teyssou E, Takeda T, Lebon V, Boillee S, Doukoure B, Bataillon G, et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol 2013;125(4):511–522.
Peng H, Yang J, Li G, You Q, Han W, Li T, et al. Ubiquitylation of p62/sequestosome1 activates its autophagy receptor function and controls selective autophagy upon ubiquitin stress. Cell Res 2017;27(5):657–674.
Gal J, Strom AL, Kilty R, Zhang F, Zhu H. p62 accumulates and enhances aggregate formation in model systems of familial amyotrophic lateral sclerosis. J Biol Chem 2007;282(15):11068–11077.
Gal J, Strom AL, Kwinter DM, Kilty R, Zhang J, Shi P, et al. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J Neurochem 2009;111(4):1062–1073.
Bandyopadhyay U, Nagy M, Fenton WA, Horwich AL. Absence of lipofuscin in motor neurons of SOD1-linked ALS mice. Proc Natl Acad Sci U S A 2014;111(30):11055–11060.
Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011;13(2):132–141.
Chen Y, Liu H, Guan Y, Wang Q, Zhou F, Jie L, et al. The altered autophagy mediated by TFEB in animal and cell models of amyotrophic lateral sclerosis. Am J Transl Res 2015;7(9):1574–1587.
Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–438.
Ito D, Suzuki N. Conjoint pathologic cascades mediated by ALS/FTLD-U linked RNAbinding proteins TDP-43 and FUS. Neurology. 2011;77(17):1636–1643.
Wang X, Fan H, Ying Z, Li B, Wang H, Wang G. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci Lett 2010;469(1):112–116.
Ryu HH, Jun MH, Min KJ, Jang DJ, Lee YS, Kim HK, et al. Autophagy regulates amyotrophic lateral sclerosis-linked fused in sarcoma-positive stress granules in neurons. Neurobiol Aging 2014;35(12):2822–2831.
Brady OA, Meng P, Zheng Y, Mao Y, Hu F. Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J Neurochem 2011;116(2):248–259.
Wang IF, Guo BS, Liu YC, Wu CC, Yang CH, Tsai KJ, et al. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci U S A 2012;109(37):15024–15029.
Barmada SJ, Serio A, Arjun A, Bilican B, Daub A, Ando DM, et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nat Chem Biol 2014;10(8):677–685.
Xia Q, Wang H, Hao Z, Fu C, Hu Q, Gao F, et al. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J 2016;35(2):121–142.
Bose JK, Huang CC, Shen CK. Regulation of autophagy by neuropathological protein TDP-43. J Biol Chem 2011;286(52):44441–44448.
Deng Z, Purtell K, Lachance V, Wold MS, Chen S, Yue Z. Autophagy receptors and neurodegenerative diseases. Trends Cell Biol 2017;27(7):491–504.
Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A 2016;113(15):4039–4044.
Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet 2015;24(15):4429–4442.
Rubino E, Rainero I, Chio A, Rogaeva E, Galimberti D, Fenoglio P, et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 2012;79(15):1556–1562.
Liu Z, Li H, Hong C, Chen M, Yue T, Chen C, et al. ALS-associated E478G mutation in human OPTN (optineurin) promotes inflammation and induces neuronal cell death. Front Immunol 2018;9:2647.
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012;11(4):323–330.
Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum Mol Genet 2014;23(13):3579–3595.
Aoki Y, Manzano R, Lee Y, Dafinca R, Aoki M, Douglas AGL, et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain. 2017;140(4):887–897.
Huotari J, Helenius A. Endosome maturation. EMBO J 2011;30(17):3481–3500.
Yang M, Liang C, Swaminathan K, Herrlinger S, Lai F, Shiekhattar R, et al. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv 2016;2(9):e1601167.
Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L, et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J 2016;35(15):1656–1676.
Ugolino J, Ji YJ, Conchina K, Chu J, Nirujogi RS, Pandey A, et al. Loss of C9orf72 enhances autophagic activity via deregulated mTOR and TFEB signaling. PLoS Genet 2016;12(11):e1006443.
Irwin DJ, Cairns NJ, Grossman M, McMillan CT, Lee EB, Van Deerlin VM, et al. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol 2015;129(4):469–491.
Olszewska DA, Lonergan R, Fallon EM, Lynch T. Genetics of frontotemporal dementia. Curr Neurol Neurosci Rep 2016;16(12):107.
Tresse E, Salomons FA, Vesa J, Bott LC, Kimonis V, Yao TP, et al. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 2010;6(2):217–227.
Urwin H, Authier A, Nielsen JE, Metcalf D, Powell C, Froud K, et al. Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum Mol Genet 2010;19(11):2228–2238.
Bayraktar O, Oral O, Kocaturk NM, Akkoc Y, Eberhart K, Kosar A, et al. IBMPFD disease-causing mutant VCP/p97 proteins are targets of autophagic-lysosomal degradation. PLoS One 2016;11(10):e0164864.
Clayton EL, Milioto C, Muralidharan B, Norona FE, Edgar JR, Soriano A, et al. Frontotemporal dementia causative CHMP2B impairs neuronal endolysosomal traffic-rescue by TMEM106B knockdown. Brain 2018;141(12):3428–3442.
Clayton EL, Mizielinska S, Edgar JR, Nielsen TT, Marshall S, Norona FE, et al. Frontotemporal dementia caused by CHMP2B mutation is characterised by neuronal lysosomal storage pathology. Acta Neuropathol 2015;130(4):511–523.
Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet 2012;90(6):1102–1107.
Zhou X, Paushter DH, Feng T, Sun L, Reinheckel T, Hu F. Lysosomal processing of progranulin. Mol Neurodegener 2017;12(1):62.
Lee CW, Stankowski JN, Chew J, Cook CN, Lam YW, Almeida S, et al. The lysosomal protein cathepsin L is a progranulin protease. Mol Neurodegener 2017;12(1):55.
Valdez C, Wong YC, Schwake M, Bu G, Wszolek ZK, Krainc D. Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum Mol Genet 2017;26(24):4861–4872.
Zhou X, Paushter DH, Feng T, Pardon CM, Mendoza CS, Hu F. Regulation of cathepsin D activity by the FTLD protein progranulin. Acta Neuropathol 2017;134(1):151–153.
Gotzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G, et al. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol 2014;127(6):845–860.
Novak MJ, Tabrizi SJ. Huntington’s disease. BMJ. 2010;340:c3109.
Croce KR, Yamamoto A. A role for autophagy in Huntington’s disease. Neurobiol Dis. 2019;122:16–22.
Martin DD, Ladha S, Ehrnhoefer DE, Hayden MR. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci 2015;38(1):26–35.
A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72(6):971–983.
Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90(3):549–558.
Bauerlein FJB, Saha I, Mishra A, Kalemanov M, Martinez-Sanchez A, Klein R, et al. In situ architecture and cellular interactions of polyQ inclusions. Cell 2017;171(1):179–187.e10.
Kim YJ, Yi Y, Sapp E, Wang Y, Cuiffo B, Kegel KB, et al. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington's disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc Natl Acad Sci U S A 2001;98(22):12784–12789.
Mende-Mueller LM, Toneff T, Hwang SR, Chesselet MF, Hook VY. Tissue-specific proteolysis of Huntingtin (htt) in human brain: evidence of enhanced levels of N- and C-terminal htt fragments in Huntington’s disease striatum. J Neurosci 2001;21(6):1830–1837.
Lunkes A, Lindenberg KS, Ben-Haiem L, Weber C, Devys D, Landwehrmeyer GB, et al. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell 2002;10(2):259–269.
Bhat KP, Yan S, Wang CE, Li S, Li XJ. Differential ubiquitination and degradation of huntingtin fragments modulated by ubiquitin-protein ligase E3A. Proc Natl Acad Sci U S A 2014;111(15):5706–5711.
Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, et al. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci 2000;20(19):7268–7278.
Petersen A, Larsen KE, Behr GG, Romero N, Przedborski S, Brundin P, et al. Expanded CAG repeats in exon 1 of the Huntington’s disease gene stimulate dopamine-mediated striatal neuron autophagy and degeneration. Hum Mol Genet 2001;10(12):1243–1254.
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004;36(6):585–595.
Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci 2010;13(5):567–576.
Wong YC, Holzbaur EL. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci 2014;34(4):1293–1305.
del Toro D, Alberch J, Lazaro-Dieguez F, Martin-Ibanez R, Xifro X, Egea G, et al. Mutant huntingtin impairs post-Golgi trafficking to lysosomes by delocalizing optineurin/Rab8 complex from the Golgi apparatus. Mol Biol Cell 2009;20(5):1478–1492.
Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet 2006;15(6):965–977.
Metzger S, Walter C, Riess O, Roos RA, Nielsen JE, Craufurd D, et al. The V471A polymorphism in autophagy-related gene ATG7 modifies age at onset specifically in Italian Huntington disease patients. PLoS One 2013;8(7):e68951.
Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science 2004;304(5667):100–104.
Gu X, Greiner ER, Mishra R, Kodali R, Osmand A, Finkbeiner S, et al. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron. 2009;64(6):828–840.
Jeong H, Then F, Melia TJ, Jr, Mazzulli JR, Cui L, Savas JN, et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 2009;137(1):60–72.
Erie C, Sacino M, Houle L, Lu ML, Wei J. Altered lysosomal positioning affects lysosomal functions in a cellular model of Huntington’s disease. Eur J Neurosci 2015;42(3):1941–1951.
Trajkovic K, Jeong H, Krainc D. Mutant Huntingtin is secreted via a late endosomal/lysosomal unconventional secretory pathway. J Neurosci 2017;37(37):9000–9012.
Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J Neurosci 2010;30(3):1166–1175.
Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A 2013;110(19):E1817–E1826.
Masini D, Bonito-Oliva A, Bertho M, Fisone G. Inhibition of mTORC1 signaling reverts cognitive and affective deficits in a mouse model of Parkinson’s disease. Front Neurol 2018;9:208-.
Siracusa R, Paterniti I, Cordaro M, Crupi R, Bruschetta G, Campolo M, et al. Neuroprotective effects of temsirolimus in animal models of Parkinson’s disease. Mol Neurobiol 2018;55(3):2403–2419.
Ghosh A, Tyson T, George S, Hildebrandt EN, Steiner JA, Madaj Z, et al. Mitochondrial pyruvate carrier regulates autophagy, inflammation, and neurodegeneration in experimental models of Parkinson’s disease. Sci Transl Med 2016;8(368):368ra174.
Suresh SN, Chavalmane AK, Dj V, Yarreiphang H, Rai S, Paul A, et al. A novel autophagy modulator 6-Bio ameliorates SNCA/alpha-synuclein toxicity. Autophagy 2017;13(7):1221–1234.
Monti B, Gatta V, Piretti F, Raffaelli SS, Virgili M, Contestabile A. Valproic acid is neuroprotective in the rotenone rat model of Parkinson’s disease: involvement of alpha-synuclein. Neurotox Res 2010;17(2):130–141.
Rodriguez-Navarro JA, Rodriguez L, Casarejos MJ, Solano RM, Gomez A, Perucho J, et al. Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis 2010;39(3):423–438.
Sarkar S, Chigurupati S, Raymick J, Mann D, Bowyer JF, Schmitt T, et al. Neuroprotective effect of the chemical chaperone, trehalose in a chronic MPTP-induced Parkinson’s disease mouse model. Neurotoxicology 2014;44:250–262.
Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTORindependent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 2007;282(8):5641–5652.
Kovács T, Billes V, Komlós M, Hotzi B, Manzéger A, Tarnóci A, et al. The small molecule AUTEN-99 (autophagy enhancer-99) prevents the progression of neurodegenerative symptoms. Sci Rep 2017;7:42014.
Bourdenx M, Daniel J, Genin E, Soria FN, Blanchard-Desce M, Bezard E, et al. Nanoparticles restore lysosomal acidification defects: implications for Parkinson and other lysosomal-related diseases. Autophagy 2016;12(3):472–483.
Migdalska-Richards A, Daly L, Bezard E, Schapira AH. Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice. Ann Neurol 2016;80(5):766–775.
Migdalska-Richards A, Ko WKD, Li Q, Bezard E, Schapira AHV. Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse (New York, NY) 2017;71(7):e21967.
Richter F, Fleming SM, Watson M, Lemesre V, Pellegrino L, Ranes B, et al. A GCase chaperone improves motor function in a mouse model of synucleinopathy. Neurotherapeutics 2014;11(4):840–856.
Sanchez-Martinez A, Beavan M, Gegg ME, Chau KY, Whitworth AJ, Schapira AH. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci Rep 2016;6:31380.
Mazzulli JR, Zunke F, Tsunemi T, Toker NJ, Jeon S, Burbulla LF, et al. Activation of beta-glucocerebrosidase reduces pathological alpha-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J Neurosci 2016;36(29):7693–7706.
Aflaki E, Borger DK, Moaven N, Stubblefield BK, Rogers SA, Patnaik S, et al. A new glucocerebrosidase chaperone reduces alpha-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and Parkinsonism. J Neurosci 2016;36(28):7441–7452.
Delphine Charvin, Rossella Medori, Robert A. Hauser, Olivier Rascol. Therapeutic strategies for Parkinson disease: beyond dopaminergic drugs. Nat Rev Drug Discov 2018;17(11):804–822.
Sardi SP, Clarke J, Kinnecom C, Tamsett TJ, Li L, Stanek LM, et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci U S A 2011;108(29):12101–12106.
Sardi SP, Viel C, Clarke J, Treleaven CM, Richards AM, Park H, et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc Natl Acad Sci 2017;114(10):2699.
Li L, Zhang S, Zhang X, Li T, Tang Y, Liu H, et al. Autophagy enhancer carbamazepine alleviates memory deficits and cerebral amyloid-beta pathology in a mouse model of Alzheimer’s disease. Curr Alzheimer Res 2013;10(4):433–441.
Steele JW, Kim SH, Cirrito JR, Verges DK, Restivo JL, Westaway D, et al. Acute dosing of latrepirdine (Dimebon), a possible Alzheimer therapeutic, elevates extracellular amyloid-beta levels in vitro and in vivo. Mol Neurodegener 2009;4:51.
Steele JW, Lachenmayer ML, Ju S, Stock A, Liken J, Kim SH, et al. Latrepirdine improves cognition and arrests progression of neuropathology in an Alzheimer’s mouse model. Mol Psychiatry 2013;18(8):889–897.
Zhang L, Wang L, Wang R, Gao Y, Che H, Pan Y, et al. Evaluating the effectiveness of GTM-1, rapamycin, and carbamazepine on autophagy and alzheimer disease. Med Sci Monit 2017;23:801–808.
Matsunaga S, Kishi T, Annas P, Basun H, Hampel H, Iwata N. Lithium as a treatment for Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis 2015;48(2):403–410.
Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem 2010;285(12):9100–9113.
Chiang MC, Nicol CJ, Cheng YC. Resveratrol activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against amyloid-beta-induced inflammation and oxidative stress. Neurochem Int 2018;115:1–10.
Turner RS, Thomas RG, Craft S, van Dyck CH, Mintzer J, Reynolds BA, et al. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015;85(16):1383–1391.
Moussa C, Hebron M, Huang X, Ahn J, Rissman RA, Aisen PS, et al. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J Neuroinflammation 2017;14(1):1.
Robinson DM, Keating GM. Memantine: a review of its use in Alzheimer’s disease. Drugs 2006;66(11):1515–1534.
van Marum RJ. Update on the use of memantine in Alzheimer’s disease. Neuropsychiatr Dis Treat 2009;5:237–247.
Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci U S A 2010;107(50):21830–21835.
Barini E, Antico O, Zhao Y, Asta F, Tucci V, Catelani T, et al. Metformin promotes tau aggregation and exacerbates abnormal behavior in a mouse model of tauopathy. Mol Neurodegener 2016;11:16.
Castillo K, Nassif M, Valenzuela V, Rojas F, Matus S, Mercado G, et al. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy. 2013;9(9):1308–1320.
Wang Y, Liu FT, Wang YX, Guan RY, Chen C, Li DK, et al. Autophagic modulation by trehalose reduces accumulation of TDP-43 in a cell model of amyotrophic lateral sclerosis via TFEB activation. Neurotox Res 2018;34(1):109–120.
Zhang X, Chen S, Song L, Tang Y, Shen Y, Jia L, et al. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 2014;10(4):588–602.
Mancuso R, del Valle J, Modol L, Martinez A, Granado-Serrano AB, Ramirez-Nunez O, et al. Resveratrol improves motoneuron function and extends survival in SOD1(G93A) ALS mice. Neurotherapeutics 2014;11(2):419–432.
Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, et al. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 2004;10(2):148–154.
Chiu CT, Liu G, Leeds P, Chuang DM. Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of Huntington’s disease. Neuropsychopharmacology 2011;36(12):2406–2421.
Pouladi MA, Brillaud E, Xie Y, Conforti P, Graham RK, Ehrnhoefer DE, et al. NP03, a novel low-dose lithium formulation, is neuroprotective in the YAC128 mouse model of Huntington disease. Neurobiol Dis 2012;48(3):282–289.
Rose C, Menzies FM, Renna M, Acevedo-Arozena A, Corrochano S, Sadiq O, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington's disease. Hum Mol Genet 2010;19(11):2144–2153.
Garriga-Canut M, Agustin-Pavon C, Herrmann F, Sanchez A, Dierssen M, Fillat C, et al. Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc Natl Acad Sci U S A 2012;109(45):E3136–E3145.
Monteys AM, Ebanks SA, Keiser MS, Davidson BL. CRISPR/Cas9 editing of the mutant Huntingtin allele in vitro and in vivo. Mol Ther 2017;25(1):12–23.
Tramutola A, Lanzillotta C, Barone E, Arena A, Zuliani I, Mosca L, et al. Intranasal rapamycin ameliorates Alzheimer-like cognitive decline in a mouse model of Down syndrome. Transl Neurodegener 2018;7:28.
Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One 2010;5(4):e9979.
Van Skike CE, Jahrling JB, Olson AB, Sayre NL, Hussong SA, Ungvari Z, et al. Inhibition of mTOR protects the blood-brain barrier in models of Alzheimer’s disease and vascular cognitive impairment. Am J Physiol Heart Circ Physiol 2018;314(4):H693-H703.
Lin AL, Zheng W, Halloran JJ, Burbank RR, Hussong SA, Hart MJ, et al. Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J Cereb Blood Flow Metab 2013;33(9):1412–1421.
Lin AL, Jahrling JB, Zhang W, DeRosa N, Bakshi V, Romero P, et al. Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. J Cereb Blood Flow Metab 2017;37(1):217–226.
Harkavyi A, Abuirmeileh A, Lever R, Kingsbury AE, Biggs CS, Whitton PS. Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson’s disease. J Neuroinflammation 2008;5(1):19.
Bertilsson G, Patrone C, Zachrisson O, Andersson A, Dannaeus K, Heidrich J, et al. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J Neurosci Res 2008;86(2):326–338.
Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, et al. Exenatide once weekly versus placebo in Parkinson's disease: a randomised, double-blind, placebocontrolled trial. Lancet (London, England) 2017;390(10103):1664–1675.
Karuppagounder SS, Brahmachari S, Lee Y, Dawson VL, Dawson TM, Ko HS. The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson’s disease. Sci Rep 2014;4:4874.
Hebron ML, Lonskaya I, Moussa CE. Tyrosine kinase inhibition facilitates autophagic SNCA/alpha-synuclein clearance. Autophagy 2013;9(8):1249–1250.
Hebron ML, Lonskaya I, Moussa CE- H. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of - synuclein in Parkinson's disease models. Hum Mol Genet 2013;22(16):3315–3328.
Pagan F, Hebron M, Valadez EH, Torres-Yaghi Y, Huang X, Mills RR, et al. Nilotinib effects in Parkinson’s disease and dementia with Lewy bodies. J Park Dis 2016;6(3):503–517.
Siddiqui A, Bhaumik D, Chinta SJ, Rane A, Rajagopalan S, Lieu CA, Lithgow GJ, Andersen JK. Mitochondrial Quality Control via the PGC1 -TFEB Signaling Pathway Is Compromised by Parkin Q311X Mutation But Independently Restored by Rapamycin. J Neurosci 2015;35(37):12833–12844.
Menzies FM, Garcia-Arencibia M, Imarisio S, O'Sullivan NC, Ricketts T, Kent BA, et al. Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ 2015;22(3):433–444.
Getaw Worku Hassen, Leo Kesner, Alfred Stracher, Abraham Shulman, Edward Rockenstein, Michael Mante, Anthony Adame, Cassia Overk, Robert A. Rissman, Eliezer Masliah. Effects of Novel Calpain Inhibitors in Transgenic Animal Model of Parkinson’s disease/dementia with Lewy bodies. Sci Rep 2018;8(1).
Hewitt G, Korolchuk VI. Repair, reuse, recycle: the expanding role of autophagy in genome maintenance. Trends Cell Biol 2017;27(5):340–351.
Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V. Secretory autophagy. Curr Opin Cell Biol 2015;35:106–116.
Clarke AJ, Simon AK. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat Rev Immunol 2019;19(3):170–183.
Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci 2011;12(8):437–452.
Zhou B, Kreuzer J, Kumsta C, Wu L, Kamer KJ, Cedillo L, et al. Mitochondrial permeability uncouples elevated autophagy and lifespan extension. Cell. 2019;177(2):299–314.e16.
Xilouri M, Brekk OR, Landeck N, Pitychoutis PM, Papasilekas T, Papadopoulou-Daifoti Z, et al. Boosting chaperone-mediated autophagy in vivo mitigates α-synuclein-induced neurodegeneration. Brain. 2013;136(7):2130–2146.
Xilouri M, Brekk OR, Polissidis A, Chrysanthou-Piterou M, Kloukina I, Stefanis L. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 2016;12(11):2230–2247.
Arrant AE, Onyilo VC, Unger DE, Roberson ED. Progranulin gene therapy improves lysosomal dysfunction and microglial pathology associated with frontotemporal dementia and neuronal ceroid lipofuscinosis. J Neurosci 2018;38(9):2341–2358.
Rocha EM, Smith GA, Park E, Cao H, Brown E, Hayes MA, et al. Glucocerebrosidase gene therapy prevents α-synucleinopathy of midbrain dopamine neurons. Neurobiol Dis 2015;82:495–503.
Steet RA, Chung S, Wustman B, Powe A, Do H, Kornfeld SA. The iminosugar isofagomine increases the activity of N370S mutant acid beta-glucosidase in Gaucher fibroblasts by several mechanisms. Proc Natl Acad Sci U S A 2006;103(37):13813–13818.
Valenzano KJ, Khanna R, Powe AC, Boyd R, Lee G, Flanagan JJ, et al. Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders. Assay and Drug Development Technologies 2011;9(3):213–235.
Patnaik S, Zheng W, Choi JH, Motabar O, Southall N, Westbroek W, et al. Discovery, structure-activity relationship, and biological evaluation of noninhibitory small molecule chaperones of glucocerebrosidase. J Med Chem 2012;55(12):5734–5748.
Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, et al. Neuronal-targeted TFEB accelerates lysosomal degradation of APP, reducing Abeta generation and amyloid plaque pathogenesis. J Neurosci 2015;35(35):12137–12151.
Wild EJ, Tabrizi SJ. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol 2017;16(10):837–847.
Tabrizi S, Leavitt B, Kordasiewicz H, Czech C, Swayze E, Norris DA, et al. Effects of IONIS-HTTRx in patients with early Huntington’s disease, results of the first HTT-lowering drug trial (CT.002). Neurology 2018;90(15 Supplement):CT.002.
Acknowledgments
We thank Dr. Yvette C. Wong and Dr. Joseph Mazzulli for their helpful advice. The authors were supported by National Institutes of Health grants as follows: D.K. by R01 NS076054 and R37 NS096241.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised to include a revised version of figure 2 overlooked by the publisher during the production process.
Electronic Supplementary Material
ESM 1
(DOCX 460 kb)
Rights and permissions
About this article

Cite this article
Peng, W., Minakaki, G., Nguyen, M. et al. Preserving Lysosomal Function in the Aging Brain: Insights from Neurodegeneration. Neurotherapeutics 16, 611–634 (2019). https://doi.org/10.1007/s13311-019-00742-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00742-3