
Abstract
We examined the neuroprotective efficacy of docosahexaenoic acid (DHA), an omega-3 essential fatty acid family member, in acute ischemic stroke; studied the therapeutic window; and investigated whether DHA administration after an ischemic stroke is able to salvage the penumbra. In each series described below, SD rats underwent 2 h of middle cerebral artery occlusion (MCAo). In series 1, DHA or saline was administered i.v. at 3, 4, 5, or 6 h after stroke. In series 2, MRI was conducted on days 1, 3 and 7. In series 3, DHA or saline was administered at 3 h, and lipidomic analysis was conducted on day 3. Treatment with DHA significantly improved behavior and reduced total infarct volume by a mean of 40% when administered at 3 h, by 66% at 4 h, and by 59% at 5 h. Total lesion volumes computed from T2-weighted images were reduced in the DHA group at all time points. Lipidomic analysis showed that DHA treatment potentiates neuroprotectin D1 (NPD1) synthesis in the penumbra 3 days after MCAo. DHA administration provides neurobehavioral recovery, reduces brain infarction and edema, and activates NPD1 synthesis in the penumbra when administered up to 5 h after focal cerebral ischemia in rats.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Each year, over 15 million people worldwide suffer a stroke, many of which are ischemic [1]. Thrombolysis is the only treatment for ischemic stroke, but due to its narrow therapeutic window and complexity of administration, only 3–5% of patients benefit from this therapy [2]. Focal cerebral ischemia produces a brain lesion composed of an irreversibly injured core and a peripheral zone (penumbra), where tissue is damaged but potentially salvageable [3]. The penumbra has a limited life span and appears to undergo irreversible damage within a few hours unless reperfusion is initiated and/or neuroprotective therapy is administered [4].
Docosahexaenoic acid (DHA; 22:6, n-3) is an essential omega-3 fatty acid concentrated in membrane phospholipids of the central nervous system [5]. DHA is a natural element of our diet and is found in cold water fatty fish, including salmon, tuna, mackerel, sardines, shellfish, and herring. DHA has been shown to be involved in brain and retinal development, aging, memory formation, synaptic membrane function, photoreceptor biogenesis and function, and neuroprotection [6–8]. The recent identification of the protective docosanoid neuroprotectin D1 (NPD1; 10R,17S-dihydroxy-docosa-4Z,7Z,11E,13E,15Z,19Z-hexaenoic acid) in brain ischemia reperfusion [9], as well as in neural cells exposed to oxidative stress [10–12], has uncovered a key survival-signaling event leading to neuroprotection. NPD1 is a pleiotropic modulator of inflammation resolution [13]. Moreover several reports show that DHA is protective in ischemia [3, 7] and in spinal cord injury [14]. However, the mechanism/s and bioactive mediator/s involved - and particularly whether or not these mechanisms/mediators target the salvageable area of the stroke penumbra - have not yet been defined. In addition, the effect of systemic administration of DHA on the ischemic brain penumbra has not been explored.
Recently, we have shown that DHA therapy in low and medium doses improves neurological and histological outcome following focal cerebral ischemia [15]. The present study investigated the therapeutic window of DHA treatment in focal cerebral ischemia in rats. In addition, we studied whether administration of DHA after an ischemic stroke is able to salvage the penumbra. We used non-invasive magnetic resonance imaging (MRI) and LC-MS/MS-mediator lipidomic analysis in conjunction with behavioral, histological, and immunostaining methods to expand our understanding of this novel therapeutic approach. No prior studies have used MRI to evaluate the temporal effects of DHA treatment on brain ischemia.
Materials and Methods
Surgical Preparation
All studies were approved by the Institutional Animal Care and Use Committees of the Louisiana State University Health Sciences Center and Loma Linda University. Male Sprague–Dawley rats (Charles River Lab., Wilmington, MA) weighing 279–340 g were fasted overnight but allowed free access to water. Anesthesia was induced with 3.5% isoflurane in a mixture of 70% nitrous oxide and 30% oxygen. All rats were orally intubated and mechanically ventilated. Temperature probes were inserted into the rectum and the left temporalis muscles to maintain temperatures at 36–37°C during surgical procedures. The right femoral artery and vein were catheterized for blood sampling for arterial gases, pH, plasma glucose, and drug infusion.
Transient Middle Cerebral Artery Occlusion
Rats underwent 2 h of right middle cerebral artery occlusion (MCAo) by an intraluminal filament, coated with poly-L-lysine as previously described [16]. After 2 h of MCAo, rats were reanesthetized with the same anesthetic combination and intraluminal sutures were carefully removed.
Behavioral Tests
Behavioral tests were performed by an observer blinded to the treatment groups at 60 min (during MCAo) and then on days 1, 2, 3, and 7 after MCAo. The battery consisted of the postural reflex test and the forelimb-placing test to visual, tactile, and proprioceptive stimuli. Neurological function was graded on a scale of 0–12 (normal score = 0; maximum score = 12), as previously described [16].
Treatment
In each series described below, rats underwent 2 h of MCAo. DHA (5 mg/kg) was dissolved in saline and administered intravenously at a constant rate over 3 min using an infusion pump according the study protocol. Vehicle-treated rats received an intravenous infusion of a comparable volume of 0.9% sodium chloride.
Study Protocols
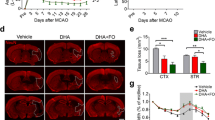
In series 1 (therapeutic window study), DHA was administered i.v. at either 3, 4, 5 or 6 h after onset of stroke (n = 6–7 per group). Vehicle group received saline at 3 h after onset of stroke. Behavioral testing was conducted on days 1, 2, 3, and 7 followed by histopathology on day 7. In series 2 (MRI study), DHA (n = 5) or saline (n = 4) was administered at 3 h after onset of stroke and MRI was conducted on days 1, 3, and 7. In series 3 (lipidomic study), DHA or saline was administered at 3 h (n = 5–6 per group) and lipidomic analysis was conducted on day 3.
Magnetic Resonance Imaging
Rats were anesthetized with isoflurane and imaged on a Bruker Avance 4.7T MRI (Bruker Biospin, Billerica MA) as previously described [17]. A thermostat-controlled heated water cushion maintained body temperature at 36–37°C and respiration was monitored using a pneumatic cushion (Biopac Model 150, Goleta CA).
Image Analysis
T2 and apparent diffusion coefficient (ADC) maps were computed from T2-weighted imaging (T2WI) and diffusion-weighted imaging (DWI), respectively, as previously described [17]. Three-dimensional images were obtained to delineate the volumetric development of ischemic injury. Analysis included extraction of lesion, non-lesioned brain, and total brain volumes. Region of interest (ROI) was drawn semi-automatically on each slice and manually checked. From these ROIs, the lesion volume was calculated over all slices (volume of lesion/total brain volume) and expressed as a percentage. Penumbral tissues were estimated by evaluating both T2 and DWI lesion volumes and comparing these to the final imaging time point, as we have previously described [17].
Histopathology and Immunostaining
Animals were perfused with 4% paraformaldehyde on day 7, and brains were removed and embedded in a gelatin matrix using MultiBrain™ Technology (NeuroScience Associates, Knoxville, TN) as previously described [18]. Coronal sections were stained with thionine (Nissl), and immunohistochemical procedures were performed on the adjacent sections. To quantitate infarct volume, histological sections were digitized at nine standardized coronal levels and analyzed (MCID™ Core imaging software, Linton, Cambridge, UK) as previously described [16]. Infarct volume was calculated as the integrated product of cross-sectional area and intersection distance and corrected for brain swelling. The following antibodies were used: glial fibrillary acid protein (GFAP, Santa Cruz, SDS Biosciences, Sweden) to label reactive astrocytes, Cd68/ED-1 (Serotec, Raleigh, NC) for activated microglia/microphages, and neuron-specific nuclear protein (NeuN, Chemicon/Millipore, Billerica, MA). The number of GFAP, ED-1, and NeuN-positive cells were counted (Zeiss Axio Imager 4.6.3) in the cortex and striatum at the level of the central lesion (bregma level −0.3 mm; magnification ×40). Data were expressed as numbers of positive cells per high-power microscopic field. Brain slices were imaged on a Zeiss LSM-510 Meta laser confocal microscope with a ×10 objective (Zeiss Plan-NEOFLUAR 10×/0.3). Fluorophore visualization (excitation/emission capture) was achieved as follows: GFAP, DyLight 488 (488 nm/505–530 nm, green); and ED1, DyLight 594 (594 nm/603–636 nm, red). The image resolution was set to 2.26 μm/pixel and the cubic voxel dimension was 129.5 μm. Computer-generated MosaiX-processed images of Nissl, GFAP, ED-1, and GFAP/ED-1 double staining from saline and DHA rats were generated.
Lipidomic Analysis
Rats were perfused with cold ice saline at 3 days after stroke. For each animal, the brain was removed and blocked into two segments that included the bregma levels +2.7 and −0.3 mm. Next, coronal blocks were divided into right and left hemispheres and cut into two regions (one for the cortex and one for the striatum). Neuroprotectin D1 (NPD1), the DHA-derived stereoselective lipid mediator, and 17-HDHA (a marker of 17H(p)DHA, the short lived-NPD1 precursor) were isolated from the penumbra as previously described [9] using a liquid chromatography-photodiode array ultraviolet detector-LTQ linear ion trap mass spectrometer (LC-UV-MS/MS; Thermo, Waltham, MA) equipped with an LC column (150 mm × 2 mm × 5 μm, LUNA C18, Phenomenex, Torrance, CA).
Statistical Analysis
Data are presented as mean values ± SEM. Repeated-measures analysis of variance (ANOVA) followed by Bonferroni procedures to correct for multiple comparisons were used for intergroup comparisons of neurobehavioral scores over time and infarct areas across coronal levels. Two-tailed Student's t tests were used for two-group comparisons. Differences at p < 0.05 were considered statistically significant.
Results
Rectal and cranial (temporalis muscle) temperatures, arterial blood gases, plasma glucose, and hematocrit showed no significant differences among groups. There were no adverse behavioral side effects observed after DHA administration to rats in the present study.
Series 1: Therapeutic Window Study
Neurological deficits were reduced by DHA, even when treatment was initiated as late as 5 h after MCAo onset (Fig. 1a): day 1 (DHA: 3 h, 7.7 ± 0.2; 4 h, 6.9 ± 0.7; 5 h, 7.2 ± 0.5; and 6 h, 8.3 ± 0.2 vs. Saline: 9.8 ± 0.2; p < 0.01), day 2 (DHA: 3 h, 7.7 ± 0.2; 5 h, 6.5 ± 0.8; and 6 h, 7.8 ± 0.3 vs. Saline: 9.8 ± 0.2; p < 0.01), day 3 (DHA: 3 h, 7.1 ± 0.6; 5 h, 6.0 ± 1.0; and 6 h, 8.2 ± 0.4 vs. Saline: 9.7 ± 0.2; p < 0.003) and day 7 (DHA: 3 h, 6.0 ± 0.6; 4 h, 5.7 ± 1.0; 5 h, 5.5 ± 0.9 vs. Saline: 9.0 ± 0.3; p < 0.0004). DHA-mediated protection was extensive in the frontal–parietal cortex (tissue salvage, 49% (p < 0.02), 77% (p < 0.002), and 71% (p < 0.004), respectively for 3 h, 4 h and 5 h; Fig. 1b) and across multiple coronal levels. DHA administration did not affect the subcortical infarct, except when it was infused at 4 h (p < 0.01; Fig. 1c). Total infarct volume, corrected for brain swelling, was reduced by 40% (p < 0.02) when DHA was administered at 3 h, by 66% (p < 0.001) at 4 h, and by 59% (p < 0.003) at 5 h (Fig. 1d). The brains of saline-treated rats exhibited a consistent pannecrotic lesion involving both cortical and subcortical (mainly striatal) regions of the right hemisphere, characterized microscopically by destruction of neuronal, glial, and vascular elements. In contrast, infarct size was dramatically reduced in rats treated with DHA at 3, 4, and 5 h after onset of ischemia, but not in 6-h group (Fig. 1e).

Therapeutic window study: a Neurological score (normal score = 0; maximum score = 12) was improved after DHA administration when administered 3, 4, and 5 h after onset of stroke. Cortical (b), subcortical (c), and total corrected infarct volumes (d) on day 7. DHA reduced cortical and total infarct volumes when administered 3, 4, and 5 h after stroke. Data are means ± SEM. Asterisk, significantly different from saline (p < 0.05; repeated-measures ANOVA followed by Bonferroni tests). e Computer-generated MosaiX-processed images of Nissl stained paraffin-embedded brain sections from rats treated with saline or DHA at 3, 4, 5, and 6 h after the onset of ischemia. Saline-treated rat shows large cortical and subcortical infarction. In contrast, rats treated with DHA at 3, 4, and 5 h show less extensive damage, mostly in the subcortical area. DHA-treated rat at 6 h shows infarct involving cortical and subcortical regions. f Computer-generated MosaiX-processed images of GFAP (green), ED-1 (red), and GFAP/ED-1 double staining (overlay) on day 7 after 2 h of MCAo at a magnification ×10. Treatment with DHA or saline was given at 3 h after onset of stroke
Saline-treated rats showed extensive neuronal loss, GFAP-positive reactive astrocytes outlining the lesion territory, and massive ED-1-positive microglia/macrophage infiltration (Fig. 1f, top). In contrast, DHA-attenuated damage as well as decreased ED-1-positive microglia/macrophages and increased GFAP-positive reactive astrocytes (Fig. 1f, bottom). Figure 2 presents GFAP-, ED-1-, and NeuN-positive cell counts. DHA treatment significantly decreased ED-1 (p < 0.01) and increased NeuN (p < 0.01) and GFAP-positive (p < 0.005) cell counts.

a Coronal brain diagram showing locations of regions for cell counts in cortex (1, 2, and 3), and striatum (S). b–d Number of GFAP-positive astrocytes, ED-1 positive microglia cells, and NeuN-positive neurons on day 7 after 2 h of MCAo. DHA or saline was given at 3 h after onset of stroke. DHA treatment decreased ED-1, increased NeuN- and GFAP-positive cell counts. Data are mean ± SEM. Asterisk, significantly different from saline (p < 0.05; repeated-measures ANOVA followed by Bonferroni tests)
Series 2: MRI Study
When DHA was given 3 h after MCAo onset, the neurological scores were significantly improved on days 3 (p < 0.01) and 7 (p < 0.02; Fig. 3a). Also, initial smaller infarcts were only localized to the striatum on day 1 and were indistinguishable from normal tissues by day 7 (Fig. 3b). T2WI revealed large cortical and striatial infarctions in saline rats that resolved over 7 days (Fig. 3b). DHA yielded reduced T2 values (decreased edema) within the lesion (p < 0.002), and there were no differences in T2 values for the contralateral tissues (Fig. 3c). There was a difference in the ADC within the infarcted lesion when comparing saline and DHA rats at days 1, 3, and 7 after MCAo. The ipsilateral ADC value was increased at days 1 and 7 (p < 0.001). However, the ADC in DHA was 21% lower at day 3. These dynamic processes reflect repair mechanisms within the DHA-rat lesion. Interestingly, ADC was increased in the DHA contralateral lesion. Three-dimensional infarct volumes were computed from T2WI (presented in Fig. 3d).

MRI study: a DHA improved neurological scores on days 3 and 7. b T2 hyperintensites were observed in the cortex and striatum of saline-treated rats, consistent with edema formation. In contrast, DHA reduced edema with little T2 hyperintensities at day 3 and not at all at day 7, resulting in an intact corpus callosum at day 7. c DHA reduced T2 values within the lesion computed from T2WI on days 1, 3, and 7, consistent with decreased edema. Data are means ± SEM. Asterisk, significantly different from saline-treated group (p < 0.05; repeated-measures ANOVA followed by Bonferroni tests). d 3D infarct volumes were computed from T2WI on days 1, 3, and 7 after MCAo. Saline-treated rats showed large cortical and subcortical infarct volumes that slowly decreased over the course of 7 days. By contrast, infarct volume was dramatically reduced in rats treated with DHA and was mostly localized in the subcortical areas. 3D reconstructions are from the same animal in each group over the 7-day time course
Series 3: Lipidomic Study
Because DHA is the precursor of NPD1 [11], we asked if NPD1 synthesis occurs in the penumbra. Figure 4a–c shows that DHA treatment potentiates NPD1 synthesis in the penumbra 3 days after MCAo. We ascertained NPD1 synthesis in the penumbra by isolating and characterizing 17-HDHA. 17-HDHA reflects the presence of the unstable 17S-HpDHA. Since 17-HDHA and NPD1 were enhanced in the penumbra by DHA, we concluded that active NPD1 synthesis occurs under these conditions. Figure 4d illustrates the release of DHA from the membrane followed by the biosynthesis of NPD1.

The characterization and quantification of 17-HDHA and NPD1 in the ipsilateral penumbra 3 days after MCAo. a–b The fragmentation pattern is depicted and c the quantification is presented. The increased content of 17-HDHA and of NPD1 in the ipsilateral penumbra in animals injected with DHA is consistent with the activation of the biosynthesis of NPD1. d Enzyme-mediated oxygenation of DHA for the biosynthesis of NPD1. Phospholipase A2 releases DHA from the second C position of phospholipids during brain ischemia-reperfusion. 15-Lipoxygenase-1 catalyzes the synthesis of 17S-H(p)DHA, which in turn is converted to a 16(17)-epoxide and then is enzymatically hydrolyzed to NPD1
Discussion
In this study, we have demonstrated that systemic administration of the DHA provides neurobehavioral recovery, reduces brain infarction and brain edema, activates neuroprotectin D1 synthesis in the penumbra, and promotes cell survival when administered up to 5 h after focal cerebral ischemia in rats.
DHA is an essential omega-3-fatty acid and is vital for the proper brain functioning. It is also necessary for the development of the nervous system, including vision. Moreover, omega-3 fatty acids are part of a healthy diet that helps lower the risk of heart disease. DHA belongs to the omega-3 essential fatty acid family. As a consequence, we depend on the dietary supply of DHA or of its precursor, linonenic acid (18:3, n-3). DHA has potent anti-inflammatory effects [5, 13, 19]. Since inflammation is at the root of many chronic diseases, DHA treatment has been widely demonstrated to have beneficial effects in patients with coronary heart disease, asthma, rheumatoid arthritis, osteoporosis, sepsis, cancer, dry eye disease, and age-related macular degeneration [19].
Cerebral ischemia is characterized by a rapid accumulation (within minutes) of free fatty acids, including arachidonic acid (AA; 20:4 n-6) and DHA (22:6, n-3), due to increases in intracellular calcium and activation of phospholipases [20]. This free pool of AA and DHA is then converted via active enzymatic processes as well as free radical-mediated lipid peroxidation into a cascade of activated pro- and anti-inflammatory mediators, the makeup of which ultimately drives the cell toward survival or programmed cell death. Specifically, after hydrolysis, free (unesterified) DHA is converted to form the stereospecific mediator NPD1 in a series of reactions beginning with lipoxygenation by 15-lipoxygenase-1 followed by epoxidation and hydrolysis reactions (Fig. 4) [11]. NPD1 acts against apoptosis, promotes cell survival, inhibits brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression, promotes neurogenesis, attenuates edema formation, and reduces stroke volume 48 h after MCAo onset [9, 21, 22]. Recently, we showed that administration of human serum albumin complexed with DHA results in increased NPD1 production in the ipsilateral hemisphere, but not the contralateral hemisphere. These results indicate that NPD1 synthesis requires available DHA as well as harmful noxious stimuli causing cellular damage [22].
Due to the challenges of treating patients in a timely manner, an extension of the therapeutic window is necessary to allow for broader appeal to clinicians treating ischemic stroke victims. We asked if DHA could prolong the therapeutic window in a setting of focal cerebral ischemia. In our previous DHA dose–response study, we showed that a low dose of DHA (3.5–7.0 mg/kg) has the greatest effect on reducing infarct volume when administered 3 h after MCAo onset [15]. We selected a dose in the middle of the effective dose range (5 mg/kg) for our therapeutic window study, which proved to be effective even with a delay of treatment up to 5 h. Treatment with DHA significantly improved the neurological score on days 1, 2, 3, and 7 compared to vehicle rats even when treatment was initiated as late as 5 h after onset of ischemia. The effect of DHA was also demonstrated clearly in the cortical (or penumbral region) of the infarct by a significant 49% reduction of cortical infarct volume when administered at 3 h, by 77% at 4 h, and by 71% at 5 h as compared to vehicle-treated rats. This effect was associated with a reduction in edema (T2 normalization). MRI studies showed that DHA treatment clearly reduced the level of edema and significantly reduced total lesion volumes compared to the saline treatment on days 1, 3, and 7.
Ischemic stroke intervention requires mixed cellular protection of the penumbra. The term neuroprotection is typically used to describe the protection of neurons from the neurotoxic environment that occur during ischemia. However, the complex processes that occur after stroke require the targeting of multiple factors and cells, including glia, vascular, and inflammatory cells. The histologic distribution of molecular markers in the infarcted regions of DHA-treated brains differed from saline-treated brains in that DHA brains showed attenuated cellular death of both astrocytes and neurons and fewer activated microglia, as compared to saline-treated brains within the infracted region. These results indicate that treatment with DHA protects not only the neurons but astrocytes, which are critical for the maintenance and protection of neurons via the secretion of growth factors and other neurotrophic mediators. Furthermore, DHA attenuated the activation of microglia. Microglia are activated by cellular damage and typically initiate an inflammatory response that results in apoptosis and debris removal. This effect demonstrates DHA's anti-inflammatory properties during cellular stress.
The next question we asked is whether DHA rescues the ischemic penumbra. The ischemic penumbra (defined as local cerebral blood flow (LCBF) of 20–40% of control) forms an irregular rim around the ischemic core and tends to be greatest in frontal and occipital cortex [23]. Although reduced LCBF is a major factor responsible for necrotic injury, other factors, including lipid peroxidation, inflammatory responses, and development of brain edema, may also contribute to either the severity or progression of penumbral injury [24]. Stroke patients (44%) may have penumbral tissue even after 18 h [25]. DHA has been shown to be neuroprotective and is the precursor of NPD1, a novel lipid mediator with powerful anti-apoptotic and pro-resolution properties during oxidative stress and cellular damage.
Our lipidomic study demonstrated that DHA treatment activates NPD1 synthesis in the salvageable penumbral region of the infarct, which typically succumbs several days after the initial ischemic insult due to the subsequent widespread inflammatory response elicited by ischemia, rather than the actual ischemic episode. DHA is converted through 15-lipoxygenase-1 into NPD1, a potent mediator that evokes counteracting cell-protective, anti-inflammatory, pro-survival repair signaling, including the induction of anti-apoptotic Bcl-2 proteins and inhibition of pro-apoptotic Bcl-2 proteins. Thus, NPD1 triggers activation of signaling pathway/s that modulate/s pro-apoptotic signals, promoting cell survival as well as inflammatory resolution [26]. Thus DHA/NPD1 is a mechanism for protection in the ischemic brain penumbra, amenable to further translational approaches.
Conclusion
We have shown that DHA administration provides neurobehavioral recovery, reduces brain infarction and brain edema, activates neuroprotectin D1 synthesis in the penumbra, and promotes cell survival when administered up to 5 h after focal cerebral ischemia in rats.
References
Feigin VL, Lawes CM, Bennett DA, Barker-Collo SL, Parag V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: a systematic review. Lancet Neurol. 2009;8:355–69.
Lyden P. Thrombolytic therapy for acute stroke—not a moment to lose. N Engl J Med. 2008;359:1393–5.
Moskowitz MA, Lo EH, ladecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–98.
Lo EH. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14:497–500.
Bazan NG. Cell survival matters: docosahexaenoic acid signaling, neuroprotection and photoreceptors. Trends Neurosci. 2006;29:263–71.
Bazan NG. Homeostatic regulation of photoreceptor cell integrity: significance of the potent mediator neuroprotectin D1 biosynthesized from docosahexaenoic acid: the Proctor Lecture. Investig Ophthalmol Vis Sci. 2007;48:4866–81.
Bazan NG. Neuroprotectin D1-mediated anti-inflammatory and survival signaling in stroke, retinal degeneration, and Alzheimer's disease. J Lipid Res. 2009;50:S400–5.
Akbar M, Calderon F, Wen Z, Kim HY. Docosahexaenoic acid: a positive modulator of Akt signaling in neuronal survival. Proc Natl Acad Sci USA. 2005;102:10858–63.
Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronert K, Musto A, et al. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem. 2003;278:43807–17.
Bazan NG. Neuroprotectin D1 (NPD1): a DHA-derived mediator that protects brain and retina against cell injury-induced oxidative stress. Brain Pathol. 2005;15:159–66.
Mukherjee PK, Marcheselli VL, Serhan CN, Bazan NG. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc Natl Acad Sci USA. 2004;101:8491–6.
Lukiw WJ, Cui JG, Marcheselli VL, Bodker M, Botkjaer A, Gotlinger K, Serhan CN, Bazan NG. A role for docosahexaenoic acid-derived neuroprotectin D1 in neural cell survival and Alzheimer disease. J Clin Invest. 2005;115:2774–83.
Serhan Cn, Yacoubian S, Yang R. Anti-inflammatory and proresolving lipid mediators. Annu Rev Pathol. 2008;3:279–312.
Ward RE, Huang W, Curran OE, Priestly JV, Michael-Titus AT. Docosahexaenoic acid prevents white matter damage following spinal cord injury. J Neurotrauma. 2010;27:1–12.
Belayev L, Khoutorova L, Atkins KD, Bazan NG. Robust docosahexaenoic acid-mediated neuroprotection in a rat model of transient, focal cerebral ischemia. Stroke. 2009;40:3121–6.
Belayev L, Alonso OF, Busto L, Zhao W, Ginsberg MD. Middle cerebral artery occlusion in the rat by intraluminal suture. Neurological and pathological evaluation of an improved model. Stroke. 1996;27:1616–22.
Obenaus A, Robbins M, Blanco G, Galloway NR, Snissarenko E, Gillard E, et al. Multi-modal magnetic resonance imaging alterations in two rat models of mild neurotrauma. J Neurotrauma. 2007;24:1147–60.
Thompson SN, Gibson TR, Thompson BM, Deng Y, Hall ED. Relationship of calpain-mediated proteolysis to the expression of axonal and synaptic plasticity markers following traumatic brain injury in mice. Exp Neurol. 2006;201:253–65.
Simopoulos AP. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med. 2008;233:674–88.
Muralikrishna AR, Hatcher JF. Phospholipase A2, reactive oxygen species, and lipid peroxidation in cerebral ischemia. Free Radic Biol Med. 2006;40:376–87.
Rodriguez de Turco EB, Belayev L, Liu Y, Busto R, Parkins N, Bazan NG, et al. Systemic fatty acid responses to transient focal cerebral ischemia: influence of neuroprotectant therapy with human albumin. J Neurochem. 2002;83:515–24.
Belayev L, Marcheselli VL, Khoutorova L, Rodriguez de Turco EB, Busto R, Ginsberg MD, et al. Docosahexaenoic acid complexed to albumin elicits high-grade ischemic neuroprotection. Stroke. 2005;36:118–23.
Hossmann KA, Traystman RJ. Chapter 4: cerebral blood flow and the ischemic penumbra. Handb Clin Neurol. 2008;92:67–92.
Liu S, Levine SR, Winn HR. Targeting ischemic penumbra. Part 1. J Exp Stroke Transl Med. 2010;3:47–55.
Ebinger M, De Silva DA, Christensen S, Parsons MW, Markus R, Donnan GA, et al. Imaging the penumbra—strategies to detect tissue at risk after ischemic stroke. J Clin Neurosci. 2009;16:178–87.
Bazan NG, Calandria JM, Serhan CN. Rescue and repair during photoreceptor cell renewal mediated by docosahexaenoic acid-derived neuroprotectin D1. J Lipid Res. 2010;51:2018–31.
Acknowledgments
We thank Pedro Hayes, Dr. Elena Titova, and Eric Knott for technical assistance; Dr. Thompson for statistical analysis; and Neuroscience Associates, Inc. for histology service.
Sources of Funding
This investigation was supported by NIH, NINDS Grant R01 NS046741 (NGB) and NIH, NCCAM Grant RC2 AT005909 (NGB). Tiffany N. Eady is a recipient of the Ruth L. Kirschstein National Research Service Awards for Individual Predoctoral MD/PhD and Other Dual Doctoral Degree Fellows (NIH, NIA Grant F30 AG032841).
Disclosures
The authors declare no conflicts of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Belayev, L., Khoutorova, L., Atkins, K.D. et al. Docosahexaenoic Acid Therapy of Experimental Ischemic Stroke. Transl. Stroke Res. 2, 33–41 (2011). https://doi.org/10.1007/s12975-010-0046-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12975-010-0046-0