
Abstract
Understanding the pathogenicity of amyloid-beta (Aβ) peptides constitutes a major goal in research on Alzheimer’s disease (AD). One hypothesis entails that Aβ peptides induce uncontrolled, neurotoxic ion flux through cellular membranes. The exact biophysical mechanism of this ion flux is, however, a subject of an ongoing controversy which has attenuated progress toward understanding the importance of Aβ-induced ion flux in AD. The work presented here addresses two prevalent controversies regarding the nature of transmembrane ion flux induced by Αβ peptides. First, the results clarify that Αβ can induce stepwise ion flux across planar lipid bilayers as opposed to a gradual increase in transmembrane current; they show that the previously reported gradual thinning of membranes with concomitant increase in transmembrane current arises from residues of the solvent hexafluoroisopropanol, which is commonly used for the preparation of amyloid samples. Second, the results provide additional evidence suggesting that Aβ peptides can induce ion channel-like ion flux in cellular membranes that is independent from the postulated ability of Αβ to modulate intrinsic cellular ion channels or transporter proteins.
Similar content being viewed by others


Introduction
One of the current hypotheses for the pathology of Alzheimer’s disease (AD) proposes that amyloid-beta (Aβ) peptides induce aberrant, neurotoxic ion flux across cellular membranes (Koh et al. 1990; Arispe et al. 1993a, b; Kawahara et al. 2000; Kourie et al. 2001; Michikawa et al. 2001; Arispe and Doh 2002; Kurganov et al. 2004; Barghorn et al. 2005; Quist et al. 2005; Baglioni et al. 2006; Devi et al. 2006; Morris and Juranka 2007; Palop et al. 2007; Simakova and Arispe 2007; Bezprozvanny and Mattson 2008). The resulting difficulty of neurons to regulate their intracellular concentration of ions, in particular calcium ions, has been associated with cell death (Khachaturian 1987; Mattson et al. 1992) and may thus contribute to cognitive impairment typical for AD. Understanding the underlying mechanisms that cause Aβ-induced ion flux may hence be crucial for developing new strategies to reduce the toxicity of Aβ in AD (Inbar and Yang 2006; Inbar et al. 2006; Stains et al. 2007).
Increasing evidence shows that exposure of cells to Aβ leads to elevated concentrations of intracellular calcium ions (Khachaturian 1987, 1989; Mattson et al. 1992; Arispe et al. 1993b; Pollard et al. 1993; Kawahara et al. 2000; Mattson and Chan 2003; Demuro et al. 2005; Tu et al. 2006; Stutzmann 2007; Cheung et al. 2008; Nimmrich et al. 2008); the predominant cellular mechanism for this disruption of Ca2+ homeostasis remains, however, a focus of intense studies. The four mechanisms that have been proposed are (1) Aβ assembles into oligomers to form Aβ ion channels in cell membranes (Arispe et al. 1993a, b, 1996, 2007, 2008; Pollard et al. 1993; Durell et al. 1994; Mirzabekov et al. 1994; Kawahara et al. 1996, 1997; Rhee et al. 1998; Hirakura et al. 1999a, b; Lin et al. 1999, 2001; Zhu et al. 2000; Kourie et al. 2001, 2002; Kagan et al. 2002, 2004; Lin and Kagan 2002; Bahadi et al. 2003; Arispe 2004; Micelli et al. 2004; Mobley et al. 2004; Quist et al. 2005; Lal et al. 2007; Jang et al. 2008), (2) Aβ interacts with membranes in such a way that it generally lowers the dielectric barrier for ions to cross the membrane (e.g. by thinning the membrane) (Kayed et al. 2004; Sokolov et al. 2004, 2006), (3) Aβ modulates, directly or indirectly, the activity of existing ion channel proteins or receptors (Yankner et al. 1990a, b; Joslin et al. 1991; Mark et al. 1995; Querfurth et al. 1997; Ueda et al. 1997; Ye et al. 1997; Morimoto et al. 1998; Wang et al. 2000; Rovira et al. 2002; Dougherty et al. 2003; Novarino et al. 2004; Verdier et al. 2004; Deshpande et al. 2006; Stutzmann et al. 2006, 2007; Cheung et al. 2008; Dreses-Werringloer et al. 2008; Fedrizzi et al. 2008; Lopez et al. 2008; Nimmrich et al. 2008), or (4) Aβ modulates, directly or indirectly, the activity of cellular ion transporters and pumps (Lam et al. 2001; Green et al. 2008). Experimental evidence for each of these mechanisms has been provided and it is possible that several mechanisms act together to disrupt Ca2+ homeostasis.
The work presented here examined the first three mechanisms. It employed electrophysiological techniques to measure ion flux by current recordings across bilayer lipid membranes (using BLM recordings) or across cellular plasma membranes (using whole-cell patch clamp recordings). This approach excluded the study of ion flux by transporter proteins or ion pumps because the ion flux generated by these proteins is too slow to be detectable by current recordings (Gennis 1989). Instead, we asked if Aβ can form ion pores in artificial lipid bilayers, in membranes of a human neuronal cell line (SH-SY5Y), and in primary neurons from transgenic mice that generate Aβ intrinsically by expressing human amyloid precursor protein (hAPP) and human presenilin 1 (hPS-1).
Materials and Methods
Chemicals
We purchased Aβ(1-40) and Aβ(1-42) from Biopeptides Inc. and from Bachem Inc., hexafluoroisopropanol (HFIP) (GC grade) from Fluka, and the following seven lipids from Avanti Polar Lipids: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-l-serine] (POPS), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dioleoyl-sn-glycero-3-phosphoserine (DOPS), 1,2-diphytanoyl-sn-glycero-3-phosphocholine (DiPhyPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (POPG). All other chemicals were purchased from Sigma–Aldrich; SH-SY5Y cells were obtained from ATCC.
Preparation of Solutions of Aβ by Purging HFIP with Nitrogen Gas
We prepared samples of Aβ by following exactly a previously reported protocol (Kayed et al. 2003, 2004; Sokolov et al. 2004, 2006; Demuro et al. 2005). Briefly, we dissolved 1 mg of Aβ peptide in 400 μl of HFIP and incubated this solution for 15 min. We mixed 100 μl of the resulting, clear Aβ solution in HFIP with 900 μl of deionized water in a siliconized 1.5 ml microtube. After incubation at room temperature for 15 min, we centrifuged the samples for 15 min at 14,000g and transferred the supernatant fraction (900–950 μl) to a new siliconized tube. To purge HFIP from these samples, we bubbled nitrogen gas for 10 min through these samples, while adjusting the gas flow to ~30 ml min−1, which was the maximum flow rate that did not result in excessive foaming and splashing of the sample. For purging times longer than 10 min, we had to reduce the flow rate of the N2 gas to 2–3 ml min−1 in order to avoid significant loss of material due to slow but steady accumulation and expulsion of foam from the sample. We analyzed aliquots of these samples for residual HFIP concentration by gas chromatography-mass spectrometry (GC-MS) and tested their ability to induce transmembrane ion flux in planar lipid bilayers and cell membranes.
Quantification of HFIP Concentrations by GC-MS Analysis
This analysis was carried out by injecting a sample volume of 2 μl into a Finnigan Trace GC-MS with the following settings: run in electron impact ionization mode using a 30 m long, 0.25 mm ID Supelco SLB-5 column, with a film thickness of 0.25 μm. Helium was used as carrier gas at a constant flow rate of 1.0 ml min−1; the injector temperature was 200°C; the run was performed in split mode with a ratio of 1:100; the interface temperature was 250°C, and the ion source block temperature was 220°C. The temperature of the GC oven was held at 40°C for 2 min and then increased to 150°C at a rate of 40°C min−1. The MS ion source filament was set to an emission current of 150 μA at 70 eV. HFIP standards and calibration curves were generated each day. Samples with unknown HFIP concentrations were measured using an external calibration curve; no internal standards were used. The quantification was based on the area under the GC peak in the chromatogram. Data were acquired and processed using Excalibur software, version 1.1.
Formation of Planar Lipid Bilayers
The two reported original procedures (Arispe et al. 1993b; Kayed et al. 2003) that are at the basis of the controversy regarding the nature of Aβ-induced ion flux across artificial lipid membranes employed different methods to generate planar lipid bilayers (Arispe et al. 1993b; Kayed et al. 2004). We, therefore, prepared planar lipid bilayers [also called black lipid membranes or bilayer lipid membranes (BLMs)] either by the so-called “folding technique” (Montal and Mueller 1972; Mayer et al. 2003; Capone et al. 2007; Blake et al. 2008), which employs apposition of lipid monolayers over a pore with a diameter of ~150 μm in a Teflon film or by the so-called “painting technique” (Mueller et al. 1962; Capone et al. 2007), which applies a solution of lipids in decane or heptane over a pore with a diameter of ~250 μm in a Delrin septum.
For the folded bilayers (Figs. 1 and 2b columns DOPC + DOPE and DiPhyPC), we pretreated the Teflon film with 2 μl of 5% (v/v) squalene in pentane and then formed the bilayers using 5 μl of either pure DiPhyPC or a 1:1 (w/w) mixture of a lipid solution in pentane that contained DOPC and DOPE. The total lipid concentration of these solutions in pentane was 20–25 mg ml−1 (Capone et al. 2007; Mayer et al. 2008). The electrolyte contained 70 mM KCl buffered with 10 mM HEPES, pH 7.4.

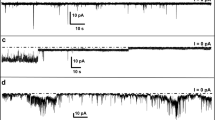
Comparison of ion flux across lipid bilayers upon exposure to samples of amyloid-β peptides (Aβ), which were prepared by two different protocols (final Aβ concentration 1 μM). a Gradual increase in transmembrane ion flux upon addition of an Aβ sample that was prepared by purging the solvent hexafluoroisopropanol (HFIP) for 10 min (Kayed et al. 2004; Sokolov et al. 2004). The “noisy” part of the current trace is a result of stirring. b Gradual increase in transmembrane ion flux following the exact same protocol as in (a) but in the absence of Aβ. Inset shows gradual increase in ion flux across the membranes of SH-SY5Y cells upon addition of HFIP (final concentration ~6 mM). c Transmembrane ion flux upon addition of an Aβ sample prepared according to previous reports (Kayed et al. 2004; Sokolov et al. 2004) but with 18 h instead of 10 min of purging to remove HFIP. d Stepwise fluctuations in transmembrane ion flux after incorporating a proteoliposome preparation of Aβ that was prepared without using HFIP (Arispe et al. 1993b). The applied voltage was −150 mV in all recordings. Membranes were prepared from DOPC:DOPE lipids at a 1:1 (w/w) ratio by the “folding method” over a pore with a diameter of 150 μm in a Teflon film that was pretreated with 5% squalene in pentane

Transmembrane ion flux induced by samples containing well-defined concentrations of HFIP as well as removal of HFIP from Aβ samples as a function of the time of purging with nitrogen gas. a Current versus voltage curves of transmembrane ion flux as a function of HFIP concentration (Aβ was not present in these experiments). The slopes of the linear best fits represent the conductance (in Siemens, S = Ω−1) of ions across a planar lipid bilayer made from a 1:1 (w/w) mixture of DOPS and POPE. The absolute magnitudes of these conductance values varied between repeated experiments and between different membrane compositions; however, the relative dose-dependent effect of increasing concentration of HFIP on the conductance remained the same. b Transmembrane conductance induced by 1 mM HFIP (equivalent to 0.01% v/v) in planar lipid bilayers of various lipid compositions and by ~6 mM HFIP in cell membranes. All indicated lipid mixtures were prepared in a 1:1 (w/w) ratio. All experiments were repeated at least three times; the error bars represent the standard deviation from the mean conductance. c Removal of HFIP from samples containing 100 μl of HFIP and 900 μl of water (concentration of HFIP ~1 M) as a function of purging time with nitrogen gas. The data indicated by red circles correspond to a flow rate of N2 gas of ~30 ml min−1. The data indicated by black squares correspond to a flow rate of 2–3 ml min−1. All points represent average values (±standard deviation) from 2 to 6 repeated experiments. Note, for purging times longer than 10 min with a flow rate of ~30 ml min−1, we observed that significant amounts of the foam that formed during purging escaped the microtube, thus rendering this fast flow rate impractical for purging times longer than 10 min
For the painted bilayers, we used a bilayer cup (Warner Instruments, Delrin perfusion cup, volume 1 ml) and the following four lipid mixtures at a 1:1 (w/w) ratio: POPG:POPE in heptane, DOPS:POPE in heptane, DOPC:DOPE in heptane, or POPE:POPS in decane. The total lipid concentration of these solutions in heptane or decane was 10–20 mg ml−1. In order to promote fusion of Aβ proteoliposomes into bilayers, we used an ionic gradient formed by filling the cis side (the side of proteoliposome addition) with 370 mM KCl and the trans side of the bilayer setup with 70 mM KCl. For these experiments, we prepared bilayers using a 1:1 (w/w) mixture of DOPC and DOPE (Fig. 1d) or POPC and POPE (Fig. 4a–c); the proteoliposomes containing Aβ were prepared as described previously using either a 1:1 (w/w) mixture of DOPC and DOPE lipids (Fig. 1d) or pure POPS lipids (Fig. 4a, b and Fig. 5a) (Arispe et al. 1993b).
Incorporation of Aβ into Lipid Membranes
In order to repeat the procedure for preparing Aβ samples that was previously described to lead to membrane thinning of planar lipid bilayers (Kayed et al. 2004; Sokolov et al. 2004, 2006; Demuro et al. 2005), we prepared Aβ samples by using the solvent HFIP exactly as described (Kayed et al. 2003) and added the resulting solutions to both compartments of the bilayer chamber. We diluted these samples more than 50-fold such that the final concentration of Aβ was 0.5–2.0 μM in each chamber of the bilayer setup.
In order to repeat the alternative Aβ procedure, which was previously described to lead to stepwise ion flux across planar lipid bilayers (Arispe et al. 1993b), we added 10–20 μl of the Aβ proteoliposome solution (this solution contained Aβ at a concentration of 0.4–0.8 mg ml−1) to the cis compartment (volume 1 ml) and stirred for 5–10 min.
Planar lipid bilayer experiments with each lipid composition were performed at least 3 times, more typically 5–7 times.
Current Recordings
Before carrying out bilayer recordings, we verified that both the painted and the folded bilayers were stable for several minutes (while applying a voltage of at least ±100 mV) and that the membrane capacitances were above 90 pF. When both criteria were fulfilled, we added Aβ (0.5–4.0 μM final concentration of Aβ) or HFIP (0.5–7.0 mM final concentration of HFIP without any Aβ present) to both chambers of the bilayer setup.
We performed all recordings in “voltage clamp mode” using Ag/AgCl electrodes. We used a filter-cutoff frequency of 2 kHz, and a sampling frequency of 15 kHz for all bilayer recordings. For representation in figures, we filtered the current traces with a digital Gaussian low-pass filter with a cutoff frequency of 100 Hz.
For whole-cell patch clamp recordings, we used the following three solutions: (1) intracellular solution containing 75 mM KCl, 10 mM NaCl, 70 mM KF, 2 mM MgCl2, 10 mM EGTA, and 10 mM HEPES buffer (pH 7.2); (2) extracellular solution containing 160 mM NaCl, 4.5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM D-glucose, and 10 mM HEPES buffer (pH 7.4); and (3) a “seal enhancer” solution containing 80 mM NaCl, 3 mM KCl, 10 mM MgCl2, 35 mM CaCl2 and 10 mM HEPES/NaOH (pH 7.4), which was added to the extracellular compartment to promote the formation of seals between the cells and the glass chip with electrical resistances higher than 1 GΩ.
In order to introduce Aβ into membranes of SH-SY5Y cells, we first established whole-cell recording conditions followed by adding ~5 μl of extracellular solution containing Aβ to a volume of ~20 μl of extracellular solution (this Aβ solution was prepared without the use of HFIP by dissolving Aβ in extracellular solution and stirring this solution for ~14 h at room temperature prior to use). The final concentration of Aβ in the extracellular solution during the recording was ~10 μM.
In order to measure the effect of Aβ and HFIP samples on SH-SY5Y cells or on primary neurons, we used a semi-automated, chip-based electrophysiology instrument (Port-a-patch, Nanion Inc.) that allowed for recordings with seal resistances of 1–6 GΩ (Schmidt et al. 2000; Bruggemann et al. 2003; Estes et al. 2008). We held the voltage at −80 mV in the intracellular compartment versus ground in the extracellular compartment to measure Aβ-induced ion flux. At this potential, SH-SY5Y cells did not show significant intrinsic ion channel activity. Samples containing Aβ or HFIP were only added after confirming the stability of the recording (i.e., after observing a quiescent baseline for 3–5 min). For all whole-cell recordings, we used a filter cutoff frequency of 1 kHz.
In order to determine the effect of Zn2+ ions or the peptide NA7 on Aβ-induced transmembrane ion flux, we added 1–10 mM (final concentration) of ZnCl2 or Zn(NO3)2 or 100–140 μM (final concentration) of the NA7 peptide to the bilayer chambers or to the extracellular solutions.
Preparation of Primary Neurons
Wild-type female mice (C57BL/6J) were crossed with double transgenic hAPPswe/hPS-1∆E9 male mice, obtained from Jackson Laboratory (Bar Harbor, Maine) to generate wild-type, and hAPPswe/hPS-1 transgenic embryos. Primary cortical neuron cultures were prepared from the brains of E16 embryos. Briefly, the cerebral cortices were dissected in calcium-free and magnesium-free Hank’s balanced salt solution and incubated with a 0.125% trypsin solution for 10 min at 37°C. The trypsin was inactivated with Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, and the cortical tissue was further dissociated by serial trituration using a Pasteur pipette. The resulting cell suspensions were diluted in neurobasal medium supplemented with B27 supplements (Gibco BRL, Grand Island, New York), and plated onto poly-d-lysine-coated Petri dishes. Neurons were maintained at 37°C in a 5% CO2 atmosphere for 4–7 days, before the experiments (Saluja et al. 2006). We carried out the planar patch clamp experiments (on the Port-a-patch instrument) between day 4 and day 7 of culture. The neurons were detached using a protocol previously described and validated by Lecoeur et al. (2004) with minor modifications; these modifications entailed (1) reducing the incubation time with trypsin-EDTA from 15 to 14 min at 37°C and (2) dissociating cell aggregates by increasing the number of successively aspirating and dispensing the cell suspension to 7 times with a 1 mL pipette tip and to 15 times with a 200 μl pipette tip.
Cytotoxicity Assay of HFIP with SH-SY5Y Cells
We cultured SH-SY5Y human neuroblastoma cells in DMEM/F-12 (1:1) medium with 10% fetal bovine serum, 4 mM glutamax, penicillin (100 units ml−1) and streptomycin (100 μg ml−1) in 5% CO2 at 37°C. We plated cells in 96-well plates overnight starting with 50,000 cells well−1. After overnight incubation at 37°C, we exchanged the media with OptiMEM, and treated cells for 24 h. We determined the cell viability using an MTT toxicology kit (Tox-1, from Sigma) according to the instructions from the supplier.
Results and Discussion
Recently, three articles highlighted an ongoing controversy with regard to the exact biophysical characteristics of Aβ-induced transmembrane ion flux across artificial lipid membranes (Eliezer 2006; Fagan et al. 2006; Marx 2007). These articles debate two mechanistic hypotheses. On the one hand, the “Aβ ion channel hypothesis” suggests that Aβ assembles into pore-like structures in lipid membranes, leading to stepwise (or spike-like) fluctuations of transmembrane current that is typical for ion channels (Arispe et al. 1993a, b, 1996, 2007, 2008; Durell et al. 1994; Mirzabekov et al. 1994; Kawahara et al. 1996, 1997; Rhee et al. 1998; Hirakura et al. 1999a, b; Lin et al. 1999, 2001; Bhatia et al. 2000; Zhu et al. 2000; Kourie et al. 2001; Kagan et al. 2002, 2004; Kourie et al. 2002; Lin and Kagan 2002; Bahadi et al. 2003; Arispe 2004; Micelli et al. 2004; Quist et al. 2005; Lal et al. 2007; Jang et al. 2008). On the other hand, the “Aβ membrane thinning hypothesis” postulates a generalized and gradually increasing ion flux as a result of Aβ-induced reduction of the dielectric barrier of membranes, for instance, by thinning of membranes (Kayed et al. 2004; Sokolov et al. 2004, 2006).
Here, we examined, in detail, the two pivotal protocols for measuring Aβ-induced conductance through artificial lipid bilayers to resolve this controversy. The protocol that leads to ion flux by gradual membrane thinning involves solubilizing Aβ(1-40) or Aβ(1-42) in the solvent hexafluoroisopropanol (HFIP), followed by dilution in water, and purging of HFIP by a stream of nitrogen gas (Kayed et al. 2003, 2004; Sokolov et al. 2004, 2006; Demuro et al. 2005; Valincius et al. 2008). Figure 1a shows the gradual increase in ion flux through a planar lipid bilayer upon exposure of membranes to these Aβ samples. Figure 1a is consistent with previous reports that used the same HFIP-based experimental procedure (Kayed et al. 2004; Sokolov et al. 2004, 2006). Figure 1b, however, shows a similar gradual increase in conductance following the exact same protocol but in the absence of Aβ. This control experiment thus shows that Aβ was not required for the observed gradual increase in transmembrane conductance but that residues of HFIP alone could be responsible for the observed ion flux.Footnote 1
In order to investigate whether this gradual increase in conductance could be reproduced by exposure of membranes to defined concentrations of HFIP (in the absence of Aβ), we examined the effect of HFIP on cell membranes (Fig. 1b inset) and on planar lipid bilayers of various lipid compositions (Fig. 2a, b). We found that, in all of these membranes, solutions containing 1–6 mM (corresponding to 0.01–0.06% v/v) concentrations of HFIP induced a gradual increase in ion flux similar to the results shown in Fig. 1b and similar to results described previously (Kayed et al. 2004; Sokolov et al. 2004, 2006).Footnote 2 Since HFIP induced a significant, gradual increase in transmembrane ion flux in all tested membranes, we investigated its toxicity on SH-SY5Y cells using the MTT viability assay. We found that HFIP was toxic to SH-SY5Y cells in a dose-dependent manner: a concentration of HFIP of ~30 mM (~0.3% v/v) reduced the viability of SH-SY5Y cells to 50% (Fig. 3). At HFIP concentrations above ~60 mM (~0.6% v/v), we observed a 90% reduction in viability of these cells.Footnote 3 Based on these results, we emphasize that it is critically important to remove HFIP completely before attempting to investigate the effect of Aβ, or other samples prepared with HFIP, on cytotoxicity or on bilayer membranes.

Viability of human SH-SY5Y neuroblastoma cells as a function of overnight exposure to various concentrations of HFIP. Data are represented relative to control cells that were treated the same way but without HFIP (corresponding to 100% viability). The solid curve represents a best fit to a first order exponential decay function of the form: Viability (%) = 100% × e−([HFIP] in mM/(41.2 ± 1.2) mM), N = 13, R 2 = 0.91
In order to examine the contribution of HFIP to the reported transmembrane ion flux across lipid bilayers in detail, we determined the residual concentration of HFIP after purging Aβ samples (Fig. 2c). In the previously described protocol of preparing Aβ samples by purging of HFIP (Kayed et al. 2003, 2004; Sokolov et al. 2004, 2006; Demuro et al. 2005; Valincius et al. 2008), the Αβ samples were subjected to a gentle stream of N2 gas for 5–10 min. We found by gas chromatography and mass spectrometry (GC-MS) analysis of Αβ samples prepared in the same fashion that the residual concentrations of HFIP exceeded 200 mM in the Aβ samples (Fig. 2c) even after purging for 30 min at a flow rate of 2–3 ml min−1 or after purging for 10 min at a fast flow rate of ~30 ml min−1 (purging times longer than 10 min at this fast flow rate were not practical since they led to loss of Aβ by excessive foam formation). Addition of these Aβ samples to planar lipid bilayers or to cell membranes led to a gradual increase in transmembrane ion flux that was similar to the one shown in Fig. 1b (the final concentration of HFIP in the bilayer chamber ranged from 5 to 20 mM). These results suggest that the reported gradual increase in conductance (Kayed et al. 2004; Sokolov et al. 2004, 2006) was due to incomplete removal of residual HFIP in these Aβ samples.
In contrast, when we purged HFIP from Aβ samples for 18 h, we found that (1) the residual concentration of HFIP in the Aβ samples was consistently below 10 mM and (2) that addition of these Aβ samples to membranes (which resulted in a final concentration of HFIP below 0.2 mM) did not result in a gradual increase in ion flux. Instead, we observed in ~75% of these experiments a stepwise ion flux as reported previously by several research groups (Fig. 1c) (Arispe et al. 1993a, b, 1996, 2007, 2008; Mirzabekov et al. 1994; Kawahara et al. 1996; Rhee et al. 1998; Hirakura et al. 1999a, b; Lin et al. 1999, 2001; Bhatia et al. 2000; Kourie et al. 2001; Kagan et al. 2002, 2004; Kourie et al. 2002; Lin and Kagan 2002; Bahadi et al. 2003; Micelli et al. 2004; Quist et al. 2005).Footnote 4 These ion channel-like current fluctuations were reminiscent of the originally reported Aβ-induced transmembrane ion flux that led to the Aβ ion channel hypothesis in AD (Arispe et al. 1993b; Pollard et al. 1993). For comparison, Fig. 1d shows such stepwise current fluctuations through planar lipid bilayers using the original protocol for preparation of Aβ samples (Arispe et al. 1993b) that did not employ HFIP (or that used a protocol for preparation of Aβ samples, which removed HFIP completely by lyophilization for 2 days prior to dissolving Aβ in aqueous solution).
The results presented here, along with evidence from other groups (Arispe et al. 1993a, b, 1996, 2007, 2008; Mirzabekov et al. 1994; Kawahara et al. 1996, 1997; Rhee et al. 1998; Hirakura et al. 1999a, b; Lin et al. 1999, 2001; Bhatia et al. 2000; Zhu et al. 2000; Kourie et al. 2001, 2002; Kagan et al. 2002, 2004; Lin and Kagan 2002; Bahadi et al. 2003; Arispe 2004; Micelli et al. 2004; Quist et al. 2005; Lal et al. 2007; Jang et al. 2008), clearly demonstrate that Aβ peptides are indeed capable of forming ion pores in artificial membranes. They also show that stepwise ion flux is the predominant mode of ion flux across artificial bilayers provided that the samples of Aβ are free of organic solvent. If gradual thinning would be the predominant mechanism of Aβ-induced ion flux, then we would have expected to detect a gradual shift of the recorded current baseline under the same conditions where we observe measurable stepwise ion flux. Since all solvent-free Aβ preparations that we tested did not lead to gradual shifts in current baseline, we conclude that the postulated effect of Aβ to lower the dielectric barrier by gradual thinning of membranes—if existent—is small compared to the ion flux induced by Aβ pores. We also note that, to the best of our knowledge, all reports that observed gradual membrane thinning employed Aβ samples that were prepared with HFIP in combination with relatively short durations of purging (<30 min) by a gentle gas stream (Kayed et al. 2004; Sokolov et al. 2004, 2006). We, therefore, attribute the reported gradual increase in ion flux to residual HFIP in Aβ samples. This conclusion resolves the controversy with regard to Aβ-induced ion flux across artificial lipid bilayers; the results presented here confirm that Aβ can form ion pores in bilayer membranes.
After establishing the stepwise nature of Aβ-induced ion flux through planar lipid bilayers, two questions remain: (1) Can Aβ form ion pores in cellular membranes? (2) What is the predominant mechanism of Aβ-induced ion flux across these cellular membranes? These questions are difficult to answer definitively because of the many direct and indirect pathways that can lead to ion flux in living cells. Here, we provide additional evidence in support of the hypothesis that Aβ is capable of forming independent pores in cellular membranes (Kawahara et al. 1997).
In order to compare ion flux induced by Αβ in artificial lipid bilayers with Αβ-induced ion flux across cellular membranes, we performed whole-cell patch clamp recordings on a human neuroblastoma cell line (SH-SY5Y cells) that we exposed to an exogenously introduced Αβ preparation (prepared after completely removing HFIP by lyophylization for 2 days). In addition, we carried out whole-cell recordings from transgenic, primary neurons that produced Αβ endogenously. In both cell types, we performed all recordings close to the resting membrane potential of the cells (i.e., at a constant applied voltage of −80 mV) in order to minimize the activity of voltage-gated ion channel proteins that are naturally (endogenously) expressed in cells.
Figure 4 shows stepwise (or spike-like) Αβ-induced transmembrane ion flux in both cell types, regardless of whether Αβ was added exogenously to SH-SY5Y cells (Fig. 4d) or produced endogenously by transgenic primary neurons (Fig. 4g). It also demonstrates that both cell types showed no (or very rare) current fluctuations in the absence of Aβ (Fig. 4f, i). We performed 16 recordings of Aβ-induced, channel-like ion flux on SH-SY5Y cells and observed ion flux in 12 experiments (~75%), while addition of Aβ induced no changes in ion flux in the remaining 4 experiments. Typically, Aβ-induced current spikes occurred 5–15 min after addition of Aβ, but in at least one recording the activity started within less than 30 s, suggesting that Aβ can induce a measurable current flux quite rapidly.

Comparison of Aβ-induced transmembrane ion flux across planar lipid bilayers, across plasma membranes of a neuroblastoma cell line, and across plasma membranes of primary cortical neurons from mouse embryos. a Aβ-induced ion flux across a planar lipid bilayer (1:1 w/w POPC:POPE). Aβ was introduced by proteoliposome fusion (final concentration of Aβ = 1 μM). The applied potential was +30 mV or −30 mV as indicated. b Effect of the addition (arrow) of ~3 mM Zn2+ on the same membrane as in (a). The small vertical arrows indicate the beginning and end of mixing. c Control experiment with a membrane of the same composition as in (a) and (b) but without Aβ present. d Whole-cell planar patch clamp recording of Aβ-induced transmembrane currents across the plasma membrane of human neuroblastoma cells (SH-SY5Y). Aβ was added to a final concentration of ~10 μM to the extracellular solution. The applied potential was −80 mV. e Recording from the same cell and under the same conditions as in (d) but after addition of ~2 mM Zn2+ to the extracellular solution. f Control experiment of a whole-cell recording from a SH-SY5Y cell in the absence of Aβ in the extracellular solution. g Whole-cell planar patch clamp recording from a primary cortical neuron of a transgenic mouse embryo that produced human Aβ endogenously. The applied potential was −80 mV. h Effect of the addition of Zn2+ on a recording from the same cell as in (g). i Control experiment with a wild-type cortical neuron that did not produce Aβ
In the case of the primary transgenic neurons, we recorded from eight cells that were extracted from three different transgenic embryos; seven of these recordings (~88%) showed a significant increase in the frequency of current spikes (Fig. 4g) compared to control recordings from wild-type neurons (Fig. 4i). Only 1 of 11 control recordings from primary, wild-type neurons showed a frequency in current fluctuations similar to transgenic neurons; the other 10 control cells displayed a significantly lower frequency of current spikes than transgenic neurons.
Since Zn2+ ions have previously been reported to block Αβ-induced stepwise ion flux (Arispe et al. 1996; Kawahara et al. 1997; Rhee et al. 1998; Lin et al. 1999; Kourie et al. 2001; Lin and Kagan 2002; Kagan et al. 2004), we added Zn2+ to determine its effect on the observed current fluctuations. In both, the SH-SY5Y and the transgenic cells, addition of Zn2+ reduced Αβ-induced ion flux significantly (Fig. 4e, h). The inhibitory effect of Zn2+ in these two cell types was similar to the Zn2+-induced inhibition of Aβ-induced, stepwise ion flux through artificial lipid bilayers (Fig. 4b).
As mentioned above, control experiments, in which Αβ was neither introduced exogenously nor produced endogenously, did not result in significant stepwise or spike-like ion flux under the same experimental conditions (Fig. 4f, i), indicating that Aβ was required to induce measurable ion flux across cellular membranes. These results hence pose the question: Did this measurable ion flux result from activation of endogenous, cellular ion channel proteins or was it a consequence of self-assembly of Aβ to pores in cellular membranes?
A comparison of Aβ-induced ion flux (Fig. 5) revealed that the average duration of stepwise (spike-like) current fluctuations was significantly longer in bilayer experiments than in cellular recordings (although bilayer experiments also revealed short-lived events). In contrast, the duration of Aβ-induced current events was similar in SH-SY5Y cells compared to transgenic primary neurons. The frequency of large-amplitude current events (i.e., events that were larger than four times the standard deviation of the current noise) ranged from 0.2 to 5 Hz in SH-SY5Y and in transgenic cells to ~10 Hz in bilayer experiments and was, thus, similar in magnitude. The single channel conductance of events ranged between 0.2 and 1.7 nS in planar lipid bilayers and between 0.2 and 0.6 nS in both SH-SY5Y cells and transgenic neurons. Although these conductance values are not directly comparable (since the ion concentrations and lipid compositions in the bilayer and cellular experiments could not be matched precisely), this comparison suggests that the conductance of Aβ-induced stepwise ion flux in bilayers and live cells was on the same order of magnitude.

Comparison of short recordings with expanded time axis of Aβ-induced ion flux across planar lipid bilayers, across plasma membranes of SH-SY5Y cells, and across plasma membranes of transgenic cortical neurons. a Aβ-induced ion flux through a planar lipid bilayer. The final concentration of Aβ was 10 μM (applied potential −30 mV). b Aβ-induced ion flux through membranes of SH-SY5Y cells. The final concentration of Aβ was ~10 μM in the extracellular solution (applied potential −80 mV). Current fluctuations significantly above baseline current noise are indicated with an asterisk. c Aβ-induced ion flux through the plasma membranes of a cortical neuron that expressed Aβ (applied potential −80 mV). Current fluctuations significantly above baseline current noise are indicated with an asterisk
In order to minimize the possibility that endogenous, cellular ion channels would be activated by Aβ, we performed all recordings close to the resting membrane potential (−80 mV) and we added a mixture of ion channel blockers to the extracellular solution. This mixture included (final concentration in the extracellular solution): 20 mM tetraethylammonium (TEA) ions to block potassium channels (Forsythe et al. 1992; Mathie et al. 1998; Tosetti et al. 1998; Guyon et al. 2005), 1 μM tetrodotoxin to block sodium channels (Forsythe et al. 1992; Toselli et al. 1996; Guyon et al. 2005), 2 μM nifedipine to block L-type calcium channels, and 2 μM ω-conotoxin to block N-type calcium channels (McDonald et al. 1994, 1995; Reeve et al. 1994, 1995; Furukawa et al. 2003; Billups et al. 2006). Figure 6a shows that Aβ-induced ion flux persisted in the presence of these blockers. Only the addition of Zn2+ caused a significant reduction in Aβ-induced, large-amplitude current fluctuations in these cells. Since Aβ-induced, channel-like ion flux through artificial bilayers is blocked effectively by Zn2+ (Fig. 4b), the Zn2+-dependent blockage of ion flux shown in Fig. 6a suggests that Aβ is also capable of inducing ion channel-like ion flux in live cells. These results do not exclude the possibility that Aβ may activate endogenous cellular channels; these channels would, however, have to fulfill at least three characteristics: (1) they would have to be activated by Aβ at the resting potential of −80 mV, (2) they would have to be insensitive to the cocktail of blockers, and (3) they would have to be sensitive to Zn2+ ions.

Reduction of Aβ-induced ion flux across the plasma membrane of SH-SY5Y cells by Zn2+ ions or NA7 peptides. a Effect of Zn2+ (final concentration ~2 mM) on Aβ-induced ion flux. Note these recordings were carried out in the presence of a mixture of ion channel blockers containing 20 mM TEA, 1 μM tetrodotoxin, 2 μM nifedipine, and 2 μM ω-conotoxin; these ion channel blockers did not affect Aβ-induced ion flux but reduced the probability that intrinsic ion channels may be responsible for the observed current spikes. Large-amplitude current spikes, i.e. spikes with amplitudes above the threshold (black solid line), are indicated with an asterisk. The vertical arrow indicates the time point when Zn2+ was added. b Effect of NA7 peptide (final concentration ~100 μM) on Aβ-induced transmembrane ion flux. Large-amplitude current spikes, i.e. spikes with amplitudes above the threshold (black solid line), are indicated with an asterisk. The vertical arrow indicates the time point when NA7 was added. Both recordings were carried out with an applied potential of −80 mV
Since it is, in principle, possible that Zn2+ ions blocked endogenous, cellular ion channels, we also tested the effect of a specific blocker of Aβ-induced transmembrane ion flux in these cell experiments. Arispe and coworkers reported that a peptide called NA7 (corresponding to the amino acid sequence EVHHQKL of residues 11 to 17 of Aβ) is an effective and specific blocker of Aβ-induced, stepwise ion flux (Simakova and Arispe 2006; Arispe et al. 2007). When we tested this peptide in SH-SY5Y cells, we found, in three of four recorded cells, up to a threefold reduction in the frequency of large-amplitude current events induced by Aβ (Fig. 6b). This result further supports the hypothesis that Aβ can independently induce stepwise (or spike-like) ion flux across cellular membranes by self-assembly to pores or by inducing membrane defects.
In conclusion, the work presented here demonstrates that Aβ can cause stepwise transmembrane ion flux through artificial membranes. The previously postulated membrane thinning that was reported to lead to gradual increase in transmembrane ion flux is likely not attributable to Aβ, but instead is due to residues of the solvent HFIP used to prepare samples of Aβ. We found that HFIP is membrane-active and cytotoxic in the low millimolar range. Therefore, it is critical to remove HFIP in any amyloid samples before they are used in membrane or cellular studies. In addition, we present evidence that Aβ is capable of inducing stepwise transmembrane ion fluctuations in living cells. Although we cannot rule out the possibility that Aβ activates intrinsic ion channels or ion pumps in cells, the results presented here suggest that Aβ is capable of self-assembling into structures that either form a pore through membranes or generate transient defects in membranes. This conclusion is based on the following four observations: (1) since Aβ clearly forms stepwise ion fluctuations in planar lipid bilayers, it is plausible that it can form similar transmembrane ion fluctuations in cellular membranes. (2) Aβ-induced stepwise ion fluctuations in cellular membranes occur at the resting potential of cells (where the activity of intrinsic ion channels is minimal) and persist in the presence of molecules that block the most common ion channel proteins. (3) In contrast, these Aβ-induced stepwise ion fluctuations in cellular membranes are blocked by Zn2+ ions and a heptapeptide derived from the sequence of Aβ (both Zn2+ ions and this heptapeptide block Aβ-induced, stepwise ion flux through artificial bilayers; Arispe et al. 2007). (4) Finally, Aβ-induced stepwise ion fluctuations in cellular membranes resemble Aβ-induced stepwise ion fluctuations in artificial membranes with regards to their frequency and amplitude of current events. Based on these observations, we conclude that stepwise ion flux through Aβ-induced pores or membrane defects could contribute to the disruption of Ca2+ homeostasis that is commonly associated with Alzheimer’s disease.
Notes
One of the previously reported results that supported the hypothesis of Aβ-induced gradual thinning of membranes was the observation that an anti-Aβ antibody could reduce the observed transmembrane ion flux caused by these Aβ preparations. These experiments were carried out with Aβ samples that were also prepared with an HFIP purging protocol (Kayed et al. 2003, 2004; Sokolov et al. 2004, 2006). We repeated this experiment with the modification that we added bovine serum albumin (BSA) instead of adding an anti-Aβ antibody. In approximately half of these trials, we found a reduction in transmembrane ion flux by adding BSA to the bilayer chamber. The results were thus similar to the effect attributed previously to the anti-Aβ antibody. Furthermore, when we induced transmembrane ion flux by adding HFIP only, we observed again a reduction in conductance by addition of BSA to bilayer chambers that did not contain Aβ. These results suggest that a specific anti-Aβ antibody was not required to reduce gradually increasing ion flux that was induced by samples containing HFIP.
These results together with previous work (Ennaceur and Sanderson 2005) show that even low concentrations of HFIP can adversely affect membranes and alter their permeability for ions. Unlike other short chain alcohols, HFIP affects membranes similarly as long chain alcohols (Gutknecht and Tosteson 1970; Ueda and Yoshida 1999; Ebihara et al. 1979).
For comparison, the ethanol concentration necessary to achieve similar cell toxicity is higher than 200 mM (Luo and Miller 1997).
In the remaining ~25% of the trials, we observed a flat baseline without any detectable transmembrane current.
References
Arispe N (2004) Architecture of the Alzheimer’s AβP ion channel pore. J Membr Biol 197:33–48
Arispe N, Doh M (2002) Plasma membrane cholesterol controls the cytotoxicity of Alzheimer’s disease AβP (1-40) and (1-42) peptides. FASEB J 16:1526–1536
Arispe N, Pollard HB, Rojas E (1993a) Giant multilevel cation channels formed by Alzheimer-disease amyloid beta-protein a-β-P-(1-40) in bilayer-membranes. Proc Natl Acad Sci USA 90:10573–10577
Arispe N, Rojas E, Pollard HB (1993b) Alzheimer-disease amyloid β-protein forms calcium channels in bilayer-membranes—blockade by tromethamine and aluminum. Proc Natl Acad Sci USA 90:567–571
Arispe N, Pollard HB, Rojas E (1996) Zn2+ interaction with Alzheimer amyloid β protein calcium channels. Proc Natl Acad Sci USA 93:1710–1715
Arispe N, Diaz JC, Simakova O (2007) Aβ ion channels. Prospects for treating Alzheimer’s disease with Aβ channel blockers. Biochim Biophys Acta 1768:1952–1965
Arispe NJ, Diaz JC, Flora M (2008) Efficiency of histidine-associating compounds for blocking the Alzheimer’s Aβ channel activity and cytotoxicity. Biophys J 95:4879–4889
Baglioni S, Casamenti F, Bucciantini M, Luheshi LM, Taddei N, Chiti F, Dobson CM, Stefani M (2006) Prefibrillar amyloid aggregates could be generic toxins in higher organisms. J Neurosci 26:8160–8167
Bahadi R, Farrelly PV, Kenna BL, Curtain CC, Masters CL, Cappai R, Barnham KJ, Kourie JI (2003) Cu2+-induced modification of the kinetics of Aβ(1-42) channels. Am J Physiol Cell Physiol 285:C873–C880
Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, Bahr M, Schmidt M, Bitner RS, Harlan J, Barlow E, Ebert U, Hillen H (2005) Globular amyloid β-peptide1-42 oligomer—a homogenous and stable neuropathological protein in Alzheimer’s disease. J Neurochem 95:834–847
Bezprozvanny I, Mattson MP (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 31:454–463
Bhatia R, Lin H, Lal R (2000) Fresh and globular amyloid β protein (1-42) induces rapid cellular degeneration: evidence for AβP channel-mediated cellular toxicity. FASEB J 14:1233–1243
Billups D, Billups B, Challiss RAJ, Nahorski SR (2006) Modulation of Gq-protein-coupled inositol trisphosphate and Ca2+ signaling by the membrane potential. J Neurosci 26:9983–9995
Blake S, Capone R, Mayer M, Yang J (2008) Chemically reactive derivatives of gramicidin A for developing ion channel-based nanoprobes. Bioconjug Chem 19:1614–1624
Bruggemann A, George M, Klau M, Beckler M, Steindl J, Behrends JC, Fertig N (2003) High quality ion channel analysis on a chip with the NPC (c) technology. Assay Drug Dev Technol 1:665–673
Capone R, Blake S, Rincon Restrepo M, Yang J, Mayer M (2007) Designing nanosensors based on charged derivatives of gramicidin A. J Am Chem Soc 129:9737–9745
Cheung K-H, Shineman D, Müller M, Cárdenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VMY, Foskett JK (2008) Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 58:871–883
Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG (2005) Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 280:17294–17300
Deshpande A, Mina E, Glabe C, Busciglio J (2006) Different conformations of amyloid β induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci 26:6011–6018
Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK (2006) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci 26:9057–9068
Dougherty JJ, Wu J, Nichols RA (2003) β-Amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J Neurosci 23:6740–6747
Dreses-Werringloer U et al (2008) A polymorphism in CALHM1 influences Ca2+ homeostasis, Aβ levels, and Alzheimer’s disease risk. Cell 133:1149–1161
Durell SR, Guy HR, Arispe N, Rojas E, Pollard HB (1994) Theoretical models of the ion channel structure of amyloid β-protein. Biophys J 67:2137–2145
Ebihara L, Hall JE, MacDonald RC, McIntosh TJ, Simon SA (1979) Effect of benzyl alcohol on lipid bilayers. A comparisons of bilayer systems. Biophys J 28:185–196
Eliezer D (2006) Amyloid ion channels: a porous argument or a thin excuse? J Gen Physiol 128:631–633
Ennaceur SM, Sanderson JM (2005) Micellar aggregates formed following the addition of hexafluoroisopropanol to phospholipid membranes. Langmuir 21:552–561
Estes DJ, Memarsadeghi S, Lundy SK, Mikol DD, Fox DA, Mayer M (2008) High-throughput profiling of ion channel activity in primary human lymphocytes. Anal Chem 80:3728–3735
Fagan TKB, Corbin D, Hwang W, Glabe C, Nault L, Lal R, Teplow D, Albensi B, Sokolov Y (2006) Alzheimer research forum live discussion: now you see them, now you don’t: the amyloid channel hypothesis. J Alzheimers Dis 9:219–224
Fedrizzi L, Lim D, Carafoli E, Brini M (2008) Interplay of the Ca2+-binding protein DREAM with presenilin in neuronal Ca2+ signaling. J Biol Chem 283:27494–27503
Forsythe ID, Lambert DG, Nahorski SR, Linsdell P (1992) Elevation of cytosolic calcium by cholinoceptor agonists in SH-SY5Y human neuroblastoma-cells—estimation of the contribution of voltage-dependent currents. Br J Pharmacol 107:207–214
Furukawa K, Wang Y, Yao PJ, Fu W, Mattson MP, Itoyama Y, Onodera H, D’Souza I, Poorkaj PH, Bird TD, Schellenberg GD (2003) Alteration in calcium channel properties is responsible for the neurotoxic action of a familial frontotemporal dementia tau mutation. J Neurochem 87:427–436
Gennis RB (1989) Biomembranes, 1st edn. Springer-Verlag, New York
Green KN, Demuro A, Akbari Y, Hitt BD, Smith IF, Parker I, LaFerla FM (2008) SERCA pump activity is physiologically regulated by presenilin and regulates amyloid β production. J Cell Biol 181:1107–1116
Gutknecht J, Tosteson DC (1970) Ionic permeability of thin lipid membranes: effects of n-alkyl alcohols, polyvalent cations, and a secondary amine. J Gen Physiol 55:359–374
Guyon A, Rovère C, Cervantes A, Allaeys I, Nahon JL (2005) Stromal cell-derived factor-1α; directly modulates voltage-dependent currents of the action potential in mammalian neuronal cells. J Neurochem 93:963–973
Hirakura Y, Lin M-C, Kagan B (1999a) Erratum: Hirakura Y, Lin M-C, Kagan BL. 1999. Alzheimer amyloid β 1-42 channels: effects of solvent, pH, and congo red. J Neurosci Res 57:458–466. J Neurosci Res 58:726
Hirakura Y, Lin M-C, Kagan BL (1999b) Alzheimer amyloid β1-42 channels: effects of solvent, pH, and congo red. J Neurosci Res 57:458–466
Inbar P, Yang J (2006) Inhibiting protein-amyloid interactions with small molecules: a surface chemistry approach. Bioorg Med Chem Lett 16:1076–1079
Inbar P, Li CQ, Takayama SA, Bautista MR, Yang J (2006) Oligo(ethylene glycol) derivatives of thioflavin T as inhibitors of protein-amyloid interactions. ChemBioChem 7:1563–1566
Jang H, Zheng J, Lal R, Nussinov R (2008) New structures help the modeling of toxic amyloidß ion channels. Trends Biochem Sci 33:91–100
Joslin G, Krause J, Hershey A, Adams S, Fallon R, Perlmutter D (1991) Amyloid-β peptide, substance P, and bombesin bind to the serpin-enzyme complex receptor. J Biol Chem 266:21897–21902
Kagan BL, Hirakura Y, Azimov R, Azimova R, Lin M-C (2002) The channel hypothesis of Alzheimer’s disease: current status. Peptides 23:1311–1315
Kagan BL, Azimov R, Azimova R (2004) Amyloid peptide channels. J Membr Biol 202:1–10
Kawahara M, Kuroda Y, Arispe N, Rojas E (1996) Direct incorporation of amyloid β-protein cation channels into excised membrane patches from GnRH neurons. Biophys J 70:S104
Kawahara M, Arispe N, Kuroda Y, Rojas E (1997) Alzheimer’s disease amyloid β-protein forms Zn(2+)-sensitive, cation-selective channels across excised membrane patches from hypothalamic neurons. Biophys J 73:67–75
Kawahara M, Kuroda Y, Arispe N, Rojas E (2000) Alzheimer’s β-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J Biol Chem 275:14077–14083
Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300:486–489
Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG (2004) Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem 279:46363–46366
Khachaturian ZS (1987) Hypothesis on the regulation of cytosol calcium concentration and the aging brain. Neurobiol Aging 8:345–346
Khachaturian ZS (1989) Introduction and overview on calcium, membranes, aging, and Alzheimer’s disease. Ann NY Acad Sci 568:1–4
Koh J-y, Yang LL, Cotman CW (1990) β-Amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage. Brain Res 533:315–320
Kourie J, Henry C, Farrelly P (2001) Diversity of amyloid β protein fragment [1-40]-formed channels. Cell Mol Neurobiol 21:255–284
Kourie J, Culverson A, Farrelly P, Henry C, Laohachai K (2002) Heterogeneous amyloid-formed ion channels as a common cytotoxic mechanism. Cell Biochem Biophys 36:191–207
Kurganov B, Doh M, Arispe N (2004) Aggregation of liposomes induced by the toxic peptides Alzheimer’s Aβs, human amylin and prion (106-126): facilitation by membrane-bound GM1 ganglioside. Peptides 25:217–232
Lal R, Lin H, Quist AP (2007) Amyloid β ion channel: 3D structure and relevance to amyloid channel paradigm. Biochim Biophys Acta 1768:1966–1975
Lam FC, Liu R, Lu P, Shapiro AB, Renoir J-M, Sharom FJ, Reiner PB (2001) β-Amyloid efflux mediated by p-glycoprotein. J Neurochem 76:1121–1128
Lecoeur H, Chauvier D, Langonne A, Rebouillat D, Brugg B, Mariani J, Edelman L, Jacotot E (2004) Dynamic analysis of apoptosis in primary cortical neurons by fixed- and real-time cytofluorometry. Apoptosis 9:157–169
Lin M-cA, Kagan BL (2002) Electrophysiologic properties of channels induced by Aβ25-35 in planar lipid bilayers. Peptides 23:1215–1228
Lin H, Zhu YJ, Lal R (1999) Amyloid β-protein (1-40) forms calcium-permeable, Zn2+-sensitive channel in reconstituted lipid vesicles. Biochemistry 38:11189–11196
Lin H, Bhatia R, Lal R (2001) Amyloid β protein forms ion channels: implications for Alzheimer’s disease pathophysiology. FASEB J 15:2433–2444
Lopez JR, Lyckman A, Oddo S, LaFerla FM, Querfurth HW, Shtifman A (2008) Increased intraneuronal resting [Ca2+] in adult Alzheimer’s disease mice. J Neurochem 105:262–271
Luo J, Miller MW (1997) Differential sensitivity of human neuroblastoma cell lines to ethanol: correlations with their proliferative responses to mitogenic growth factors and expression of growth factor receptors. Alcohol Clin Exp Res 21:1186-1194
Mark R, Hensley K, Butterfield D, Mattson M (1995) Amyloid β-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci 15:6239–6249
Marx J (2007) Alzheimer’s disease—fresh evidence points to an old suspect: calcium. Science 318:384–385
Mathie A, Wooltorton JRA, Watkins CS (1998) Voltage-activated potassium channels in mammalian neurons and their block by novel pharmacological agents. Gen Pharmacol 30:13–24
Mattson MP, Chan SL (2003) Calcium orchestrates apoptosis. Nat Cell Biol 5:1041–1043
Mattson M, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel R (1992) β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci 12:376–389
Mayer M, Kriebel JK, Tosteson MT, Whitesides GM (2003) Microfabricated teflon membranes for low-noise recordings of ion channels in planar lipid bilayers. Biophys J 85:2684–2695
Mayer M, Semetey V, Gitlin I, Yang J, Whitesides GM (2008) Using ion channel-forming peptides to quantify protein-ligand interactions. J Am Chem Soc 130:1453–1465
McDonald RL, Vaughan PFT, Peers C (1994) Muscarinic (M(1)) receptor-mediated inhibition of K+-evoked [H-3] noradrenaline release from human neuroblastoma (SH-SY5Y) cells via inhibition of L-type and N-type Ca2+ channels. Br J Pharmacol 113:621–627
McDonald RL, Vaughan PFT, Beck-Sickinger AG, Peers C (1995) Inhibition of Ca2+ channel currents in human neuroblastoma (SH-SY5Y) cells by neuropeptide Y and a novel cyclic neuropeptide Y analogue. Neuropharmacology 34:1507–1514
Micelli S, Meleleo D, Picciarelli V, Gallucci E (2004) Effect of sterols on β-amyloid peptide (AβP 1-40) channel formation and their properties in planar lipid membranes. Biophys J 86:2231–2237
Michikawa M, Gong J-S, Fan Q-W, Sawamura N, Yanagisawa K (2001) A novel action of Alzheimer’s amyloid β-protein (Aβ): oligomeric Aβ promotes lipid release. J Neurosci 21:7226–7235
Mirzabekov T, Lin MC, Yuan WL, Marshall PJ, Carman M, Tomaselli K, Lieberburg I, Kagan BL (1994) Channel formation in planar lipid bilayers by a neurotoxic fragment of the β-amyloid peptide. Biochem Biophys Res Commun 202:1142–1148
Mobley DL, Cox DL, Singh RRP, Maddox MW, Longo ML (2004) Modeling amyloid β-peptide insertion into lipid bilayers. Biophys J 86:3585–3597
Montal M, Mueller P (1972) Formation of bimolecular membranes from lipid monolayers and a study of their electrical properties. Proc Natl Acad Sci USA 69:3561–3566
Morimoto T, Ohsawa I, Takamura C, Ishiguro M, Nakamura Y, Kohsaka S (1998) Novel domain-specific actions of amyloid precursor protein on developing synapses. J Neurosci 18:9386–9393
Morris CE, Juranka PF (2007) Lipid stress at play: mechanosensitivity of voltage-gated channels. In: Mechanosensitive ion channels, Pt B, pp 297–338
Mueller P, Rudin DO, Tien HT, Wescott WC (1962) Reconstitution of cell membrane structure in vitro and its transformation into an excitable system. Nature 194:979–980
Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, Hillen H, Gross G, Ebert U, Bruehl C (2008) Amyloid β oligomers (Aβ1-42 globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci 28:788–797
Novarino G, Fabrizi C, Tonini R, Denti MA, Malchiodi-Albedi F, Lauro GM, Sacchetti B, Paradisi S, Ferroni A, Curmi PM, Breit SN, Mazzanti M (2004) Involvement of the intracellular ion channel CLIC1 in microglia-mediated β-amyloid-induced neurotoxicity. J Neurosci 24:5322–5330
Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu G-Q, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L (2007) Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55:697–711
Pollard HB, Rojas E, Arispe N (1993) A new hypothesis for the mechanism of amyloid toxicity, based on the calcium-channel activity of amyloid-beta protein (a-β-P) in phospholipid-bilayer membranes. In: Alzheimer’s disease: amyloid precursor proteins, signal transduction, and neuronal transplantation, pp 165–168
Querfurth HW, Jiang J, Geiger JD, Selkoe DJ (1997) Caffeine stimulates amyloid β-peptide release from β-amyloid precursor protein-transfected HEK293 cells. J Neurochem 69:1580–1591
Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R (2005) Amyloid ion channels: a common structural link for protein-misfolding disease. Proc Natl Acad Sci USA 102:10427–10432
Reeve HL, Vaughan PFT, Peers C (1994) Calcium-channel currents in undifferentiated human neuroblastoma (SH-SY5Y) cells—actions and possible interactions of dihydropyridines and omega-conotoxin. Eur J Neurosci 6:943–952
Reeve Hl, Vaughan PFT, Peers C (1995) Inhibition of N-type Ca2+ channel currents in human neuroblastoma (SH-SY5Y) cells by muscarine via stimulation of M3 receptors. Neuropharmacology 34:319–326
Rhee SK, Quist AP, Lal R (1998) Amyloid β protein-(1-42) forms calcium-permeable, Zn2+-sensitive channel. J Biol Chem 273:13379–13382
Rovira C, Arbez N, Mariani J (2002) Aβ(25-35) and Aβ(1-40) act on different calcium channels in CA1 hippocampal neurons. Biochem Biophys Res Commun 296:1317–1321
Saluja I, Saunders TL, Turner RS (2006) Modulation of neuronal APP metabolism by X11α/mint-1/APBA1. In: Alzheimer’s disease: new advances. Medimond international proceedings
Schmidt C, Mayer M, Vogel H (2000) A chip-based biosensor for the functional analysis of single ion channels. Angew Chem Int Ed 39:3137–3140
Simakova O, Arispe NJ (2006) Early and late cytotoxic effects of external application of the Alzheimer’s Aβ result from the initial formation and function of Aβ ion channels. Biochemistry 45:5907–5915
Simakova O, Arispe NJ (2007) The cell-selective neurotoxicity of the Alzheimer’s A peptide is determined by surface phosphatidylserine and cytosolic ATP levels. Membrane binding is required for Aβ toxicity. J Neurosci 27:13719–13729
Sokolov YV, Kayed R, Kozak A, Edmonds B, McIntire TM, Milton S, Cahalan M, Glabe CG, Hall JE (2004) Soluble amyloid oligomers increase lipid bilayer conductance by increasing the dielectric constant of the hydrocarbon core. Biophys J 86:382A–382A
Sokolov Y, Kozak JA, Kayed R, Chanturiya A, Glabe C, Hall JE (2006) Soluble amyloid oligomers increase bilayer conductance by altering dielectric structure. J Gen Physiol 128:637–647
Stains CI, Mondal K, Ghosh I (2007) Molecules that target beta-amyloid. ChemMedChem 2:1674–1692
Stutzmann GE (2007) The pathogenesis of Alzheimers disease—is it a lifelong “Calciumopathy”? Neuroscientist 13:546–559
Stutzmann GE, Smith I, Caccamo A, Oddo S, LaFerla FM, Parker I (2006) Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci 26:5180–5189
Stutzmann GE, Smith I, Caccamo A, Oddo S, Parker I, Laferla F (2007) Enhanced ryanodine-mediated calcium release in mutant PS1-expressing Alzheimer’s mouse models. Ann NY Acad Sci 1097:265–277
Toselli M, Tosetti P, Taglietti V (1996) Functional changes in sodium conductances in the human neuroblastoma cell line SH-SY5Y during in vitro differentiation. J Neurophysiol 76:3920–3927
Tosetti P, Taglietti V, Toselli M (1998) Functional changes in potassium conductances of the human neuroblastoma cell line SH-SY5Y during in vitro differentiation. J Neurophysiol 79:648–658
Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee S-F, Hao Y-H, Serneels L, De Strooper B, Yu G, Bezprozvanny I (2006) Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 126:981–993
Ueda I, Yoshida T (1999) Hydration of lipid membranes and the action mechanisms of anesthetics and alcohols. Chem Phys Lipids 101:65–79
Ueda K, Shinohara S, Yagami T, Asakura K (1997) Amyloid β protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J Neurochem 68:265–271
Valincius G, Heinrich F, Budvytyte R, Vanderah DJ, McGillivray DJ, Sokolov Y, Hall JE, Losche M (2008) Soluble amyloid β oligomers affect dielectric membrane properties by bilayer insertion and domain formation: implications for cell toxicity. Biophys J 95:4845–4861
Verdier Y, Zarándi M, Penke B (2004) Amyloid β-peptide interactions with neuronal and glial cell plasma membrane: binding sites and implications for Alzheimer’s disease. J Pept Sci 10:229–248
Wang H-Y, Lee DHS, Davis CB, Shank RP (2000) Amyloid peptide Aβ1-42 binds selectively and with picomolar affinity to α7 nicotinic acetylcholine receptors. J Neurochem 75:1155–1161
Yankner BA, Duffy LK, Kirschner DA (1990a) Neurotrophic and neurotoxic effects of amyloid β protein: reversal by tachykinin neuropeptides. Science 250:279–282
Yankner BA, Caceres A, Duffy LK (1990b) Nerve growth factor potentiates the neurotoxicity of β amyloid. Proc Natl Acad Sci USA 87:9020–9023
Ye C, Ho-Pao CL, Kanazirska M, Quinn S, Rogers K, Seidman CE, Seidman JG, Brown EM, Vassilev PM (1997) Amyloid-β; proteins activate Ca2+-permeable channels through calcium-sensing receptors. J Neurosci Res 47:547–554
Zhu YJ, Lin H, Lal R (2000) Fresh and nonfibrillar amyloid β protein (1-40) induces rapid cellular degeneration in aged human fibroblasts: evidence for AβP-channel-mediated cellular toxicity. FASEB J 14:1244–1254
Acknowledgments
This work was supported by the Wallace H. Coulter Foundation and a NSF Career Award 02-111 (M.M.). M.R.B and J.Y. acknowledge support from the Alzheimer’s Disease Research Center (3P50 AG005131) and a grant from the Alzheimer’s Association (NIRG-08-91651). P.P. acknowledges a fellowship from the government of Thailand. A.M.S. acknowledges a scholarship from Solvay Inc. We thank James Windak from the University of Michigan, Instrument Services, for measurements of HFIP concentrations by GC-MS.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Capone, R., Quiroz, F.G., Prangkio, P. et al. Amyloid-β-Induced Ion Flux in Artificial Lipid Bilayers and Neuronal Cells: Resolving a Controversy. Neurotox Res 16, 1–13 (2009). https://doi.org/10.1007/s12640-009-9033-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-009-9033-1