
Abstract
Species (or cryptic species) identification in microbial eukaryotes often requires a combined morphological and molecular approach, and if possible, mating reaction tests that confirm, for example, that distant populations are in fact one species. We used P. biaurelia (one of the 15 cryptic species of the P. aurelia complex) collected worldwide from 92 sampling points over 62 years and analyzed with the three above mentioned approaches as a model for testing protistan biogeography hypotheses. Our results indicated that despite the large distance between them, most of the studied populations of P. biaurelia do not differ from each other (rDNA fragment), or differ only slightly (COI mtDNA fragment). These results could suggest that in the past, the predecessors of the present P. biaurelia population experienced a bottleneck event, and that its current distribution is the result of recent dispersal by natural or anthropogenic factors. Another possible explanation for the low level of genetic diversity despite the huge distances between the collecting sites could be a slow rate of mutation of the studied DNA fragments, as has been found in some other species of the P. aurelia complex. COI haplotypes determined from samples obtained during field research conducted in 2015–2016 in 28 locations/374 sampling points in southern Poland were shared with other, often distant P. biaurelia populations. In the Kraków area, we found 5 of the 11 currently known COI P. biaurelia haplotypes. In 5 of 7 reservoirs from which P. biaurelia was obtained, two different COI haplotypes were identified.
Similar content being viewed by others

Introduction
Although members of genera like Paramecium or Tetrahymena have become objects of research in many fields of life sciences (Kodama and Fujishima 2014; Picariello et al. 2014; Tomazic et al. 2014; Benbouzid et al. 2015), data from populations of particular species living in natural habitats remain scarce (Katz and Turkewitz 2013). Therefore estimating and evaluating the species composition, biodiversity and distribution of a variety of ecosystems is crucial for generating hypotheses on the course of microevolution, ecology and potential protection of ciliates and other microbial eukaryotes. As other free-living protists, ciliates play a key ecological role in various types of environment (Weisse 2006; Pawlowski et al. 2012). However, in many cases determining the boundaries between eukaryotic microbial species is difficult due to the lack of a strict species definition (Boenigk et al. 2012), scarce data on natural populations (Heger et al. 2014) as well as existence of cryptic species (Nanney and McCoy 1976; Gentekaki and Lynn 2010; McManus et al. 2010; Katz et al. 2011).
In the genus Paramecium, strain crosses and molecular analyses have revealed the occurrence of cryptic differentiation (for example Sonneborn 1975; Greczek-Stachura et al. 2012; Tarcz et al. 2014; Przyboś and Tarcz 2016) in the majority of the 19 valid morphospecies (Fokin 2010/2011; Krenek et al. 2015). One of these is Paramecium aurelia, which is in fact a complex of 15 sibling species (Sonneborn 1975; Aufderheide et al. 1983) that are morphologically indistinguishable but sexually isolated. However reciprocal relationships within the P. aurelia species complex are still not clear because in some strains inconsistencies have been found between DNA fragment comparisons and mating test classifications (Catania et al. 2009), which may be the result of incomplete lineage sorting or hybridization-introgression processes (Tarcz et al. 2013). Although the latter issue is rare in microbial eukaryotes and rather concerns parasitic species (Messenger et al. 2012), interspecies mating reactions between some species of the P. aurelia complex have been reported (Sonneborn 1975). In order to avoid misidentification during species delimitation, a combination of different (molecular, morphological and physiological) approaches should be applied (Duff et al. 2008; Caron 2013; Agatha and Strüder-Kypke 2014). Thus, in P. aurelia complex for proper cryptic species designation different approaches are used: comparing cell morphology features, characteristics of the nuclear apparatus, testing genetic isolation by strain crosses, and analysis of DNA fragment variability. Until now two DNA regions have most frequently been used in P. aurelia studies: ribosomal (ITS1-5.8S-ITS2-5’LSU rDNA), and mitochondrial (a part of COI gene) regions (Tarcz et al. 2013). Moreover these regions have been proposed as a DNA barcoding tool for other ciliates (Strüder-Kypke and Lynn 2010; Stoeck et al. 2014b), and have been used as a markers for population studies in particular ciliates (Gentekaki and Lynn 2009; Zufall et al. 2012) as well as in several Paramecium species (Snoke et al. 2006; Zhao et al. 2013).
Faunistic data on the P. aurelia complex (Sonneborn 1975; Przyboś and Surmacz 2010) can be used to recognize the existence of cryptic species with either a narrow (for example P. undecaurelia) or wide range of occurrence (for example P. biaurelia). The latter seem to be ideal objects of research in the fields of wide microbial biogeography and population genetics. Paramecium biaurelia, according to Sonneborn (1975), is found in Europe, Asia, Australia and New Zealand, North and South America and is generally common in cold to moderate climates and never found in the tropics. The cell length of P. biaurelia is about 133 μm (Fokin and Chivilev 2000) and its characteristic feature, which distinguishes it from the other P. aurelia species, is that it carries out mating reactions below 21 °C and between 9 PM and 7 AM (Sonneborn 1975). Importantly, there is no evidence for gene flow between P. biaurelia and other P. aurelia species (Sonneborn 1975). A preliminary molecular survey of 12 strains originating from distant sampling sites (Tarcz et al. 2013) showed that P. biaurelia forms a well separated clade, distinct form other species of the complex and reveals very low genetic variability.
Due to the sparse data on the spatial variability of other free-living, freshwater microbial eukaryotes it could be supposed that more accurate sampling of distant as well as local populations may reveal previously hidden genetic variation in P. biaurelia.
Materials and methods
Strain maintenance
Most strains used in the present study are part of the strain collection of the Department of Experimental Zoology, Institute of Systematics and Evolution of Animals, Polish Academy of Sciences. Ciliates were collected between 1954 and 2016 and identified as P. biaurelia according to the Sonneborn method (1970) by researchers working on Paramecium (E. Przyboś and others (see supplementary Table S1), sent by collaborators, or donated (mainly standard strains) by Prof. T. M. Sonneborn (Department of Biology, Indiana University, Bloomington, USA) and Prof. G. H. Beale (Institute of Genetics, Edinburgh University, Scotland, UK)). Strains were cultivated on a lettuce medium inoculated with Enterobacter aerogenes according to Sonneborn’s method (1950, 1970) and supplemented with 0.8 mg/ml ß sitosterol (Merck, Darmstadt, Germany). For phylogenetic analysis, we used 139 strains of P. biaurelia originating from Europe, Asia, Australia, and North and South America (Fig. 1).

The origin (N = 92) of Paramecium biaurelia strains used in present studies
Molecular techniques
Paramecium genomic DNA was isolated from vegetative cells at the end of the exponential phase (approx. 1000 cells were used for DNA extraction) using a NucleoSpin Tissue Kit (Macherey-Nagel, Germany), according to the manufacturer’s instructions for DNA isolation from cell cultures. The only modification was that the cell culture was centrifuged for 20 min at 13,200 rpm. The supernatant was then removed and the remaining cells were suspended in lysis buffers and proteinase K. Fragments of rDNA (1062 bp) and COI (638 bp) genes were amplified, sequenced and analyzed. The rDNA fragment was amplified with an ITS1 universal eukaryotic primer (5′-TCCGTAGGTGAACCTGCGG-3′ (White et al. 1990)) and a 3pLSU primer (5′-CAAGACGGGTCAGTAGAAGCC-3′ using the protocol previously described by (Tarcz et al. 2012)). The COI fragment of mitochondrial DNA was amplified with the primer pair F388dT (5′-TGTAAAACGACGGCCAGTGGwkCbAAAGATGTwGC-3′) and R1184dT (5′-CAGGAAACAGCTATGACTAdACyTCAGGGTGACCrAAAAATCA-3′) using the protocol previously described by (Strüder-Kypke and Lynn 2010). The composition of the reaction mixtures, PCR programs, and purification procedure were presented in detail in Tarcz et al. (2012). Cycle sequencing was done in both directions with BigDye Terminator v3.1 chemistry (Applied Biosystems, USA). The primers used in the PCR reactions were again used for sequencing the rDNA region and the primer pair M13F (5′-TGTAAAACGACGGCCAGT-3′) and M13R (5′-CAGGAAACAGCTATGAC-3′) (Strüder-Kypke and Lynn 2010) was used for sequencing the COI fragment. The details of the sequencing procedure were as in Tarcz et al. (2012). The sequences of both studied fragments are available in the NCBI GenBank database (see supplementary Table S1).
Data analysis
Sequences were examined using Chromas Lite (Technelysium, Australia) to evaluate and correct chromatograms. Alignments of the studied sequences were performed using BioEdit software (Hall 1999) and checked manually. All the obtained sequences were unambiguous and were used in the later analyses. Phylograms were constructed for the studied fragments in Mega v6.0 (Tamura et al. 2013), using neighbor-joining (NJ), maximum parsimony (MP), and maximum likelihood (ML). All positions containing gaps and missing data were eliminated. NJ analysis was performed in Mega 6.0 by bootstrapping with 1000 replicates. MP analysis was evaluated with a min-min heuristic parameter (at level 2) and bootstrapping with 1000 replicates. Mega 6.0 identified the T92 model for the ITS1-5.8S-ITS2-5′ end of LSU rDNA and for COI mtDNA as the best nucleotide substitution model for maximum likelihood tree reconstruction. Bayesian inference (BI) was performed in MrBayes 3.1.2 (Ronquist and Huelsenbeck 2003); the analysis was run for 5,000,000 generations and trees were sampled every 100 generations. All trees for BI analysis were visualized in TreeView 1.6.6 (Page 1996). Analysis of haplotype diversity (Hd) and its sampling variance (SD), nucleotide diversity (π), and polymorphic sites was done in DnaSP v5.10.01 (Librado and Rozas 2009). Haplotype networks, which presented the distribution and relationships among haplotypes of P. biaurelia strains, were reconstructed with the Median Joining method (Bandelt et al. 1999), as implemented in PopART v. 1.7 (Leigh and Bryant 2015).
Results
Variability of the studied DNA fragments within the Paramecium biaurelia strains
In the present study, we sampled 123 strains (from the ISEA PAS collection) in terms of both analyzed DNA fragments (12 of them were studied previously in Tarcz et al. 2013). Moreover for the molecular analysis we included an extra 2 (in the case of rDNA fragment) and 16 (in the case of COI mtDNA fragment) sequences from GenBank. Altogether 92 sampling points were tested, with most of them from the following ecozones: the Western Palearctic (67), Central Palearctic (10), Nearctic (8), Eastern Palearctic (4), Australasia (2) and finally one location in the Neotropic (Supplementary Table S1).
Studies on the ITS1-5.5S-ITS2-5’LSU rDNA fragment showed that all (N = 125) studied P. biaurelia strains were almost identical—only three haplotypes (Pa2RD_01, Pa2RD_02 and Pa2RD_03) were identified (Table S1). Intraspecific haplotype diversity among the studied P. biaurelia strains (N = 125) was Hd = 0.032 (0.00048), and nucleotide diversity was π = 0.00003. The mean divergence over all P. biaurelia (N = 125) sequence pairs was 0.000 (ranging from 0.000 to 0.001), and only 2 variable positions (0 parsimony informative) were detected in the ITS1-5.5S-ITS2-5’LSU rDNA fragment (1062 bp).
In the COI mtDNA dataset intraspecific haplotype diversity among the studied P. biaurelia strains (N = 139) was Hd = 0.626 (0.00144), which indicates that the studied DNA fragment has rather low variability. Nucleotide diversity amounted to π = 0.00248. The mean divergence over all P. biaurelia (N = 139) sequence pairs was 0.002 (ranging from 0.000 to 0.016) and only 18 variable positions (7 parsimony informative) were detected in the COI fragment (638 bp). A total of 11 COI haplotypes were found in the studied dataset (Table S1). Notably, 6 of the 11 were private haplotypes (Pa2COI_01, Pa2COI_07, Pa2COI_08, Pa2COI_09, Pa2COI_10, Pa2COI_11) and the remaining 5 (Pa2COI_02, Pa2COI_03, Pa2COI_04, Pa2COI_05, Pa2COI_06) were shared between 133 of 139 strains of P. biaurelia. The most frequent haplotype was Pa2COI_03, observed in 79 isolates, followed by Pa2COI_02 in 28 P. biaurelia strains, Pa2COI_04 in 15 strains, Pa2COI_06 in 7 strains, and finally Pa2COI_05, which was characteristic for 4 strains from one location (Table S1). For better understanding and to preserve continuity between previous and future Paramecium studies, we decided to number the individual haplotypes according to the formula first used by Barth et al. (2006).
Inter- and intraspecific relationships of P. biaurelia species based on the comparison of ITS1-5.8S-ITS2-5’LSU rDNA and COI mtDNA sequences
Based on the obtained rDNA and COI sequences and the constructed tree we found that P. aurelia, P. jenningsi complexes together with P. schewiakoffi form a monophyletic clade distant from P. caudatum and P. multimicronucleatum, which were used as an outgroup in the present study (Figs. 2 and 3). Similarly, P. biaurelia forms a well-defined monophyletic clade within the P. aurelia complex and the other species of the subgenus Paramecium (Figs. 2 and 3). In the case of ribosomal data, P. biaurelia is situated close to P. tredecaurelia and P. quadecaurelia (Fig. 2). The intraspecific relationships of P. biaurelia rDNA haplotypes (Figs. 4a and 5a) indicate the existence of one dominating haplotype Pa2RD_01 (N = 123) and two private haplotypes Pa2RD_02, Pa2RD_03, which are different from Pa2RD_01 by one nucleotide substitution.

Phylogenetic tree constructed for Paramecium aurelia complex, P. jenningsi complex and P. schewiakoffi (two species: P. caudatum and P. multimicronucleatum were used as an outgroup). The tree was constructed on the basis of a comparison of sequences from the ribosomal ITS1-5.8S-ITS2-5’LSU fragment using the maximum likelihood method. Bootstrap values for neighbor joining, maximum parsimony, maximum likelihood, and posterior probabilities for Bayesian inference are presented. Bootstrap values smaller than 50% (posterior probabilities < 0.50) are not shown. Dashes represent no bootstrap or posterior value at a given node. All positions containing gaps and missing data were eliminated. Phylogenetic analyses were conducted using MEGA 6.0 (NJ/ML) and MrBayes 3.1.2 (BI). The analysis involved 145 nucleotide sequences. There was a total of 1062 positions in the final dataset

Phylogenetic tree constructed for Paramecium aurelia complex, P. jenningsi complex and P. schewiakoffi (two species: P. caudatum and P. multimicronucleatum were used as an outgroup). The tree was constructed on the basis of a comparison of sequences from the mitochondrial COI fragment using the maximum likelihood method. Bootstrap values for neighbor joining, maximum parsimony, maximum likelihood, and posterior probabilities for Bayesian inference are presented. Bootstrap values smaller than 50% (posterior probabilities < 0.50) are not shown. Dashes represent no bootstrap or posterior value at a given node. All positions containing gaps and missing data were eliminated. Phylogenetic analyses were conducted using MEGA 6.0 (NJ/ML) and MrBayes 3.1.2 (BI). The analysis involved 159 nucleotide sequences. There was a total of 638 positions in the final dataset

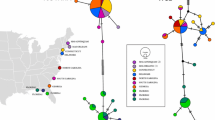
Haplotype network of Paramecium biaurelia constructed using the 123 sequences of ribosomal ITS1-5.8S-ITS2-5’LSU fragments (a) and 139 of mitochondrial COI fragments (b). The network presents reciprocal relationships between, and the origin of P. biaurelia haplotypes identified in current study. Black dashes on particular branches represent nucleotide substitutions between particular haplotypes. Analyses were conducted using the Median Joining method in PopART software v. 1.7

Haplotype network of Paramecium biaurelia constructed using the 123 sequences of ribosomal ITS1-5.8S-ITS2-5’LSU fragments (a) and 139 of mitochondrial COI fragments (b). The network presents a comparison of haplotypes obtained in the Kraków area vs. the other localities, where molecular data for P. biaurelia is available. Black dashes on particular branches represent nucleotide substitutions between particular haplotypes. Analyses were conducted using the Median Joining method in PopART software v. 1.7
In turn, the COI dataset revealed that P. biaurelia is rather distant from the other P. aurelia species and is situated between the P. sexaurelia and P. tredecaurelia branches (Fig. 3). The intraspecific relationships of P. biaurelia COI haplotypes (Figs. 4b and 5b) showed the existence of a dominating haplotype located in the central part of the network—Pa2COI_03 (N = 79). Most P. biaurelia COI haplotypes are directly connected with Pa2COI_03 and differ from it by one to three substitutions. It is worth noting that there are only 10 nucleotide substitutions between the most distant haplotypes (Pa2COI_10 and Pa2COI_11) (Figs. 4b and 5b).
The biogeographical pattern within the studied P. biaurelia strains
The most frequent haplotype, Pa2RD_01 was observed in 123 isolates (Palearctic, Nearctic, Australasia ecozones), while haplotypes Pa2RD_02 and Pa2RD_03 were characteristic for MbIII.3 (Münster, Botanical Garden) and US_Bl 11III1 (Indiana, Bloomington, Miller Showers Park) strains, respectively (Fig. 4a, Table S1). The COI fragment median-joining network showed that haplotype Pa2COI_03 (N = 79) was found in the Palearctic, Nearctic and Australasia ecozones (Fig. 4b, Table S1). The second most common haplotype (Pa2COI_02; N = 28) was from Europe and the Central Palearctic. The remaining haplotypes with broader geographical range (Pa2COI_04; N = 15) were identified from four sampling points in the Eastern and Western Palearctic, Nearctic and Australasia, as well as (PaCOI_06; N = 7) from the Western Palearctic and Nearctic realms. Finally there were seven rare haplotypes with restricted distributions: Pa2COI_05 (four sampling points in Boston, USA), Pa2COI_01 (one sampling point in Scotland, Great Britain), Pa2COI_07, Pa2COI_08, Pa2COI_10 (three different sampling points in Poland), Pa2COI_09 from North Carolina, USA and Pa2COI_11 from Chile. The constructed COI (similarly as rDNA) network revealed no median vectors or reticulate patterns between the obtained haplotypes. Despite intercontinental sampling, most (8 of 11 haplotypes) of the identified P. biaurelia COI haplotypes were only present in strains originating from Europe (except Pa2COI_05, 09, 11), and (7 of 11 haplotypes) Poland (Fig. 4b, Table S1).
Analysis of P. biaurelia natural population variability
In order to more accurately analyze molecular variability of P. biaurelia populations, we conducted field research (seasons 2015–2016) by sampling 49 water reservoirs in 28 locations (Southern Poland, mainly Kraków), which finally gave 177 sampling points altogether. Some reservoirs were sampled 2–6 times, which consequently led to 374 examined sampling points; P. biaurelia was detected in 3.74% (N = 14) of them (Table S2). In the studied area (within the administrative boundaries of Kraków, and one sampling point in Pieskowa Skała about 20 km northwest of Kraków), based on genetic crosses (the biological species concept) and COI mtDNA analysis (the phylogenetic species concept), we identified 4 out of the 11 known P. biaurelia COI haplotypes (no differences were detected in the rDNA fragment). All identified haplotypes were shared with the other, often distant P. biaurelia populations. A comparison of the obtained COI data with the other studied P. biaurelia strains showed that 5 of the 11 known haplotypes of the global COI variability were detected within the Kraków area (Fig. 5b, Table S2). In 5 of the 7 reservoirs in which P. biaurelia was identified, two different COI haplotypes were detected (at different sampling points) (Fig. 6, Table S2).

Map of sampling sites of Paramecium biaurelia strains collected during field research in the Kraków area. a Kraków, “At the brickyard” pond, 1 sampling point. b Kraków, Zaczarowana Dorożka Park (pond), 2 sampling points. c Pieskowa Skała (pond), 1 sampling point. d Kraków, Botanical Garden (3 ponds), 6 sampling points. e Kraków, Królówka (pond), 2 sampling points. f Kraków, Opatkowice (pond), 2 sampling points. g Kraków, Rajsko (pond), 2 sampling points
The occurrence of different COI haplotypes in one sampling point or even in one water sample has been observed before at locations outside the Kraków area—for example in a small river near Moscow three COI haplotypes were identified (Table S1).
Discussion
Paramecium biaurelia as a good model for testing microbial biogeography
Despite ciliates from the Paramecium genus being one of the most characteristic microorganisms of aquatic ecosystems, known since the seventeenth century thanks to Leeuwenhoek’s observations, their biogeography and distribution remain poorly understood compared with animals or plants (Azovsky et al. 2016). This problem is similar among most other protists. Currently two different models describe the observed distribution patterns of microbial eukaryotes—the “ubiquity model” (UM) (Fenchel and Finlay 2004; Finlay et al. 2006) and the “moderate endemicity model” (MEM) (Foissner 2006; Foissner et al. 2008). At present, despite intensive debate over these hypotheses neither has been fully confirmed. Reasons for this are twofold: firstly, the difficulty of defining a species in protistology (Caron 2013), which is crucial for shaping protist biogeography (Finlay 1998; Dijoux et al. 2014), and secondly—an under-sampling of particular species, which precludes determining whether they have wide or narrow ranges (Foissner 2006; Caron et al. 2009; Fokin 2010/2011).
Recent developments in molecular analyses have given rise to new possibilities not only for testing the UM and MEM biogeography models (Darling et al. 2007; Watts et al. 2011), but also for more easily designating protist species (Chantangsi et al. 2007; Gentekaki and Lynn 2010), testing their phylogenetic relationships (Kosakyan et al. 2013; Lahr et al. 2013) or even assessing the biodiversity of entire ecosystems (Sogin et al. 2006; Stoeck et al. 2014a; Lara et al. 2015; Pawlowski 2015). The latter issue, based on next generation sequencing technology allows for the most accurate examination of protistan diversity (Mahe et al. 2015) through the detection of species or molecular operational taxonomic units (MOTU) composition. However, for the obtained data to be useful for identifying new species, they must be matched with previously described taxons by comparing them with sequences deposited in databases like GenBank or BOLD (Thomsen and Willerslev 2015). Moreover, there exists the potential for the accidental sequencing of paralogs of study genes (West et al. 2014) as well as of orthologs from environmental samples (Thomsen and Willerslev 2015), which would make it almost impossible to study the biogeographical occurrence of some protist species. An alternative to the above and proposed in the current study may be the collection of samples of particular, well-defined species from as many sampling points as possible. Species (or cryptic species) identification in microbial eukaryotes often requires at least a morpho-molecular approach (Boenigk et al. 2012), and if possible, mating reaction tests that confirm distant populations to be in fact one species. Thus, we suppose that P. biaurelia collected worldwide from 92 sampling points over 62 years and then analyzed with the three above presented approaches seems to be an appropriate model for testing protistan biogeography hypotheses.
The reasons for the wide distribution of Paramecium biaurelia
The occurrence of several species from the P. aurelia complex may be restricted by temperature barriers, as P. quadecaurelia is restricted to the tropical zone (Przyboś et al. 2013), and P. sexaurelia, P. octaurelia as well as P. sonneborni to the warm zone (Przyboś et al. 2014, 2015; Przyboś and Prajer 2015). On the other hand, Paramecium biaurelia is one of the species of the P. aurelia complex that is characterized by wide distribution in areas with colder climates. Moreover, many years of sampling (since 1959) of different regions of Poland have revealed that it is a common species, recorded in 88 habitats (40.36%) among 218 studied (Przyboś et al. 2011). P. biaurelia’s frequent occurrence could be explained by fact that it is capable of invading marginal habitats, and adapts to circumstances that enforce inbreeding, such as intraclonal mating. Gill and Hairston (1972) found that P. biaurelia inhabits nearly all parts of two investigated woodland seeps at very low density over the whole year, in contrast to other species that occurred only at various locations at different times. Furthermore, this species is less sensitive to low temperatures (Hairston 1958), low food levels (Hairston and Kellermann 1965) and bacterial endosymbionts (Beale and Preer 2008) that may be toxic to other species of the complex. Therefore, the role of competition between species from the same ecological niche may be an important factor shaping the observed diversity of occurrence of particular P. aurelia species (Hairston 1958; Hairston and Kellermann 1965). It has been shown that P. biaurelia can exert a dominating or inhibitory effect on other paramecia (P. triaurelia), probably caused by the presence of killer strains in this species (P. biaurelia) (Beale and Preer 2008). In fact this species is characterized by the greatest variety of endosymbionts among the whole P. aurelia complex (Sonneborn 1974). The above characters lead to the conclusion that P. biaurelia is a species with superior fitness for inhabiting new, unpromising ecological conditions and could be called a pioneer species. In summary, a high adaptive capacity for new environmental conditions (Gill and Hairston 1972) as well as a vast population size (Finlay 2002) may be key factors for the wide distribution of P. biaurelia. In contrast, small population size causes restricted gene flow and restricted distribution in the other flagship ciliate—Tetrahymena thermophila (Zufall et al. 2012).
Explanation of the observed low genetic variation of P. biaurelia
The comparison of COI sequences from 139 P. biaurelia strains (Table S1) revealed the existence of 11 haplotypes of which only 4 were frequent in the studied dataset and observed on different continents whereas 6 of them appeared only once in the studied dataset and finally one haplotype has been identified in 4 strains in one location (Fig. 4b). Moreover, the number of nucleotide differences between particular haplotypes did not exceed four (usually only one or two) substitutions.
An explanation of the obtained results may be that the P. biaurelia population (predecessors of the present population) went through a bottleneck in the past, and its current distribution is the result of recent dispersal by natural or anthropogenic factors. This hypothesis is supported by fact that the P. biaurelia effective population size is smaller than that of the other widespread Paramecium species: P. primaurelia and P. tetraurelia (Snoke et al. 2006). At present, the dispersal of the P. aurelia complex (among them P. biaurelia) as well as other paramecia is still not completely solved despite several suggestions and attempts at explanation. It is worth noting that the existence of cysts has never been reported in the Paramecium genus (Landis 1988; Gutierrez et al. 1988; Beale and Preer 2008). Therefore the passive dispersal of paramecia is possible, with some drops of water on migrating birds, even over long non-stop routes (Hedenström 2010), mammals over further distances (Sonneborn 1975; Coleman 2005) or by insects over shorter distances (Razowski 1996). Furthermore, Foissner (2006) suggested an important role for human activities in the dispersal of microeukaryotes. However, the bottleneck explanation for low molecular variability contradicts the suggestion that P. biaurelia is a pioneer species.
In our opinion extraordinary genome stability is a more likely explanation for the observed low molecular diversity in P. biaurelia, and has been reported previously in P. tetraurelia (Sung et al. 2012). This hypothesis could be supported by the fact that P. biaurelia diverged from the same lineage as P. tetraurelia (McGrath et al. 2014) and therefore similarly to P. tetraurelia could be characterized by a low mutational rate (Sung et al. 2012) that led to low molecular diversity between strains belonging to the same species. Moreover, it has been supposed that whole genome duplication (WGD) has seemingly not influenced phenotypic innovations in some P. aurelia species (McGrath et al. 2014). In contrast, in other taxa WGD events have played an important role in increasing their complexity and molecular diversity, for example in land plants (Van de Peer et al. 2009) or yeast (Thomson et al. 2005).
Sampling the neighborhood—can almost the entire global diversity of P. biaurelia be observed in a nearby pond?
As was mentioned above, under-sampling (in all ecozones as well as particular ecosystems/ecological niches) negatively affects biogeographical studies of protists species (Foissner 2006; Fokin 2010/2011), especially in the case of freshwater habitats (Filker et al. 2016). Moreover, it has been shown that detailed sampling of particular species can reveal previously hidden molecular variability. This happened recently both in the model algae species of the genus Klebsormidium, where 66% of rbcL genotypes from nearctic and palaearctic colonies were identified for the first time (Ryšánek et al. 2015), and in the ciliate Tetrahymena thermophila, which is restricted to east coast of the USA (Zufall et al. 2012). Similarly, in five Paramecium species (7 populations from the Qingdao neighborhood) as many as 90 COI haplotypes were recently identified (Zhao et al. 2013). Studies carried out recently (2015) and previously (1992, 1999) in ponds of the botanical garden in Kraków revealed two different P. biaurelia COI haplotypes (Pa2COI_02 and Pa2COI_03) (Przyboś et al. 2016). However, it is worth noting that in the case of the botanical gardens, the observed molecular variability could have been connected with new populations (not only microorganisms but also aquatic invertebrates) having been transported to the water bodies with tropical plants (Kolicka et al. 2015).
The present P. biaurelia study revealed that 6 of 11 COI haplotypes occurred only once in the analyzed dataset (N = 139), and three of them (Pa2COI_07, Pa2COI_08, Pa2COI_10) were obtained from Poland (Table S1). Due to the observed high incidence in the various ecosystems tested to date (Przyboś et al. 2011) and the ability to invade new habitats (Gill and Hairston 1972), perhaps more accurate sampling of small areas (i.e., one or several ponds) will reveal previously hidden genetic variation of the studied microorganism.
Generally, during the recent sampling (2015–2016) of 28 locations in southern Poland (374 sampling points), we confirmed that P. biaurelia is one of the most common species from the P. aurelia complex (24% among eight P. aurelia species identified) in the studied reservoirs. Interestingly, no new COI haplotypes were revealed, and haplotypes Pa2COI_02, Pa2COI_03 dominated within the studied area, which in turn is in concordance with worldwide P. biaurelia analysis (Figs. 5b and 6). Further to the above, in the Kraków area we detected the Pa2COI_04, Pa2COI_06 haplotypes. Therefore it may be concluded that every COI haplotype in the studied dataset (N = 139) that appeared in more than one location was observed in the Kraków area (Figs. 5b and 6, Table S1), confirming the global ubiquity of frequent haplotypes and local endemism of private haplotypes as in Ryšánek et al. (2015). On the other hand, the less frequent haplotypes in current dataset (Pa2COI_05, Pa2COI_09, Pa2COI_11) may turn out to be widespread in the Nearctic and Neotropical ecozones (Fig. 4) after careful sampling of these areas. The observed distinctiveness of P. biaurelia collected in both Americas (haplotypes Pa2COI_05, Pa2COI_09, Pa2COI_11) is in concordance with a previous analysis of genomic data (883 genes) of 11 strains that suggested a USA-Europe separation (Johri et al. 2017). However, our results, similarly to (Pawlowski et al. 2007), showed that despite isolation by distance, gene flow is still possible between populations from different ecozones.
Another finding of the present sampling of the Kraków area is that in most of the reservoirs in which P. biaurelia was detected, two different COI haplotypes were identified (Fig. 6, Table S2). The most plausible explanation of this finding is independent colonization of the reservoirs by different P. biaurelia populations, as was observed in Coleps sp. (Barth et al. 2008).
We are aware that the majority of the studied P. biaurelia COI variability is based on strains from the Palearctic ecozone. However, we expect that more accurate sampling of reservoirs in Australasia, the Southern part of the Ethiopian and Neotropical ecozones as well as the Nearctic ecozone where P. biaurelia is very common (Hairston 1958) will bring new data on the distribution of this microbial eukaryote.
References
Agatha, S., & Strüder-Kypke, M. C. (2014). What morphology and molecules tell us about the evolution of Oligotrichea (Alveolata, Ciliophora). Acta Protozoologica, 53, 77–99.
Aufderheide, K. J., Daggett, P.-M., & Nerad, T. A. (1983). Paramecium sonneborni n. sp., a new member of the Paramecium aurelia species complex. Journal of Protozoology, 30, 128–131.
Azovsky, A. I., Tikhonenkov, D. V., & Mazei, Y. A. (2016). An estimation of the global diversity and distribution of the smallest eukaryotes: biogeography of marine benthic heterotrophic flagellates. Protist, 167, 411–424.
Bandelt, H., Forster, P., & Röhl, A. (1999). Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution, 16(1), 37–48.
Barth, D., Krenek, S., Fokin, S. I., & Berendonk, T. U. (2006). Intraspecific genetic variation in Paramecium revealed by mitochondrial cytochrome c oxidase I sequences. Journal of Eukaryotic Microbiology, 53, 20–25.
Barth, D., Tischer, K., Berger, H., Schlegel, M., & Berendonk, T. U. (2008). High mitochondrial haplotype diversity of Coleps sp. (Ciliophora: Prostomatida). Environmental Microbiology, 10, 626–634.
Beale, G. H., & Preer, J. R., Jr. (2008). Paramecium. Genetics and epigenetics. Boca Raton: CRC Press, Taylor & Francis Group.
Benbouzid, H., Berrebbah, H., & Djebar, M. R. (2015). Toxicity of the chlorfenapyr: growth inhibition and induction of oxidative stress on a freshwater protozoan: Paramecium sp. Advances in Environmental Biology, 9(3), 281–285.
Boenigk, J., Ereshefsky, M., Hoef-Emden, K., Mallet, J., & Bass, D. (2012). Concepts in protistology: species definitions and boundaries. European Journal of Protistology, 48, 96–102.
Caron, D. A. (2013). Towards a molecular taxonomy for protists: benefits, risks, and applications in plankton ecology. Journal of Eukaryotic Microbiology, 60, 407–413.
Caron, D. A., Gast, R. J., Countwaym, P. D., & Heidelberg, K. B. (2009). Microbial eukaryote diversity and biography. Microbe, 4, 71–77.
Catania, F., Wűrmser, F., Potekhin, A. A., Przyboś, E., & Lynch, M. (2009). Genetic diversity in the Paramecium aurelia species complex. Molecular Biology and Evolution, 26, 421–431.
Chantangsi, C., Lynn, D. H., Brandl, M. T., Cole, J. C., Hetrick, N., & Ikonomi, P. (2007). Barcoding ciliates: a comprehensive study of 75 isolates of the genus Tetrahymena. International Journal of Systematic and Evolutionary Microbiology, 57, 2412–2425.
Coleman, A. W. (2005). Paramecium aurelia revisited. Journal of Eukaryotic Microbiology, 52, 68–77.
Darling, K. F., Kucera, M., & Wade, C. M. (2007). Global molecular phylogeography reveals persistent Arctic circumpolar isolation in a marine planktonic protist. Proceedings of the National Academy of Sciences of the United States of America, 104, 5002–5007.
Dijoux, L., Viard, F., & Payri, C. (2014). The more we search, the more we find: discovery of a new lineage and a new species complex in the genus Asparagopsi. PLoS One, 9, e103826.
Duff, R. J., Ball, H., & Lavrentyev, P. J. (2008). Application of combined morphological-molecular approaches to the identification of planktonic protists from environmental samples. Journal of Eukaryotic Microbiology, 55, 306–312.
Fenchel, T., & Finlay, B. J. (2004). The ubiquity of small species: patterns of local and global diversity. Bioscience, 54, 777–784.
Filker, S., Sommaruga, R., Vila, I., & Stoeck, T. (2016). Microbial eukaryote plankton communities of high-mountain lakes from three continents exhibit strong biogeographic patterns. Molecular Ecology, 25, 2286–2301.
Finlay, B. J. (1998). The global diversity of protozoa and other small species. International Journal for Parasitology, 28, 29–48.
Finlay, B. J. (2002). Global dispersal of free-living microbial eukaryote species. Environmental Microbiology, 296, 1061–1064.
Finlay, B. J., Esteban, G. F., Brown, S., Fenchel, T., & Hoef-Emden, K. (2006). Multiple cosmopolitan ecotypes within a microbial eukaryote morphospecies. Protist, 157, 377–390.
Foissner, W. (2006). Biogeography and dispersal of micro-organisms: a review emphasizing protists. Acta Protozoologica, 45, 111–136.
Foissner, W., Chao, A., & Katz, L. A. (2008). Diversity and geographic distribution of ciliates (Protista: Ciliophora). Biodiversity and Conservation, 17, 345–363.
Fokin, S. I. (2010/2011). Paramecium genus: biodiversity, some morphological features and the key to the main morphospecies discrimination. Protistology, 6, 227–235.
Fokin, S. I., & Chivilev, S. M. (2000). Paramecium. Morphometric analysis and taxonomy. Acta Protozoologica, 39, 1–14.
Gentekaki, E., & Lynn, D. H. (2009). High-level genetic diversity but no population structure inferred from nuclear and mitochondrial markers of the peritrichous ciliate Carchesium polypinum in the Grand River basin (North America). Applied and Environmental Microbiology, 75, 3187–3195.
Gentekaki, E., & Lynn, D. H. (2010). Evidence for cryptic speciation in Carchesium polypinum Linnaeus, 1758 (Ciliophora: Peritrichia) inferred from mitochondrial, nuclear, and morphological markers. Journal of Eukaryotic Microbiology, 57, 508–519.
Gill, E. D., & Hairston, N. G. (1972). The dynamics of natural population of Paramecium and the role of interspecific competition in community structure. Journal of Animal Ecology, 41, 81–96.
Greczek-Stachura, M., Potekhin, A., Przyboś, E., Rautian, M., Skoblo, I., & Tarcz, S. (2012). Identification of Paramecium bursaria syngens through molecular markers—comparative analysis of three loci in the nuclear and mitochondrial DNA. Protist, 163, 671–685.
Gutierrez, J. C., Martin-Gonzales, A., & Callejas, S. (1988). Nuclear changes, macronuclear chromatin reorganization and DNA modifications during ciliate encystment. European Journal of Protistology, 34, 97–103.
Hairston, N. G. (1958). Observations on the ecology of Paramecium, with comments on the species problem. Evolution, 12, 440–450.
Hairston, N. G., & Kellermann, S. L. (1965). Competition between varieties 2 and 3 of Paramecium aurelia: the influence of temperature in a food limited system. Ecology, 46, 134–139.
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98.
Hedenström, A. (2010). Extreme endurance migration: what is the limit to non-stop flight? PLoS Biology, 8(5), e1000362.
Heger, T. J., Edgcomb, V. P., Kim, E., Lukeš, J., Leander, B. S., & Yubuki, N. (2014). A resurgence in field research is essential to better understand the diversity, ecology, and evolution of microbial eukaryotes. Journal of Eukaryotic Microbiology, 61, 214–223.
Johri, P., Krenek, S., Marinov, G. K., Doak, T. G., Berendonk, T. U., & Lynch, M. (2017). Population genomics of paramecium species. Molecular Biology and Evolution, 34, 1194–1216.
Katz, L. A., & Turkewitz, A. P. (2013). Stalking the wild Tetrahymena. Molecular Ecology, 22, 912–914.
Katz, L. A., Deberardinis, J., Hall, M. S., Kovner, A. M., Dunthorn, M., & Muse, S. V. (2011). Heterogeneous rates of molecular evolution among cryptic species of the ciliate morphospecies Chilodonella uncinata. Molecular Biology and Evolution, 73, 266–272.
Kodama, Y., & Fujishima, M. (2014). Symbiotic Chlorella variabilis incubated under constant dark condition for 24 hours loses ability to avoid digestion by host lysosomal enzymes in digestive vacuoles of host ciliate Paramecium bursaria. FEMS Microbiology Ecology, 90, 946–955.
Kolicka, M., Dziuba, M. K., Zawierucha, K., Kuczyńska-Kippen, N., & Kotwicki, L. (2015). Palm house—biodiversity hot spot or risk of invasion? Aquatic invertebrates: the special case of Monogononta (Rotifera) under greenhouse conditions. Biologia, Section Zoology, 70, 94–103.
Kosakyan, A., Gomaa, F., Mitchell, E. A. D., Heger, T. J., & Lara, E. (2013). Using DNA-barcoding for sorting out protist species complexes: a case study of the Nebela tincta-collaris-bohemica group (Amoebozoa; Arcellinida, Hyalospheniidae). European Journal of Protistology, 49, 222–237.
Krenek, S., Berendonk, T. U., & Fokin, S. I. (2015). New Paramecium (Ciliophora, Oligohymenophorea) congeners shape our view on its biodiversity. Organism Diversity and Evolution, 15, 215–233.
Lahr, D. J. G., Grant, J. R., & Katz, L. A. (2013). Multigene phylogenetic reconstruction of the Tubulinea (Amoebozoa) corroborates four of the six major lineages, while additionally revealing that shell composition does not predict phylogeny in the Arcellinida. Protist, 164, 323–339.
Landis, W. G. (1988). The interplay among ecology, breeding systems, and genetics in the Paramecium aurelia and Paramecium bursaria complexes. In J. O. Corliss & D. J. Patterson (Eds.), Progress in Protistology (Vol. 1, pp. 287–307). Bristol: Biopress.
Lara, E., Mataloni, G., Seppey, C. V. M., Singer, D., Schiaffino, M. R. & Izaguire, I. (2015). Freshwater plankton community ecology under high throughput sequencing perspective. VII European Congress of Protistology, the International Society of Protistologists, 5–10 September 2015, Seville. Abstracts, p. 145.
Leigh, J. W., & Bryant, D. (2015). PopART: full-feature software for haplotype network construction. Methods in Ecology and Evolution, 6(9), 1110–1116.
Librado, P., & Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25, 1451–1452.
Mahe, F., Mayor, J., Bunge, J., Chi, J., Siemensmeyer, T., Stoeck, T., Wahl, B., Paprotka, T., Filker, S., & Dunthorn, M. (2015). Comparing high-throughput platforms for sequencing the V4 region of SSU-rDNA in environmental microbial eukaryotic diversity surveys. Journal of Eukaryotic Microbiology, 62, 338–345.
McGrath, C. L., Gout, J. F., Johri, P., Doak, T. G., & Lynch, M. (2014). Differential retention and divergent resolution of duplicate genes following whole-genome duplication. Genome Research, 24(10), 1665–1675.
McManus, G. B., Xu, D., Costas, B. A., Katz, L., & A. (2010). Genetic identities of cryptic species in the Strombidium stylifer/apolatum/oculatum cluster, including a description of Strombidium rassoulzadegani n. sp. Journal of Eukaryotic Microbiology, 57, 369–378.
Messenger, L. A., Llewellyn, M. S., Bhattacharyya, T., Franzen, O., Lewis, M. D., Ramirez, J. D., Carrasco, H. J., Andersson, B., & Miles, M. A. (2012). Multiple mitochondrial introgression events and heteroplasmy in Trypanosoma cruzi revealed by maxicircle MLST and next generation sequencing. PLoS Neglected Tropical Diseases, 6, e1584.
Nanney, D. L., & McCoy, J. W. (1976). Characterization of the species of the Tetrahymena pyriformis complex. Transactions of the American Microscopical Society, 95, 664–682.
Page, R. D. M. (1996). TreeView: an application to display phylogenetic tress on personal computers. Bioinformatics, 12, 357–358.
Pawlowski, J. (2015). Protist metabarcoding and its applications. VII European Congress of Protistology in partnership with the International Society of Protitologists, 5–10 September 2015, Seville. Abstracts, p. 60.
Pawlowski, J., Fahrni, J., Lecroq, B., Longet, D., Cornelius, N., Excoffier, L., Cedhagen, T., & Gooday, A. J. (2007). Bipolar gene flow in deep-sea benthic foraminifera. Molecular Ecology, 16, 4089–4096.
Pawlowski, J., Audic, S., Adl, S., Bass, D., Belbahri, L., et al. (2012). CBOL protist working group: barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms. PLoS Biology, 10(11), e1001419.
Picariello, T., Valentine, M. S., Yano, J., & Van Houten, J. (2014). Reduction of meckelin leads to general loss of cilia, ciliary microtubule misalignment and distorted cell surface organization. Cilia, 3, 2.
Przyboś, E., & Prajer, M. (2015). New stands of species of the Paramecium aurelia complex: is the occurrence of particular species limited by temperature barriers? Folia Biologica (Kraków), 63, 215–220.
Przyboś, E., & Surmacz, M. (2010). New, world-wide data on the distribution of species of the Paramecium aurelia complex (Ciliophora, Protozoa). Folia biologica (Kraków,), 58, 185–188.
Przyboś, E., & Tarcz, S. (2016). Paramecium jenningsi complex: existence of three cryptic species confirmed by multi-locus analysis and strain crosses. Systematics and Biodiversity, 14, 140–154.
Przyboś, E., Tarcz, S., Greczek-Stachura, M., & Surmacz, M. (2011). Seasonal and spatial variability of species occurrence of the Paramecium aurelia complex in a single natural pond (Protista, Ciliophora). Hydrobiologia, 663, 233–244.
Przyboś, E., Tarcz, S., & Dusi, E. (2013). New Paramecium quadecaurelia strains (P. aurelia spp. complex, Ciliophora) identified by molecular markers (rDNA and mtDNA). European Journal of Protistology, 49, 477–486.
Przyboś, E., Tarcz, S., Rautian, M., & Lebedeva, N. (2014). The first European stand of Paramecium sonneborni (P. aurelia complex), a species known only from the northern America (USA, Texas). Insights from molecular, genetic, and cytological studies. European Journal of Protistology, 50, 236–247.
Przyboś, E., Tarcz, S., Rautian, M., & Sawka, N. (2015). Delimiting species boundaries within a paraphyletic species complex: insights from morphological, genetic, and molecular data on Paramecium sonneborni (Paramecium aurelia species complex, Ciliphora, Protozoa). Protist, 166, 438–456.
Przyboś, E., Surmacz, M., & Tarcz, S. (2016). Seasonal variability of the Paramecium aurelia complex in the botanical garden of the Jagiellonian University, Kraków—in the light of species composition and COI haplotype variation. Folia Biologica (Kraków), 64, 253–265.
Razowski, J. (1996). Dictionary of insect morphology (in Polish) (p. 140). Warszawa-Kraków: Wydawnictwo Naukowe PWN.
Ronquist, F., & Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics, 19, 1572–1574.
Ryšánek, D., Hrčková, K., & Skaloud, P. (2015). Global ubiquity and local endemism of free-living terrestrial protists: phylogeographic assessment of the streptophyte alga Klebsormidium. Environmental Microbiology, 17, 689–698.
Snoke, M. S., Berendonk, T. U., Barth, D., & Lynch, M. (2006). Large global effective population sizes in Paramecium. Molecular Biology and Evolution, 23, 2474–2479.
Sogin, M. L., Morrison, H. G., Huber, J. A., Welch, D. M., Huse, S. M., Neal, P. R., Arrieta, J. M., & Herndl, G. J. (2006). Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proceedings of the National Academy of Sciences of the United States of America, 103, 12115–12120.
Sonneborn, T. M. (1950). Methods in the general biology and genetics of Paramecium aurelia. Journal of Experimental Zoology, 113, 87–148.
Sonneborn, T. M. (1970). Methods in Paramecium research. In D. M. Prescott (Ed.), Methods cell Physiol (Vol. 4, pp. 242–339). New York: Academic Press.
Sonneborn, T. M. (1974). Paramecium aurelia. In R. C. King (Ed.), Handbook of genetics (Vol. 2, pp. 469–594). New York: Plenum Press.
Sonneborn, T. M. (1975). The Paramecium aurelia complex of fourteen sibling species. Transactions of the American Microscopical Society, 94, 155–178.
Stoeck, T., Breiner, H.-W., Filker, S., Ostermaier, V., Kammerlander, B., & Sonntag, B. (2014a). A morphogenetic survey on ciliate plankton from a mountain lake pinpoints the necessity of lineage-specific barcode markers in microbial ecology. Environmental Microbiology, 16, 430–444.
Stoeck, T., Przyboś, E., & Dunthorn, M. (2014b). The D1-D2 region of the large ribosomal DNA as barcode for ciliates. Molecular Ecology, 14, 458–468.
Strüder-Kypke, M. C., & Lynn, D. L. (2010). Comparative analysis of the mitochondrial cytochrome c oxidase subunit I (COI) gene in ciliates (Alveolata, Ciliophpra) and evaluation of its suitability as a biodiversity marker. Systematics and Biodiversity, 8, 131–148.
Sung, W., Tucker, A. E., Doak, T. G., Choi, E., Thomas, W. K., & Lynch, M. (2012). Extraordinary genome stability in the ciliate Paramecium tetraurelia. Proceedings of the National Academy of Sciences of the United States of America, 109, 19339–19344.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., & Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30, 2725–2729.
Tarcz, S., Pothekin, A., Rautian, M., & Przyboś, E. (2012). Variation in ribosomal and mitochondrial DNA sequences demonstrates the existence of intraspecific groups in Paramecium multimicronucleatum (Ciliophora, Oligohymenophorea). Molecular Phylogenetics and Evolution, 63, 500–509.
Tarcz, S., Przyboś, E., & Surmacz, M. (2013). An assessment of haplotype variation in ribosomal and mitochondrial DNA fragments suggests incomplete lineage sorting in some species of the Paramecium aurelia complex (Ciliophora, Protozoa). Molecular Phylogenetics and Evolution, 67, 255–265.
Tarcz, S., Rautian, M., Potekhin, A., Sawka, N., Beliavskaya, A., Kiselev, A., Nekrasova, I., & Przyboś, E. (2014). Paramecium putrinum (Ciliophora, Protozoa): the first insight into the variation of two DNA fragments—molecular support for the existence of cryptic species. Molecular Phylogenetics and Evolution, 73, 140–145.
Thomsen, P. F., & Willerslev, E. (2015). Environmental DNA—an emerging tool in conservation for monitoring past and present biodiversity. Biological Conservation, 183, 4–18.
Thomson, J. M., Gaucher, E. A., Burgan, M. F., De Kee, D. W., Li, T., Aris, J. P., et al. (2005). Resurrecting ancestral alcohol dehydrogenases from yeast. Nature Genetics, 37, 630–635.
Tomazic, M. L., Poklepovich, T. J., Nudel, C. B., & Nusblat, A. D. (2014). Incomplete sterols and hopanoids pathways in ciliates: gene loss and acquisition during evolution as a source of biosynthetic genes. Molecular Phylogenetics and Evolution, 74, 122–134.
Van de Peer, Y., Maere, S., & Meyer, A. (2009). The evolutionary significance of ancient genome duplications. Nature Reviews Genetics, 10, 725–732.
Watts, P. C., Martin, L. E., Kimmance, S. E., Montagnes, D. J. S., & Lowe, C. D. (2011). The distribution of Oxyrrhis marina—a global wanderer or poorly characterised endemic? Journal of Plankton Research, 33, 579–589.
Weisse, T. (2006). Freshwater ciliates as ecophysiological model organisms—lessons from Daphnia, major achievements, and future perspectives. Archiv für Hydrobiologie, 167, 371–402.
West, C., James, S. A., Davey, R. P., Dicks, J., & Roberts, I. N. (2014). Ribosomal DNA sequence heterogeneity reflects intraspecies phylogenies and predicts genome structure in two contrasting yeast species. Systematic Biology, 63, 543–554.
White, T. J., Bruns, T., Lee, S., & Taylor, J. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In M. A. Innis, D. H. Gelfand, J. J. Sninsky, & T. J. White (Eds.), PCR protocols: a guide to methods and applications (pp. 315–322). San Diego: Academic Press.
Zhao, Y., Gentekaki, E., Yi, Z., & Lin, X. (2013). Genetic differentiation of the mitochondrial cytochrome oxidase c subunit I gene in genus Paramecium (Protista, Ciliophora). PLoS One, 8(10), e77044.
Zufall, R. A., Dimond, K. L., & Doerder, F. P. (2012). Restricted distribution and limited gene flow in the model ciliate Tetrahymena thermophila. Molecular Ecology, 22, 1081–1109.
Acknowledgements
Molecular analyses of P. biaurelia strains from the ISEA PAS collection were supported by grant Iuventus Plus No. IP2011 055971 awarded to ST by the Ministry of Science and Higher Education, Warsaw, Poland.
Field research, species designation and molecular studies of strains collected in 2015–2016 were supported by a grant awarded to ST by the National Science Centre, Poland, no. 2014/15/B/NZ8/00258.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tarcz, S., Sawka-Gądek, N. & Przyboś, E. Worldwide sampling reveals low genetic variability in populations of the freshwater ciliate Paramecium biaurelia (P. aurelia species complex, Ciliophora, Protozoa). Org Divers Evol 18, 39–50 (2018). https://doi.org/10.1007/s13127-017-0357-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13127-017-0357-z