
Abstract
The linker of nucleoskeleton and cytoskeleton (LINC) complex couples the nuclear lamina to the cytoskeleton. The LINC complex and its associated proteins play diverse roles in cells, ranging from genome organization, nuclear morphology, gene expression, to mechanical stability. The importance of a functional LINC complex is highlighted by the large number of mutations in genes encoding LINC complex proteins that lead to skeletal and cardiac myopathies. In this review, the structure, function, and interactions between components of the LINC complex will be described. Mutations that are known to cause cardiomyopathy in patients will be discussed alongside their respective mouse models. Furthermore, future challenges for the field and emerging technologies to investigate LINC complex function will be discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The nuclear envelope (NE) plays a critical role in dividing the cytoplasm from the nucleus. The NE is comprised of the inner nuclear membrane (INM) and the outer nuclear membrane (ONM), which is contiguous with the endoplasmic reticulum (Fig. 1). These membranes are separated by the perinuclear space (PNS) and are periodically joined by nuclear pore complexes (NPC) that allow bidirectional transport of macromolecules across the NE (Brohawn et al. 2009; Gorlich and Kutay 1999; Grossman et al. 2012). Underlying the INM is an interconnected meshwork of intermediate filaments collectively known as the nuclear lamina, which is made up of A-type and B-type lamins. Lamins play essential roles in maintaining nuclear structure, gene expression, and chromatin organization (Burke and Stewart 2013; Gerace and Tapia 2018). Furthermore, the nuclear lamina is functionally coupled to the cytoskeleton via connections mediated by INM and ONM proteins, termed the linker of nucleoskeleton and cytoskeleton (LINC) complex (Crisp et al. 2006; Sosa et al. 2013; Stroud et al. 2014) (Fig. 1). The LINC complex structurally supports the nucleus and plays a mechanosensory role to translate mechanical cues and alterations in the extracellular matrix into biochemical signals (Banerjee et al. 2014; Haque et al. 2006; Padmakumar et al. 2005; Sosa et al. 2012; Starr and Fridolfsson 2010; Starr and Han 2002; Swift et al. 2013), thereby allowing the cell to adapt to its surrounding environment by modulation of cytoskeleton organization, gene expression, nuclear organization, and structure (Jaalouk and Lammerding 2009; Lombardi and Lammerding 2011).

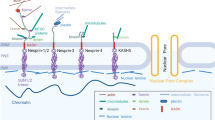
The cardiomyocyte LINC complex: Magnified view of cardiac myocyte nuclei (blue) in the circled region highlighting the nuclear envelope (NE) and linker between nucleoskeleton and cytoskeleton (LINC) complex. The LINC complex forms a continuous network of protein-protein interactions between the nuclear lamins; lamins A/C, B1, B2; and the various cytoskeletons. Inner nuclear membrane (INM) proteins SUN1 and SUN2 form heterotrimeric complexes that interact via their SUN domain with the KASH domain of the Nesprins that reside in the outer nuclear membrane (ONM). The giant isoforms of Nesprins 1 and 2 may directly link the NE to the sarcomere by interacting with the Z-disk (Z) or indirectly through intermediate binding partners(s). Nesprin 2 is reported at A/I junctions in the sarcomere. Nesprin 1α2 interacts with kinesin 1, thereby linking the NE to the microtubule cytoskeleton and Nesprin 3 indirectly links to the desmin intermediate filaments through plectin. Many other proteins are associated with the LINC complex that interact with lamins or SUN proteins including LAP2α, Emerin, MAN1, LEM2, Luma, and LAP1 and have been shown to play important roles in cardiomyocytes. Heterochromatin directly interacts with lamin A/C and indirectly with LEM domain proteins via barrier to autointegration factor (BAF). Emerin binds the histone modification enzyme histone deacetylase 3 (HDAC3). Lamin B receptor (LBR) interacts with lamin B and heterochromatin via heterochromatin protein 1 (HP1). LAP2α interacts with nucleoplasmic lamin A/C. PNS indicates perinuclear space and M, M-band
The importance of the LINC complex has been highlighted by the plethora of mutations in LINC complex-encoding genes that are associated with skeletal and cardiac myopathies, including dilated cardiomyopathy (DCM), arrhythmogenic cardiomyopathy (AC), and Emery-Dreifuss muscular dystrophy (EDMD) (Dellefave and McNally 2010; McNally and Mestroni 2017; Meinke and Schirmer 2016; Mejat and Misteli 2010; Mendez-Lopez and Worman 2012; Worman et al. 2010). Here, we will describe the roles of the LINC complex and its associated proteins that have been identified in the heart. The structural and functional interactions made between nuclear envelope spectrin repeat proteins (Nesprins), SUN proteins, Emerin, LAP1, LAP2, MAN1, LEM2, Luma, and nuclear lamins will be discussed, and mutations that lead to disease highlighted.
Nuclear envelope spectrin repeat proteins
Nesprins form the ONM component of the LINC complex that connects the nuclear lamina to the cytoskeleton (Apel et al. 2000; Crisp et al. 2006; Mislow et al. 2002b; Padmakumar et al. 2004; Shanahan et al. 1993; Zhang et al. 2001). The Nesprin family are comprised of four members (Nesprins 1–4) (Zhang et al. 2002), of which Nesprins 1, 2, and 3 have been detected in the heart (Banerjee et al. 2014; Ketema et al. 2013; Postel et al. 2011; Wilhelmsen et al. 2005) (Fig. 2).

Nesprin family members and isoforms reported in cardiomyocytes. Nesprin 1α2 contains an isoform-specific spectrin repeat (pink) followed by 6 other spectrin repeats (red) and a KASH domain (yellow). Nesprin 1 giant contains N-terminal tandem calponin homology (CH) domains (purple) that bind actin, spectrin repeats (orange), and a C-terminal KASH domain. Nesprin 2 giant contains tandem CH domains, abutted to spectrin repeats (orange) and a KASH domain. Nesprin 3α is comprised of an isoform-specific spectrin repeat (green) that binds plectin followed by 7 spectrin repeats (blue) and a KASH domain. Nesprin 3β is organized similarly to Nesprin 3α, but the isoform-specific spectrin repeat found in 3α
Nesprins 1 and 2
Nesprin 1 is alternatively named synaptic NE-1 (Syne-1) (Apel et al. 2000), Enaptin (Padmakumar et al. 2004), and myocyte NE protein-1 (Myne-1) (Mislow et al. 2002b) because of its simultaneously discovery by independent groups. Nesprin 2 is also known as Syne-2, or nucleus and actin connecting element (NUANCE) (Zhen et al. 2002). Many potential isoforms of Nesprins 1 and 2 exist, and they vary greatly in size due to alternative transcription initiation, termination, and RNA splicing of SYNE-1 and SYNE-2 genes, respectively (Rajgor et al. 2012). The largest, or giant (G), isoforms of Nesprins contain N-terminally paired calponin homology (CH) domains that bind actin, a spectrin repeat-containing rod domain, and a C-terminal Klarsicht, ANC-1, and Syne homology (KASH) domain that interacts with Sad1/UNC-84 (SUN) proteins (Padmakumar et al. 2004; Sosa et al. 2012) (Fig. 2). Other Nesprin isoforms exist that lack either the N-terminal CH domains, C-terminal KASH domain, or have a varying number of spectrin repeats (Warren et al. 2005).
Many antibodies have been raised against Nesprins 1 and 2 to different regions and domains (Potter et al. 2017; Randles et al. 2010; Razafsky and Hodzic 2015). Nesprin 1 epitopes have been reported at the NE and the Z-disc in both human and mouse skeletal and cardiac tissue (Apel et al. 2000; Holt et al. 2016; Mislow et al. 2002b; Nikolova-Krstevski et al. 2011; Zhang et al. 2002). Nesprin 2 epitopes have been reported at the NE, Z-disc, and A/I junction in human skeletal muscle, and at both NE and striations in mouse cardiac tissue (Banerjee et al. 2014; Zhang et al. 2005). However, it should be noted that the specificities of some of the antibodies described to date have not been thoroughly validated in suitable knockout animal models. Furthermore, the lack of isoform-specific sequences in the various Nesprin proteins make the generation of specific antibodies challenging.
In humans, several mutations in Nesprins 1 and 2 are associated with cardiomyopathy (Haskell et al. 2017; Puckelwartz et al. 2009, 2010; Zhang et al. 2007a; Zhou et al. 2017). However, the limited number of patients and small family pedigrees make it difficult to ascertain the critical Nesprin isoforms and whether the identified mutations in Nesprins 1 and 2 are the unequivocal cause of disease. Therefore, to understand the role of Nesprins 1 and 2 in the mammalian heart, several mouse models have been developed.
The initial Nesprin 1 mutant mouse models to study Nesprin 1 function either replaced the KASH domain (Nesprin 1rKASH) or deleted it (Nesprin 1ΔKASH) (Puckelwartz et al. 2009; Zhang et al. 2007b). Around half of Nesprin 1rKASH mutant mice died perinatally due to respiratory failure and the surviving mice developed progressive muscle weakness and cardiomyopathy including cardiac conduction defects (Puckelwartz et al. 2009, 2010). The replacement of the KASH domain with 61 unrelated C-terminal amino acids prevents Nesprin 1 from binding SUN proteins, thereby disrupting the LINC complex. Notably, cardiomyocyte nuclei were elongated and heterochromatin levels were reduced in the Nesprin 1rKASH mice. These results mirrored what had been shown previously in lamin A/C mutant cardiomyocytes and point to the importance of LINC complex integrity (Nikolova et al. 2004).
In contrast to Nesprin 1rKASH mice, Nesprin 1ΔKASH mutants were viable and able to breed as normal. However, mice in which Nesprin 2’s KASH domain was also deleted (Nesprin 1ΔKASH/Nesprin 2ΔKASH) died of respiratory failure 20 min after birth. The cardiac function of the Nesprin 1ΔKASH mice was not reported, and it was unclear whether KASH domain-less Nesprin 1 isoforms were expressed in these mice that may have obscured the phenotype (Zhang et al. 2007b). Similarly to Nesprin 1rKASH, Nesprin 1ΔKASH mice had altered nuclear positioning and shape in skeletal muscle, but these parameters were not reported in cardiomyocytes.
As noted above, Nesprin 1 isoforms lacking KASH domains exist; therefore, strategies to replace or delete the KASH domain may still be able to express these Nesprin 1 isoforms. To overcome this, Zhang et al. inserted LoxP sites either side of an exon in SYNE1 that is common to both KASH domain-containing and KASH domain-less isoforms to allow generation of Nesprin 1−/− mice (Zhang et al. 2010). Akin to the Nesprin 1rKASH model, Nesprin 1−/− mice had reduced survival rates, exhibiting 60% perinatal lethality, growth restriction, and compromised exercise capacity. In agreement with the other studies of Nesprin 1 function, nuclear morphology and positioning in skeletal muscle were abnormal. However, no changes in cardiac contractile function were observed up to 12 months of age, suggesting that there may be some compensatory mechanism occurring in these mice.
Along these lines, Banerjee et al. reasoned that Nesprin 2 may be compensating for loss of Nesprin 1 in the heart and therefore took an approach to conditionally ablate Nesprin 1 expression in cardiomyocytes on a Nesprin 2-null background (hereafter called Nesprin 1/2 cKO) (Banerjee et al. 2014). Cardiac-specific deletion of Nesprin 1 was achieved by crossing Nkx2.5-Cre mice (McFadden et al. 2005) with the floxed Nesprin 1 mouse line used to generate Nesprin 1−/− mice. Conditional ablation of Nesprin 1 or global deletion of Nesprin 2 alone did not alter cellular or cardiac function. Conversely, Nesprin 1/2 cKO mice developed cardiomyopathy at 10 weeks and displayed altered nuclear positioning, shape, and chromatin positioning. Furthermore, Nesprin 1/2 cKO mice had impaired gene expression changes in response to biomechanical load. These data suggest that Nesprins 1 and 2 play critical roles in normal cardiomyocyte function, thereby enabling the heart to adapt appropriately to mechanical demands. Failure to do so resulted in aberrant apoptosis, cardiac remodeling, fibrosis, and eventually heart failure, which are also observed in patients with mutations in Nesprins 1 and 2 (Haskell et al. 2017; Puckelwartz et al. 2010).
These studies highlight the power of using mouse models to understand mammalian cardiac function and the importance of a functional LINC complex. However, because the Nesprin 1 floxed allele (Zhang et al. 2010) would remove all Nesprin 1 isoforms, a key question that remained unanswered in the field was which of the Nesprin 1 isoforms was important for normal cardiac function?
Because Nesprin 1G and Nesprin 1α2 are the predominant isoforms of Nesprin 1 expressed in striated muscle (Duong et al. 2014; Padmakumar et al. 2004; Randles et al. 2010), we generated two novel mouse lines in which either the actin-binding domain of Nesprin 1G or the actin-binding domain of Nesprin 1α2 was ablated (Nesprin 1ΔCH and Nesprin 1α2−/−, respectively) (Stroud et al. 2017). Surprisingly, Nesprin 1ΔCH mice were born at Mendelian ratios, displayed no perinatal lethality nor loss in bodyweight compared to wildtype littermates. Furthermore, they lived normal lifespans, with no evidence of cardiac dysfunction up to 18 months of age. These data demonstrate that actin binding by Nesprin 1 is dispensable for viability and that the lack of actin binding in Nesprin 1G is not sufficient to explain the perinatal lethality observed in Nesprin 1−/− mice. We therefore postulated that Nesprin 1α2 might be the critical isoform. Similarly to the Nesprin 1−/− mice, Nesprin 1α2−/− mice exhibited perinatal lethality and nuclear mispositioning in skeletal muscle fibers. Components of the LINC complex were largely unaffected, apart from SUN1, which was mislocalized from the NE and its levels slightly reduced, suggesting Nesprin 1α2 may preferentially bind to SUN1. Isolation of embryonic hearts from Nesprin 1α2−/− at embryonic day 18.5 (E18.5) revealed normal heart weight/body weight ratios and histological analyses revealed no gross morphological defects. To understand whether Nesprin 1α2 was critical for normal heart development and function, cardiac-specific knockout mice were generated, but they exhibited normal heart function up to 14 months of age (unpublished data). These data are in agreement with previous findings that showed simultaneous deletion of Nesprins 1 and 2 is required to perturb cardiac function and suggest that Nesprin 2 expression in the heart may compensate for loss of Nesprin 1α2 (Banerjee et al. 2014).
It would be interesting to investigate whether Nesprin 1G and Nesprin 1α2 play similar or different roles in the heart by crossing the Nesprin 1CH f/f and Nesprin 1α2f/f with a cardiac-specific Cre on a Nesprin 2-null background. Alternatively, approaches to induce physiological or pathological cardiac hypertrophy in the Nesprin 1ΔCH or Nesprin 1α2 cKO mice may be necessary to elicit a phenotype. Clearly, further work is needed to understand whether the cardiomyopathy-associated mutations in Nesprin 1 actually lead to cardiac dysfunction in mammalian hearts. With the advent of CRISPR/Cas9 technology to generate point mutations, knock-in mice that mimic human mutations will enable further exploration of Nesprins 1 and 2 function.
Nesprin 3
In contrast to the many Nesprin 1 and 2 isoforms, Nesprin 3 exists as two isoforms; 3α (108 kDa) or 3β (99 kDa), with 3α the predominant isoform in the heart (Wilhelmsen et al. 2005) (Fig. 2). Both Nesprin 3 isoforms contain a C-terminal KASH domain that binds SUN 1/2 and localizes Nesprin 3 to the nuclear envelope in cardiac tissue (Ketema et al. 2013). The N-terminus of Nesprin 3α interacts with the ABD domain of plectin, which in turn interacts with intermediate filaments (Ketema et al. 2007).
In terms of Nesprin 3 function in vivo, there are currently no mutations described in Nesprin 3 associated with cardiomyopathy. Furthermore, ablation of Nesprin 3 expression in both zebrafish and mouse models resulted in no baseline phenotype (Ketema et al. 2013; Postel et al. 2011). It would be interesting to test whether Nesprin 3 KO mice have an abnormal cardiac response to stress because knockdown of Nesprin 3 in vitro leads to defects in cell migration and morphology (Khatau et al. 2012; Morgan et al. 2011; Petrie et al. 2014). However, these effects may be masked by the presence of Nesprins 1 and 2 and therefore may require ablation of all three Nesprin genes.
SUN proteins
SUN proteins are named after their C-terminal SUN domain that was first identified in fission yeast protein Sad1 and C. elegans protein UNC-84 (Hagan and Yanagida 1995; Malone et al. 1999). In mammals, five SUN proteins have been identified (Starr and Fridolfsson 2010). SUN1 and SUN2 were cloned from human brain cDNA (Malone et al. 1999) and are ubiquitously expressed, whereas SUN3, SUN4, and SUN5 are tissue restricted in the testes (Frohnert et al. 2011; Gob et al. 2010; Xing et al. 2004). Importantly, SUN1 and SUN2 are expressed in the heart and skeletal muscle (Crisp et al. 2006; Puckelwartz et al. 2009; Zhang et al. 2010) and therefore will be discussed in more detail in this review. We refer readers elsewhere for a broader overview of SUN domain proteins in other cell types and tissues (Rothballer et al. 2013).
At the mRNA level, six different isoforms of SUN1 are potentially expressed in the heart (Gob et al. 2011; Nishioka et al. 2016). All of these isoforms are predicted to contain the SUN domain, stalk region, transmembrane (TM) domain, but vary in the length that the N-terminus protrudes into the nucleoplasm. There are potentially 6 and 12 isoforms of SUN2 that are expressed in mouse and human heart, respectively (www.ensembl.org). However, it remains to be elucidated whether the shorter SUN1 and SUN2 isoforms are translated into protein, and indeed whether they play different functional roles. Both SUN1 and SUN2 are type 2 membrane proteins and have similar domain architectures. The largest isoform of human SUN 1 is around 90 kDa and SUN2 is ~80 kDa. Overall, they are highly similar and share 64% homology (Haque et al. 2006). SUN1 and SUN2 comprise an N-terminal region that interacts with lamins inside the nucleus (Crisp et al. 2006; Haque et al. 2006; Nishioka et al. 2016; Sosa et al. 2013), followed by single TM domain that spans the INM. Adjacent to the TM domain is the ‘stalk’ region that is structurally made up of coiled-coil repeats necessary for trimerization (Hennen et al. 2018; Jahed et al. 2018; Sosa et al. 2012; Wang et al. 2012; Zhou et al. 2012). The most conserved region between SUN proteins is the C-terminal SUN domain that interacts with the KASH domain of Nesprins (Sosa et al. 2012; Wang et al. 2012; Zhou et al. 2012).
The integral nature of SUN proteins in the LINC complex makes it surprising that compared to other LINC components, relatively few mutations in SUN1 and SUN2 are associated with cardiomyopathy (Meinke et al. 2014). It appears that SUN proteins play a role as genetic modifiers because disease phenotypes are only observed in patients when mutations in SUN proteins are combined with other mutations in cardiomyopathy-associated genes. For example, the R453W mutation in LMNA normally results in a mild form of EDMD (Brown et al. 2001; Colomer et al. 2002; Fidzianska and Hausmanowa-Petrusewicz 2003; Raffaele Di Barletta et al. 2000; Voit et al. 1988; Vytopil et al. 2003); however, when combined with a W377C mutation in SUN1, it resulted in cardiac abnormalities followed by heart failure at age 34 (Meinke et al. 2014). Further evidence of their role as genetic modifiers comes from dystrophic (LMNA−/−) and progeric (LMNAΔ9) laminopathy mouse models, as well as from patient-derived fibroblasts. SUN1 was shown to be dramatically upregulated in these mouse models. Interestingly, ablation of SUN1 in both of these models extended lifespan and ameliorated the phenotypes observed (Chen et al. 2012). Specifically, cardiac function as assessed by ejection fraction was restored to near wildtype levels of ~ 70% in DKO mice compared to ~ 50% in LMNA−/− mice. Furthermore, the noticeable increase of sarcoplasmic vacuoles and number of inflammatory cells in the LMNA−/− mice were reduced in DKO mice. The underlying molecular mechanisms behind the pathogenesis are unknown, but the authors noted that the cytotoxicity may be due to the excessive accumulation of SUN1 in the Golgi apparatus.
In mice, global ablation of SUN1 expression resulted in sterility due to an essential role of SUN1 in gametogenesis (Ding et al. 2007); however, no changes in cardiac or skeletal muscle function were noted. Unlike SUN1 KO mice, SUN2 KO mice were normal presumably due to functional redundancy between SUN proteins (Lei et al. 2009). Conversely, global ablation of SUN1 and SUN2 resulted in perinatal lethality, likely because of respiratory defects. Interestingly, the perinatal lethality observed in these mice could be rescued by overexpressing SUN1 with a neural-specific promoter. Furthermore, it is intriguing that the DKO mice have a similar phenotype to the Nesprin 1α2 KO and Nesprin 1/2 DKO mice. Taken together, these data suggest that SUN1 may play unique, non-redundant roles in neurons and the reproductive organs. Whether SUN1 and SUN2 play essential, non-redundant roles in the heart remain unknown.
The generation of floxed alleles of these genes will enable targeted deletion of specific isoforms in specific tissues to further our understanding in cardiomyocytes. Furthermore, armed with the knowledge that SUN proteins likely play key roles as genetic modifiers, it would be interesting to sequence SUN1 and SUN2 genes in patients with cardiomyopathy in which other LINC complex mutations have been identified that may not fully explain the resulting phenotype. This may reveal clues as to why certain strong cardiomyopathy/EDMD phenotypes in the clinic are not well recapitulated in mouse models that are designed to mimic the exact mutation found in humans (Ozawa et al. 2006; Stroud et al. 2018).
Emerin
Emerin is ubiquitously expressed in tissues and localizes to the NE in both cardiac and skeletal muscle (Manilal et al. 1996; Nagano et al. 1996; Stroud et al. 2018, 2017). Emerin is a type II membrane protein in which the nucleoplasmic N-terminus is adjacent to a TM domain that traverses the INM followed by a short luminal region that resides in the PNS (Manilal et al. 1996; Nagano et al. 1996). The interaction between Emerin and A-type lamins is thought to be important for its retention at the INM (Cartegni et al. 1997; Holaska et al. 2002; Manilal et al. 1996; Nagano et al. 1996). Other binding partners exist and include SUN1, SUN2, Nesprin 1α, HDAC3, and BAF, the latter two providing a link between Emerin and heterochromatin (Berk et al. 2014; Demmerle et al. 2012; Haque et al. 2010; Lee et al. 2001; Mislow et al. 2002a). For an extensive list of Emerin-binding partners, we refer the reader here (Berk et al. 2013).
At the cellular level, Emerin-null mouse embryonic fibroblasts (MEFs) have abnormal nuclear shape, altered NE plasticity, and impaired response to mechanical stimulation (Lammerding et al. 2005; Rowat et al. 2006). These data suggest that Emerin may regulate gene expression to enable the cell to adapt to mechanical load. In support of this, Emerin has been shown to indirectly regulate the localization of the mechanosignalling transcription factor megakaryoblastic leukaemia 1 (MKL1) by modulating actin dynamics (Ho et al. 2013). MKL1, also known as myocardin-related transcription factor a (MRTF-A), is a co-activator of serum response factor, which is a master regulator of cytoskeletal proteins such as vinculin and actin.
Mutations in Emerin lead to X-linked EDMD, cardiac conduction defects, DCM, and skeletal muscle defects (Bione et al. 1994, 1995; Nagano et al. 1996; Nigro et al. 1995; Vohanka et al. 2001; Yamada and Kobayashi 1996). For an extensive overview of disorders caused by Emerin mutations, we refer the reader to recent reviews (Astejada et al. 2007; Holaska 2008). The majority of these mutations are predicted to result in loss of Emerin expression (Manilal et al. 1996; Nagano et al. 1996). Therefore, Emerin KO mice were generated to study its function in the heart and skeletal muscle (Lammerding et al. 2005; Melcon et al. 2006; Ozawa et al. 2006; Stubenvoll et al. 2015). Surpisingly, Emerin-null mice have no overt skeletal myopathy and a mild atrioventricular conduction defect that is age-related (Melcon et al. 2006; Ozawa et al. 2006). Interestingly, a recent study revealed that Emerin KO mice have a mild, albeit significant reduction in ejection fraction that was observed using functional magnetic resonance imaging (MRI) at 12 weeks (Stubenvoll et al. 2015). Furthermore, Emerin KO mice challenged with transverse aortic constriction (TAC)-induced pressure overload showed a decline in ejection fraction and increase in end-systolic volume at 6 months after TAC compared to wildtype controls (Rockman et al. 1991; Stubenvoll et al. 2015). This study revealed that Emerin-null hearts contain an increased number of small cardiomyocytes that was likely due to aberrant β-catenin signaling during development, which resulted in an increase in cell proliferation at embryonic day 12.5 (E12.5). These data suggest that Emerin plays a role in the heart under baseline conditions and is necessary to cope with the consequences of pressure-induced cardiac overload.
Despite these conflicting findings about Emerin’s function in the heart, it is clear that Emerin-null mice do not develop a severe skeletal muscle myopathy as might be expected from patient data. To explain this difference, Shin et al. recently showed that the disparities observed between mouse and humans are likely due to the differential expression of Emerin between the two species. Specifically, Emerin levels in human skeletal muscle were found to be much higher than in the equivalent mouse tissue, suggesting that other protein(s) may be able to functionally compensate (Shin et al. 2013). Indeed, the authors found the expression levels of Emerin’s binding partner, lamina-associated polypeptide 1 (LAP1), were higher in mouse than in human. In line with LAP1 playing an important role in muscle, conditional ablation of LAP1 in mouse skeletal muscle (LAP1 scKO) led to muscular dystrophy resulting in premature death.
Furthermore, the authors wanted to understand whether there was a functional interaction between Emerin and LAP1, and therefore crossed LAP1 scKO with Emerin-null mice to generate LAP1 scKO/Emerin−/y (herein named DKO). Strikingly, DKO mice had an exacerbated myopathy and their median survival was reduced to more than half compared to LAP1 scKO mice. These data suggest that Emerin and LAP1 both physically and functionally interact in skeletal muscle (Shin et al. 2013).
Lamina-associated polypeptide 1
Lamina-associated polypeptide 1 (LAP1) consists of three isoforms (A, B, C) that are the result of alternative mRNA splicing of the TOR1AIP1 gene. Initially discovered in rat liver nuclear envelope extracts (Senior and Gerace 1988), they have recently been described in mouse cardiac and skeletal muscle (Shin et al. 2014, 2013). LAP1B and LAP1C are expressed in human hearts, with LAP1B thought to be the predominant isoform (Rebelo et al. 2015; Santos et al. 2014).
LAP1A, LAP1B, and LAP1C have predicted molecular weights of 75, 68, and 55 kDa, respectively (Senior and Gerace 1988). All LAP1 isoforms are type II membrane proteins. The N-terminus is located on the nucleoplasmic side of the INM and interacts with nuclear lamins and Emerin (Foisner and Gerace 1993; Shin et al. 2013). Emerin binding has been shown to be essential for normal skeletal muscle function (Shin et al. 2013). The C-terminus interacts with the Torsin AAA+ ATPase family members Torsin A, B, 2, and 3 (Goodchild and Dauer 2005; Jungwirth et al. 2010; Kim et al. 2010). Interestingly, global ablation of LAP1 was sufficient to exactly phenocopy both the nuclear blebbing and perinatal lethality observed in Torsin A KO mice, which suggests the interaction may have functional importance in neurons (Kim et al. 2010). Whether there is a similar functional interaction between the Torsin family member enriched in the heart, Torsin B, and LAP1 remains to be elucidated.
In humans, several mutations in the LAP1 gene are associated with cardiac defects (Dorboz et al. 2014; Kayman-Kurekci et al. 2014). For example, a nonsense frameshift mutation c.186delG in the TOR1AIP1 gene was identified in a three-generation family diagnosed with autosomal recessive limb-girdle muscular dystrophy with joint contractures (Kayman-Kurekci et al. 2014). Notably, one of the family members presented with reduced cardiac function as measured by echocardiography with an ejection fraction of 57% but had normal sinus rhythm as assessed by electrocardiogram analysis. Ultrastructural analysis of muscle biopsies revealed that the sarcomeres were intact, but the nuclei were fragmented and nuclear envelope disrupted. As expected, Western blot analysis of muscle tissue from the affected patient revealed the loss of the LAP1B band at 68 kDa, but noted the upregulation of a band at ~ 50 kDa. With the recent identification of LAP1C in humans (Santos et al. 2014), others have speculated that this ~ 50 kDa band might be LAP1C (Rebelo et al. 2015). Whether this band is indeed LAP1C remains to be determined. Nevertheless, it is apparently unable to completely rescue the phenotype caused by loss of LAP1B. It is also possible that the 50 kDa protein may partially compensate for the loss of LAP1B because deletion of all LAP1 isoforms in mice results in a much more severe form of skeletal myopathy with cardiac involvement (Shin et al. 2013). Further support for this hypothesis is from evidence of a missense mutation in LAP1 that changes glutamic acid 482 to alanine (E482A) and was identified in a patient with progressive dystonia, cerebellar atrophy, and DCM (Dorboz et al. 2014). Unlike the nonsense mutation c.186delG described above, the E482A mutation led to a more severe form of DCM that resulted in death at age 17. Western blot and immunofluorescence analysis of patient fibroblasts revealed a drastic reduction of all three LAP1 isoforms. Interestingly, the ultrastructure of the nuclear envelope in fibroblasts appeared normal. Whole exome sequencing was used to reveal this mutation, but it was unclear whether other mutations in genes that are strongly associated with DCM, such as LMNA (encoding lamin A/C), were investigated.
LAP1 function has also been investigated using mouse models (Kim et al. 2010; Shin et al. 2014, 2013). As mentioned above, deletion of all LAP1 isoforms in mouse striated muscle resulted in severe skeletal and cardiac myopathy (Shin et al. 2013). However, the specific role of LAP1 in the heart remained elusive. To investigate this, Shin et al. made use of their LAP1 floxed mice to generate a cardiomyocyte-specific knockout of LAP1 (herein referred to as LAP1 cKO) (Shin et al. 2014). LAP1 cKO mice were born at the expected Mendelian ratios and appeared overtly normal at 20 weeks; however, echocardiography revealed a significant reduction in cardiac function as measured by fractional shortening. Furthermore, levels of the fetal genes ANP and BNP as well as the pro-fibrotic markers collagen 1a1, collagen 1a2, and fibronectin 1 were all significantly upregulated in LAP1 cKO hearts. Mutant mouse models generated to study the function of LAP1’s interaction partners lamin A/C (LMNAH222P/H222P) and Emerin (EMD−/−) have previously shown activated extracellular signal-regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK), and p38alpha branches of the mitogen-activated protein kinase (MAPK) signaling pathways (Muchir et al. 2007a, b). Therefore, the authors investigated whether these were affected in the LAP1 cKO mice. Interestingly, they found that ERK1/2 and JNK pathways were activated, but not p38alpha. The authors speculate that the extent of activation of these pathways may depend on the severity of the phenotype. For example, the LMNAH222P/H222P mice (which develop significant DCM at 20 weeks) have all three pathways activated, whereas only ERK1/2 were activated in Emerin-null mice.
With regard to recent evidence from human data, suggesting that LAP1C might be upregulated and partially compensate for LAP1B loss, it would be interesting to generate LAP1 isoform-specific knockout mice. This would shed light on which LAP1 isoform(s) are critical for heart development and function.
Lamina-associated polypeptide 2
Due to alternative splicing of the lamina-associated polypeptide 2 (LAP2) gene, there are six potential isoforms of LAP2. Five of which have been observed in both mouse and human hearts (LAP2α, LAP2β, LAP2γ, LAP2δ, LAP2ε) (Gotic et al. 2010; Harris et al. 1994; Taylor et al. 2005).
LAP2, also known as thymopoietin (TMPO), is one of the founding members of the LAP2, Emerin, and MAN1 (LEM) domain-containing family of proteins (Lin et al. 2000). All LAP2 isoforms contain an N-terminal LEM-like domain followed by the LEM domain that interacts with barrier to autointegration factor (BAF), which in turn binds to heterochromatin and double-stranded DNA (Montes de Oca et al. 2011). Whereas the N-termini of the LAP2 isoforms share high degrees of homology, their C-termini vary both in terms of length and the domains contained (Berger et al. 1996). Compared to the other isoforms of LAP2, LAP2α lacks a TM domain but contains an extended C-terminus that is important for its interaction with lamin A/C and mostly localizes to the nucleoplasm (Dechat et al. 1998, 2000). Conversely, LAP2β contains a TM domain and binds to B-type lamins at the nuclear envelope (Foisner and Gerace 1993; Furukawa et al. 1998). Presumably, the other TM domain-containing isoforms of LAP2 localize similarly to LAP2β.
LAP2α is the best characterized isoform of LAP2 and has been studied in both human and mouse hearts and will therefore be discussed in detail. In terms of LAP2α function, a missense mutation in LAP2α has been associated with a severe form of DCM that was diagnosed in two brothers at ages 33 and 22 (Taylor et al. 2005). The mutation identified changes arginine 690 to cysteine (R690C) in the C-terminus of LAP2α in a region that interacts with lamin A/C. Functional analysis of the R690C mutant in vitro revealed that the ability of LAP2α to interact with lamin A/C was reduced by around 50–75% compared to wildtype.
Since no patient biopsies were available, it was unclear whether LAP2α was expressed at normal levels in the myocardium. Therefore, to understand LAP2α’s function in vivo, loss of function mouse models were generated (Gotic et al. 2010; Naetar et al. 2008). Although the LAP2α−/− mice were grossly indistinguishable from their littermates and lived normal lifespans, they had significantly reduced cardiac function as measured by fractional shortening at 10 weeks of age. LAP2α−/− mice had normal heart weight:body weight ratios, but 18% of older mice developed extensive subendocardial fibrosis of the left ventricle. At the molecular level, master cardiac transcription factors MEF2c and GATA4 as well as their downstream targets were deregulated in LAP2α−/− mice. LAP2α−/− mice showed a blunted response to chronic infusion of isoproterenol to induce hypertrophy likely due to downregulation of the beta-adrenergic receptor β2AR. To further investigate the role of LAP2α in striated muscle cells (including cardiomyocytes), LAP2α floxed mice were crossed with mice expressing Cre recombinase under control of the muscle creatine kinase promoter. However, these mice did not develop systolic dysfunction, suggesting that LAP2α may play a more critical role during early stages of heart development. In support of this, LAP2α expression levels were higher during embryonic heart development (E12 and E14) and were downregulated at postnatal day 2 (Gotic et al. 2010).
Given the timing of LAP2α expression in embryonic cardiomyocytes, it would be interesting to ablate LAP2α expression using an earlier cardiomyocyte-specific Cre, such as XMLC2v that is expressed at E7.5 (Breckenridge et al. 2007). This will be important to understand whether the cardiac defects in the germline deleted LAP2α−/− mice were cardiomyocyte-autonomous or due to loss of LAP2α expression in other cell types. In addition, the role of other LAP2 isoforms remains unexplored in terms of cardiac function.
MAN1
Human MAN1, also known as LEM domain-containing protein 3 (Lemd3), is an 82.3 kDa INM protein that comprises an N-terminal LEM domain, two TM domains followed by MAN1-Src1-p C-terminal (MSC) domain and an RNA recognition motif (RRM) (Lin et al. 2000). MAN1 is ubiquitously expressed and has been found at the RNA level in human hearts (Lin et al. 2000). It has been shown to interact with lamin A/C, BAF, Smads 2 and 3, PPM1A (Smad2/3 phosphatase) and to regulate transforming growth factor β (TGF-β) and bone morphogenetic protein (BMP) signaling pathways through its interaction with Smads (Bourgeois et al. 2013; Cohen et al. 2007; Ishimura et al. 2006; Lin et al. 2005; Osada et al. 2003; Pan et al. 2005; Raju et al. 2003). Specifically, Smads 2 and 3 are phosphorylated downstream of TGF-β/BMP receptor signaling and translocate to the nucleus (Bourgeois et al. 2013; Lin et al. 2005; Osada et al. 2003; Pan et al. 2005; Raju et al. 2003). Once inside the nucleus, the C-terminus of MAN1 binds to Smads 2 and 3 and sequesters them at the nuclear envelope. This interaction is thought to compete for binding with transcription activator complexes, thereby regulating expression of gene downstream of TGF-β signaling.
In humans, several mutations have been described in MAN1 that lead to defects in the bone and skin (Hellemans et al. 2004). To our knowledge, no mutations have been described that result in skeletal or cardiac myopathy. However, several studies have been performed to uncover the role of MAN1 in model organisms. Global deletion of MAN1 using a genetrap approach led to embryonic lethality at E10.5 in mice. This was likely due to aberrant TGF-β signaling and vasculogenesis in the embryonic yolk sac (Ishimura et al. 2006; Cohen et al. 2007). Further analysis of the mutant hearts revealed pericardial edema in 31 of 32 embryos and defects in left-right axis formation of the heart (Ishimura et al. 2008). Further adding to a potential role of MAN1 in the heart, knockdown of MAN1 using a morpholino approach in Xenopus also led to cardiac defects (Reil and Dabauvalle 2013). Whether these findings were due to altered cardiomyocyte function or other confounding factors such as knocking down MAN1 in all tissues remain to be explored. Nevertheless, the expression of MAN1 during early stages of heart development makes studying MAN1 function in cardiomyocytes an interesting prospect.
LEM domain-containing 2
LEM domain-containing 2 (Lem2) localizes to the INM and interacts with heterochromatin, A-type lamins, and BAF (Brachner et al. 2005; Cai et al. 2001, 2007; Liu et al. 2003). Lem2 is widely expressed in adult human and mouse tissues, with levels enriched in both the heart and skeletal muscle (Brachner et al. 2005; Chen et al. 2006) Similarly to MAN1, Lem2 contains an N-terminal LEM domain followed by a TM domain and a MSC domain at its C-terminus. Both LEM and MSC domains are reported to bind to heterochromatin in yeast (Barrales et al. 2016). These domains are highly conserved, suggesting they play an important role in Lem2 function. Lem2 interacts with the repressive histone modifications H3K9Me2 and H3K9Me3 in nematodes (Towbin et al. 2012), and in yeast, the loss of Lem2 led to the reduction of H3K9Me2, suggesting that it may play a role in establishing or maintaining this repressive mark (Barrales et al. 2016). As well as interacting with histone modifications, the LEM domain binds to the chromatin protein BAF, which itself directly binds to double-stranded DNA and directly to histones and influences histone modifications (Margalit et al. 2007; Montes de Oca et al. 2011, 2005, 2009; Tapia et al. 2015). Given the localization of Lem2 to the INM and proximity to heterochromatin as well as its interaction with A-type lamins, these properties make Lem2 a good candidate for regulating gene expression. In support of this, Lem2 has been shown to regulate ERK signaling in mouse myoblasts (Huber et al. 2009), is critical for nematode development (Barkan et al. 2012; Liu et al. 2003), and was found to be important for skeletal muscle regeneration after cardiotoxin injury (Tapia et al. 2015).
In humans, an autosomal recessive mutation in the LEM domain of Lem2 was recently identified that substitutes leucine 13 to arginine and leads to juvenile cataracts (Boone et al. 2016). Notably, some of these patients also died from sudden cardiac death; however, the cause of this was unclear and warrants further investigation.
In terms of mouse models to investigate Lem2 function, global loss of Lem2 resulted in embryonic lethality between E10.5 and E11.5. Although many normal hallmarks were observed in E10.5 knockout embryos, most of the tissues were substantially smaller in size. Furthermore, multiple MAP kinase pathways were upregulated, including ERK1/2, JNK, and p38 as well as AKT. As noted earlier, MAN1 KO mice display a similar timepoint of lethality at E10.5. However, the Lem2 KO embryos show a distinct phenotype from the MAN1 KO with no defects in vasculogenesis of the yolk sac, or changes in TGF-β signaling. Furthermore, Lem2 lacks the region used by MAN1 to interact with Smads (Caputo et al. 2006).
Luma
Luma, alternatively known as transmembrane protein 43 (TMEM43), is associated with the LINC complex and was initially identified with a proteomics screen for INM proteins in neuroblastoma cells (Dreger et al. 2001). Luma is widely expressed in tissues including the heart (Bengtsson and Otto 2008; Dreger et al. 2001; Schirmer et al. 2003; Stroud et al. 2018) and is highly conserved throughout metazoans. It contains four TM domains and localizes to the INM, where it interacts with lamins A/C, B1, Emerin, and SUN2 (Bengtsson and Otto 2008; Liang et al. 2011).
Several mutations in Luma have been identified as the cause of arrhythmogenic cardiomyopathy (AC) (Christensen et al. 2011; Haywood et al. 2013; Hodgkinson et al. 2013; Honda et al. 2016; Merner et al. 2008; Milting et al. 2015). The best characterized is a missense, autosomal dominant mutation that changes serine 358 to leucine and was identified as the unequivocal cause of arrhythmogenic right ventricle cardiomyopathy type 5 (ARVC5) in humans (Christensen et al. 2011; Hodgkinson et al. 2013; Merner et al. 2008). The S358L mutation is in the third TM domain of Luma and was originally mapped in an extended eight-generation family in Newfoundland, Canada. The S358L mutation was subsequently identified in a German family, which shares a common haplotype with those from Newfoundland, suggesting that the mutation originated in Europe (Milting et al. 2015).
ARVC5 is a fully penetrant, lethal, sex-influenced disorder, in which men have a median lifespan of 41 compared to 83 in the control group and women have a median lifespan of 71. The disorder is fully penetrant at the age of 63 and 76 in men and women, respectively. The prominent features of ARVC5 are ventricular tachycardia, fibrofatty replacement of cardiomyocytes, premature ventricular contractions (PVCs), and left ventricular dilation resulting in heart failure and sudden cardiac death. The available therapies for ARVC5 patients are limited and include the use of implantable cardioverter defibrillator (ICD) devices. ICDs have shown to be more effective in ARVC5 patients particularly when used in men as a primary prophylaxis (before incidence of sustained ventricular tachycardia) versus secondary prophylaxis (after sustained ventricular tachycardia) (Hodgkinson et al. 2016).
Luma function was originally assessed in vitro using cardiomyocytes and non-cardiomyocytes (Franke et al. 2014; Jiang et al. 2017; Rajkumar et al. 2012; Siragam et al. 2014). However, there was limited information as to the role of Luma is cardiac development and function in vivo. Therefore, we took genetic approaches to uncover the role of Luma in cardiac function by generating several novel mouse models (Stroud et al. 2018). Using a series of cell-type specific antibodies and indicator mouse lines, Luma was expressed sporadically in cardiomyocytes, pericytes, and endothelial cells, whereas it was highly and uniformly expressed in fibroblasts and vascular smooth muscle cells (VSMCs). Furthermore, Luma localization was consistent between mouse and human myocardium as human myocardium from non-failing adults showed the same localization to the nuclear envelope. Importantly, the specificity of the Luma antibody was validated using Luma KO mouse tissue for both Western blots and immunofluorescence.
Because Luma was found in most cardiac cell types, a global knockout approach was taken to try and understand Luma’s role in heart function. Given the strength of data suggesting that Luma as the unequivocal cause of AC in humans, it was surprising that Luma KO mice appeared normal and had no abnormalities in cardiac function at baseline up to almost 2 years of age. Furthermore, no changes in the fetal gene programme or pro-fibrotic genes were observed, further supporting the notion that Luma KO hearts were normal. We hypothesized that other LINC complex proteins may compensate for loss of Luma, but the localization and levels of lamins A/C, B1, Emerin, Sun1, and Sun2 were unchanged (Stroud et al. 2018).
The LINC complex has been suggested to play a role in mediating response to hypertrophic stimuli (Cupesi et al. 2010; Stubenvoll et al. 2015); therefore, Luma KO mice were challenged with pressure overload using TAC. Similarly to their wildtype littermates, Luma KO mice displayed a normal hypertrophic response at 1–2 weeks, then progressively went into failure by 4 weeks post-TAC. Furthermore, the contractile function of Luma KO mice was unaffected in response to beta-adrenergic stimulation.
From these data, we concluded that ablation of Luma does not affect cardiac development or function, and that the S358L mutation that causes ARVC5 might be a gain of function mutation. To test this hypothesis, Luma S358L knock-in mice were generated using CRISPR/Cas9 technology to mimic the mutation described in humans with ARVC5. Mice heterozygous for the S358L mutation displayed no changes in cardiac function up to 1 year of age. Other mouse models of human autosomal dominant mutations exhibit a phenotype when both alleles are mutated in mice (Arimura et al. 2005; Mounkes et al. 2005). In contrast to other models, cardiac function in Luma S358L homozygous mice was normal and mutant Luma remained localized to the NE. A previous report suggested that Luma expression was downregulated in human myocardium from patients with the S358L mutation (Christensen et al. 2011). However, immunofluorescence and Western blotting revealed no changes between wildtype and both S358L heterozygous and homozygous mice.
An intriguing finding from this study was that both in human and mouse myocardium, Luma is predominantly expressed in fibroblasts and VSMCs. ARVC is normally considered a ‘disease of the desmosome’ (Zhang et al. 2015); therefore, it is unusual for a protein that is expressed at low levels in cardiomyocytes to result in ARVC. We therefore speculate that paracrine crosstalk between fibroblasts, VSMCs, and cardiomyocytes might be perturbed by the Luma mutation in humans. However, the pathophysiological mechanisms that occur as a result of the S358L mutation remain unclear. Because patients with the mutation develop ARVC5 later in life, we speculated that aging the S358L mice for longer might be necessary to elicit a phenotype. Alternatively, stressing the S358L knock-in mice using approaches to induce physiological or pathological hypertrophy may be required, as was shown for Luma’s binding partner, Emerin (Stubenvoll et al. 2015). Another possibility is that other yet-to-be-identified mutation(s) exist in modifier genes that are within the vicinity of the Luma gene and are therefore inherited in the same manner. Identification of these will require whole genome sequencing.
Nuclear lamins
Nuclear lamins were first described in the 1970s and are type V intermediate filaments that readily self-associate to form parallel coil-coil homodimers (Gerace et al. 1978). The homodimers assemble higher-order filamentous structures to form the nuclear lamina that resides beneath the INM. The major components of the nuclear lamina in cardiomyocytes are A-type lamins A and C (encoded by the LMNA gene) and B-type lamins 1 and 2 (encoded by LMNB1 and LMNB2, respectively) (Fisher et al. 1986; Hoger et al. 1990; Lin and Worman 1995; McKeon et al. 1986). Lamins share a similar structure that is comprised of an alpha helical rod that is abutted to non-helical globular domains at the N- and C-termini (Stuurman et al. 1998).
The LMNA gene is alternatively spliced to generate lamins A and C, which are the major isoforms found in cardiomyocytes (Fisher et al. 1986; McKeon et al. 1986). Expression of A-type lamins is ubiquitous and developmentally regulated as they are only detected in differentiated cells (Broers et al. 1997; Rober et al. 1989; Solovei et al. 2013). Conversely, B-type lamins are constitutively expressed and are found in multiple tissues (Worman and Courvalin 2000). For a comprehensive review of nuclear lamin discovery and characterization, we refer readers elsewhere (Gerace and Huber 2012).
Nuclear lamins have been shown to play multiple roles in the cell, which are mediated via interactions at the nuclear periphery and nucleoplasm (Prokocimer et al. 2009). They are important for maintenance of nuclear structural integrity, chromatin organization, gene expression regulation, nuclear positioning, and cytoskeletal organization (Andres and Gonzalez 2009; Broers et al. 2004; Dechat et al. 2008; Folker et al. 2011; Lammerding et al. 2004) and interact with a plethora of different binding partners that enable direct or indirect interaction with chromatin through histones, lamin B receptor, heterochromatin protein 1, Emerin, and BAF (Burke and Stewart 2013; Mattout-Drubezki and Gruenbaum 2003; Wagner and Krohne 2007; Wilson and Foisner 2010).
As intermediate filament proteins, lamins provide mechanical stability to the nucleus (Lammerding et al. 2004). In support of this, nuclei from lamin A/C-null mouse embryonic fibroblasts have increased nuclear deformation, defective mechanotransduction, and impaired viability under mechanical strain (Lammerding et al. 2004). These data fit with in vivo data from lamin A/C-null mice in which cardiomyocyte nuclei are misshapen (Nikolova et al. 2004). Lamin A/C levels have been shown to scale with collagen 1 levels in tissues, whereas lamin B levels remain constant (Swift et al. 2013). These data suggest that lamin A/C may act as a ‘mechanostat’ and its levels are regulated with regard to tissue stiffness. It is tempting to speculate that a more fibrotic and stiffer myocardium may have elevated lamin A/C levels. However, the results from meta-analysis of existing proteomic datasets from cardiac tissue was inconclusive as to whether this is the case (Cho et al. 2017). Further work using defined animal models of cardiac fibrosis that can be tightly controlled and manipulated will be required to understand if there is a correlation between fibrosis and lamin A/C levels in the heart.
As well as providing mechanical stability, lamin A/C also plays a role in responding to mechanical force stimulation. For example, lamin A/C hemizygous mice have an attenuated response to TAC-induced pressure overload and impaired activation of the mechanosensitive gene EGR-1 (Lammerding et al. 2004). Further evidence comes from MEFs derived from lamin A/C-null or LMNAN195K/N195K mice that showed impaired nuclear translocation of the mechanosensitive transcription factor MKL1 (Ho et al. 2013). Cardiac sections from both mouse lines had significantly reduced fractions of cardiomyocytes with nuclear MKL1. MKL1 is a co-activator of SRF, which is a master regulator of genes encoding cytoskeletal proteins.
These data suggest that lamin A/C not only plays a mechanical role but also may regulate gene expression. In support of this, hearts from the LMNAH222P/H222P mouse model for EDMD and cardiomyopathy have upregulated MAPK signaling pathways, ERK 1/2, JNK, and p38-MAPK (Muchir et al. 2007b, 2012). Notably, these signaling pathways are activated prior to the onset of cardiomyopathy, suggesting that targeting these pathways with inhibitors may ameliorate cardiac dysfunction in the LMNAH222P/H222P laminopathy model. Indeed, beneficial effects to the heart have been observed in mice when treated with various inhibitors of these pathways (Muchir et al. 2009; Wu et al. 2011, 2010).
Other potential roles of lamins include regulating muscle differentiation by differential binding of lamina-associated domain (LAD) regions of chromatin (Perovanovic et al. 2016). LADs are typically gene-poor regions that are tethered to the lamina in a constitutive or facultative manner (Pickersgill et al. 2006; van Steensel and Belmont 2017). Little is known about LADs in the heart, but work from Eric Schirmer’s lab suggests that tissue-specific nuclear envelope transmembrane proteins may play a role in striated muscle differentiation by regulating LADs at the NE (Robson et al. 2016). Whether LADs play a similar role in the heart remain to be elucidated.
Since the identification of mutations in lamin genes that cause cardiac and skeletal myopathies in the late 1990s and early 2000s, coined ‘laminopathies’, the nuclear lamin field has received considerable attention using cellular and mouse models to try and uncover the pathophysiological mechanisms (Bonne et al. 1999, 2000; Fatkin et al. 1999; Worman 2012; Worman et al. 2009). Laminopathies present as a myriad of different disorders including DCM (Fatkin et al. 1999), heart-hand syndrome (Renou et al. 2008), autosomal dominant (AD) EDMD (Bonne et al. 1999), limb-girdle muscular dystrophy (Muchir et al. 2000), lipodystrophies (Shackleton et al. 2000), and LMNA-related congenital muscular dystrophy (Quijano-Roy et al. 2008). For an extensive list of laminopathies we refer readers here (Captur et al. 2018). With regard to cardiomyopathy, mutations in the LMNA gene account for 5–8% of patients with genetic DCM (Hershberger et al. 2013; McNally and Mestroni 2017). In contrast, no mutations in B-type lamins have been identified that lead to cardiomyopathy. This may be because the severity of the phenotype is not tolerated in mammals. Indeed, loss of function models of both lamins B1 and B2 in mice results in embryonic lethality (Coffinier et al. 2010; Padiath et al. 2006; Vergnes et al. 2004).
As mentioned above, many mouse models have been generated to understand the function of lamin A/C in the heart (Arimura et al. 2005; Cattin et al. 2013; Kubben et al. 2011; Lu et al. 2010; Mewborn et al. 2010; Mounkes et al. 2005; Nikolova et al. 2004; Sullivan et al. 1999; Wang et al. 2006). Interestingly, some of these mouse models (e.g. H222P, N195K) need to be homozygous for the LMNA mutation to elicit a phenotype, whereas in humans, the disease is normally dominant. This could be due to unidentified mutations in modifier genes or greater dosage sensitivity in humans. Thus far, no approaches have been taken to ablate lamin A/C, B1, or B2 expression in a cardiomyocyte-specific manner. This is somewhat surprising given the robust phenotypes observed with the global knockout mice. Combined with the data from the global knockout mice, these data would lend new insights on which cell-type expression of lamins A/C, B1, and B2 is critical for normal heart function. Furthermore, the original lamin A/C knockout mice produce a 54 kDa truncated version of lamin A/C that arises as a result of an unforeseen splicing event (Jahn et al. 2012; Nikolova et al. 2004). The truncated version contains the N-terminal globular domain and rod domains, whereas a large proportion of the C-terminal globular domain is missing. Therefore, generation of LMNA floxed mice would not only enable the original model to be validated as a loss of function model but also allow tissue-specific ablation.
Future directions
The importance of the LINC complex in fundamental cellular processes has become apparent since the discovery of mutations in LINC complex-encoding genes that cause cardiomyopathy (McNally and Mestroni 2017; Meinke and Schirmer 2016; Mejat and Misteli 2010; Worman et al. 2010). The LINC complex structurally supports the nucleus and physically couples the nucleoskeleton to the cytoskeleton (Crisp et al. 2006; Sosa et al. 2013). Furthermore, it is hypothesized to act as a mechanosensor that translates mechanical stimuli into biochemical cues that mediate gene expression changes and affect chromatin organization (Jaalouk and Lammerding 2009; Lombardi and Lammerding 2011). It is clear that the structural and gene expression roles are not mutually exclusive and that modulating one aspect of LINC complex function will likely influence the other. As a consequence, disruption of the LINC complex and its associated proteins affects cellular function and contributes to cardiomyopathy as described in this review.
Integrated approaches that combine mouse models, cell-based assays, and biophysical analyses have contributed to our progress in understanding LINC complex function. Armed with the knowledge of the mutations in the LINC complex that cause cardiomyopathy, the next steps will be to uncover the pathophysiological molecular mechanism underlying disease.
As described in this review, many of the LINC complex and its associated proteins have been studied using global knockout mouse approaches, including Nesprins, SUN proteins, Emerin, LAP1, LAP2, MAN1, LEM2, Luma, and nuclear lamins. Therefore, a lot remains to be uncovered in terms of the tissue-specific roles in cardiomyocytes as well as the function of the different isoforms. Another challenge will be to identify the interaction networks of the LINC complex proteins. Until recently, this remained elusive, partly due to the insolubility of the nuclear lamina and chromatin (Roux et al. 2012). However, with the advent of proximity proteomic approaches, such as Bio-ID and APEX, the LINC complex interactome can now be explored (Kim et al. 2016; Kim and Roux 2016; Rhee et al. 2013; Roux et al. 2012). This approach has been mostly applied in cell culture, but mouse lines are currently being engineered to identify binding partners in an in vivo setting.
As well as taking these approaches, complementary assessment of LINC complex function in human cardiomyocytes generated using induced pluripotent stem cell (iPSC) technology will be essential. Indeed, next steps have been taken using this protocol from laminopathy patients (Shimojima et al. 2017; Siu et al. 2012). Despite the question of cardiomyocyte maturity in iPSC-derived cardiomyocytes, the field is rapidly progressing thanks to the technology to generate engineered heart tissue (Ronaldson-Bouchard et al. 2018; Zimmermann et al. 2006). Furthermore, the use of CRISPR/Cas9 machinery to correct patient mutations and generate isogenic controls will further strengthen this technology. Not only will this approach enhance our understanding of the fundamental biology of the LINC complex, it will enable the development of novel therapeutics to target the devastating effects of LINC complex-associated cardiomyopathies.
Abbreviations
- AC:
-
Arrhythmogenic cardiomyopathy
- CH:
-
Calponin homology
- DCM:
-
Dilated cardiomyopathy
- EDMD:
-
Emery-Dreifuss muscular dystrophy
- INM:
-
Inner nuclear membrane
- KASH:
-
Klarsicht, ANC-1, and Syne homology
- KO:
-
Knockout
- LAD:
-
Lamina-associated domain
- LAP:
-
Lamina-associated peptide
- LINC:
-
Linker of nucleoskeleton and cytoskeleton
- MEF:
-
Mouse embryonic fibroblast
- NE:
-
Nuclear envelope
- NPC:
-
Nuclear pore complex
- ONM:
-
Outer nuclear membrane
- PNS:
-
Perinuclear space
- SUN:
-
Sad1/UNC-84
- TAC:
-
Transverse aortic constriction
- TM:
-
Transmembrane
- VSMC:
-
Vascular smooth muscle cell
References
Andres V, Gonzalez JM (2009) Role of A-type lamins in signaling, transcription, and chromatin organization. J Cell Biol 187:945–957. https://doi.org/10.1083/jcb.200904124
Apel ED, Lewis RM, Grady RM, Sanes JR (2000) Syne-1, a dystrophin- and Klarsicht-related protein associated with synaptic nuclei at the neuromuscular junction. J Biol Chem 275:31986–31995. https://doi.org/10.1074/jbc.M004775200
Arimura T et al (2005) Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet 14:155–169. https://doi.org/10.1093/hmg/ddi017
Astejada MN et al (2007) Emerinopathy and laminopathy clinical, pathological and molecular features of muscular dystrophy with nuclear envelopathy in Japan. Acta Myol 26:159–164
Banerjee I et al (2014) Targeted ablation of nesprin 1 and nesprin 2 from murine myocardium results in cardiomyopathy, altered nuclear morphology and inhibition of the biomechanical gene response. PLoS Genet 10:e1004114. https://doi.org/10.1371/journal.pgen.1004114
Barkan R et al (2012) Ce-emerin and LEM-2: essential roles in Caenorhabditis elegans development, muscle function, and mitosis. Mol Biol Cell 23:543–552. https://doi.org/10.1091/mbc.E11-06-0505
Barrales RR, Forn M, Georgescu PR, Sarkadi Z, Braun S (2016) Control of heterochromatin localization and silencing by the nuclear membrane protein Lem2. Genes Dev 30:133–148. https://doi.org/10.1101/gad.271288.115
Bengtsson L, Otto H (2008) LUMA interacts with emerin and influences its distribution at the inner nuclear membrane. J Cell Sci 121:536–548. https://doi.org/10.1242/jcs.019281
Berger R et al (1996) The characterization and localization of the mouse thymopoietin/lamina-associated polypeptide 2 gene and its alternatively spliced products. Genome Res 6:361–370
Berk JM, Tifft KE, Wilson KL (2013) The nuclear envelope LEM-domain protein emerin. Nucleus 4:298–314. https://doi.org/10.4161/nucl.25751
Berk JM, Simon DN, Jenkins-Houk CR, Westerbeck JW, Gronning-Wang LM, Carlson CR, Wilson KL (2014) The molecular basis of emerin-emerin and emerin-BAF interactions. J Cell Sci 127:3956–3969. https://doi.org/10.1242/jcs.148247
Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D (1994) Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet 8:323–327. https://doi.org/10.1038/ng1294-323
Bione S et al (1995) Identification of new mutations in the Emery-Dreifuss muscular dystrophy gene and evidence for genetic heterogeneity of the disease. Hum Mol Genet 4:1859–1863
Bonne G et al (1999) Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet 21:285–288. https://doi.org/10.1038/6799
Bonne G et al (2000) Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol 48:170–180
Boone PM et al (2016) Hutterite-type cataract maps to chromosome 6p21.32-p21.31, cosegregates with a homozygous mutation in LEMD2, and is associated with sudden cardiac death. Mol Genet Genomic Med 4:77–94. https://doi.org/10.1002/mgg3.181
Bourgeois B et al (2013) Inhibition of TGF-beta signaling at the nuclear envelope: characterization of interactions between MAN1, Smad2 and Smad3, and PPM1A. Sci Signal 6:ra49. https://doi.org/10.1126/scisignal.2003411
Brachner A, Reipert S, Foisner R, Gotzmann J (2005) LEM2 is a novel MAN1-related inner nuclear membrane protein associated with A-type lamins. J Cell Sci 118:5797–5810. https://doi.org/10.1242/jcs.02701
Breckenridge R, Kotecha S, Towers N, Bennett M, Mohun T (2007) Pan-myocardial expression of Cre recombinase throughout mouse development. Genesis 45:135–144. https://doi.org/10.1002/dvg.20275
Broers JL, Machiels BM, Kuijpers HJ, Smedts F, van den Kieboom R, Raymond Y, Ramaekers FC (1997) A- and B-type lamins are differentially expressed in normal human tissues. Histochem Cell Biol 107:505–517
Broers JL et al (2004) Decreased mechanical stiffness in LMNA-/- cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Hum Mol Genet 13:2567–2580. https://doi.org/10.1093/hmg/ddh295
Brohawn SG, Partridge JR, Whittle JR, Schwartz TU (2009) The nuclear pore complex has entered the atomic age. Structure 17:1156–1168. https://doi.org/10.1016/j.str.2009.07.014
Brown CA et al (2001) Novel and recurrent mutations in lamin A/C in patients with Emery-Dreifuss muscular dystrophy. Am J Med Genet 102:359–367
Burke B, Stewart CL (2013) The nuclear lamins: flexibility in function. Nat Rev Mol Cell Biol 14:13–24. https://doi.org/10.1038/nrm3488
Cai M, Huang Y, Ghirlando R, Wilson KL, Craigie R, Clore GM (2001) Solution structure of the constant region of nuclear envelope protein LAP2 reveals two LEM-domain structures: one binds BAF and the other binds DNA. EMBO J 20:4399–4407. https://doi.org/10.1093/emboj/20.16.4399
Cai M, Huang Y, Suh JY, Louis JM, Ghirlando R, Craigie R, Clore GM (2007) Solution NMR structure of the barrier-to-autointegration factor-Emerin complex. J Biol Chem 282:14525–14535. https://doi.org/10.1074/jbc.M700576200
Captur G et al (2018) Lamin and the heart. Heart 104:468–479. https://doi.org/10.1136/heartjnl-2017-312338
Caputo S et al (2006) The carboxyl-terminal nucleoplasmic region of MAN1 exhibits a DNA binding winged helix domain. J Biol Chem 281:18208–18215. https://doi.org/10.1074/jbc.M601980200
Cartegni L et al (1997) Heart-specific localization of emerin: new insights into Emery-Dreifuss muscular dystrophy. Hum Mol Genet 6:2257–2264
Cattin ME et al (2013) Heterozygous LmnadelK32 mice develop dilated cardiomyopathy through a combined pathomechanism of haploinsufficiency and peptide toxicity. Hum Mol Genet 22:3152–3164. https://doi.org/10.1093/hmg/ddt172
Chen IH, Huber M, Guan T, Bubeck A, Gerace L (2006) Nuclear envelope transmembrane proteins (NETs) that are up-regulated during myogenesis. BMC Cell Biol 7:38. https://doi.org/10.1186/1471-2121-7-38
Chen CY et al (2012) Accumulation of the inner nuclear envelope protein Sun1 is pathogenic in progeric and dystrophic laminopathies. Cell 149:565–577. https://doi.org/10.1016/j.cell.2012.01.059
Cho S, Irianto J, Discher DE (2017) Mechanosensing by the nucleus: from pathways to scaling relationships. J Cell Biol 216:305–315. https://doi.org/10.1083/jcb.201610042
Christensen AH, Andersen CB, Tybjaerg-Hansen A, Haunso S, Svendsen JH (2011) Mutation analysis and evaluation of the cardiac localization of TMEM43 in arrhythmogenic right ventricular cardiomyopathy. Clin Genet 80:256–264. https://doi.org/10.1111/j.1399-0004.2011.01623.x
Coffinier C et al (2010) Abnormal development of the cerebral cortex and cerebellum in the setting of lamin B2 deficiency. Proc Natl Acad Sci U S A 107:5076–5081. https://doi.org/10.1073/pnas.0908790107
Cohen TV, Kosti O, Stewart CL (2007) The nuclear envelope protein MAN1 regulates TGFbeta signaling and vasculogenesis in the embryonic yolk sac. Development 134:1385–1395. https://doi.org/10.1242/dev.02816
Colomer J et al (2002) Autosomal dominant Emery-Dreifuss muscular dystrophy: a new family with late diagnosis. Neuromuscul Disord 12:19–25
Crisp M et al (2006) Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol 172:41–53. https://doi.org/10.1083/jcb.200509124
Cupesi M, Yoshioka J, Gannon J, Kudinova A, Stewart CL, Lammerding J (2010) Attenuated hypertrophic response to pressure overload in a lamin A/C haploinsufficiency mouse. J Mol Cell Cardiol 48:1290–1297. https://doi.org/10.1016/j.yjmcc.2009.10.024
Dechat T, Gotzmann J, Stockinger A, Harris CA, Talle MA, Siekierka JJ, Foisner R (1998) Detergent-salt resistance of LAP2alpha in interphase nuclei and phosphorylation-dependent association with chromosomes early in nuclear assembly implies functions in nuclear structure dynamics. EMBO J 17:4887–4902. https://doi.org/10.1093/emboj/17.16.4887
Dechat T, Korbei B, Vaughan OA, Vlcek S, Hutchison CJ, Foisner R (2000) Lamina-associated polypeptide 2alpha binds intranuclear A-type lamins. J Cell Sci 113(Pt 19):3473–3484
Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD (2008) Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev 22:832–853. https://doi.org/10.1101/gad.1652708
Dellefave L, McNally EM (2010) The genetics of dilated cardiomyopathy. Curr Opin Cardiol 25:198–204. https://doi.org/10.1097/HCO.0b013e328337ba52
Demmerle J, Koch AJ, Holaska JM (2012) The nuclear envelope protein emerin binds directly to histone deacetylase 3 (HDAC3) and activates HDAC3 activity. J Biol Chem 287:22080–22088. https://doi.org/10.1074/jbc.M111.325308
Ding X, Xu R, Yu J, Xu T, Zhuang Y, Han M (2007) SUN1 is required for telomere attachment to nuclear envelope and gametogenesis in mice. Dev Cell 12:863–872. https://doi.org/10.1016/j.devcel.2007.03.018
Dorboz I et al (2014) Severe dystonia, cerebellar atrophy, and cardiomyopathy likely caused by a missense mutation in TOR1AIP1. Orphanet J Rare Dis 9:174. https://doi.org/10.1186/s13023-014-0174-9
Dreger M, Bengtsson L, Schoneberg T, Otto H, Hucho F (2001) Nuclear envelope proteomics: novel integral membrane proteins of the inner nuclear membrane. Proc Natl Acad Sci U S A 98:11943–11948. https://doi.org/10.1073/pnas.211201898
Duong NT, Morris GE, Lam le T, Zhang Q, Sewry CA, Shanahan CM, Holt I (2014) Nesprins: tissue-specific expression of epsilon and other short isoforms. PLoS One 9:e94380. https://doi.org/10.1371/journal.pone.0094380
Fatkin D et al (1999) Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med 341:1715–1724. https://doi.org/10.1056/NEJM199912023412302
Fidzianska A, Hausmanowa-Petrusewicz I (2003) Architectural abnormalities in muscle nuclei. Ultrastructural differences between X-linked and autosomal dominant forms of EDMD. J Neurol Sci 210:47–51
Fisher DZ, Chaudhary N, Blobel G (1986) cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc Natl Acad Sci U S A 83:6450–6454
Foisner R, Gerace L (1993) Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell 73:1267–1279
Folker ES, Ostlund C, Luxton GW, Worman HJ, Gundersen GG (2011) Lamin A variants that cause striated muscle disease are defective in anchoring transmembrane actin-associated nuclear lines for nuclear movement. Proc Natl Acad Sci U S A 108:131–136. https://doi.org/10.1073/pnas.1000824108
Franke WW, Dorflinger Y, Kuhn C, Zimbelmann R, Winter-Simanowski S, Frey N, Heid H (2014) Protein LUMA is a cytoplasmic plaque constituent of various epithelial adherens junctions and composite junctions of myocardial intercalated disks: a unifying finding for cell biology and cardiology. Cell Tissue Res 357:159–172. https://doi.org/10.1007/s00441-014-1865-1
Frohnert C, Schweizer S, Hoyer-Fender S (2011) SPAG4L/SPAG4L-2 are testis-specific SUN domain proteins restricted to the apical nuclear envelope of round spermatids facing the acrosome. Mol Hum Reprod 17:207–218. https://doi.org/10.1093/molehr/gaq099
Furukawa K, Fritze CE, Gerace L (1998) The major nuclear envelope targeting domain of LAP2 coincides with its lamin binding region but is distinct from its chromatin interaction domain. J Biol Chem 273:4213–4219
Gerace L, Huber MD (2012) Nuclear lamina at the crossroads of the cytoplasm and nucleus. J Struct Biol 177:24–31. https://doi.org/10.1016/j.jsb.2011.11.007
Gerace L, Tapia O (2018) Messages from the voices within: regulation of signaling by proteins of the nuclear lamina. Curr Opin Cell Biol 52:14–21. https://doi.org/10.1016/j.ceb.2017.12.009
Gerace L, Blum A, Blobel G (1978) Immunocytochemical localization of the major polypeptides of the nuclear pore complex-lamina fraction. Interphase and mitotic distribution. J Cell Biol 79:546–566
Gob E, Schmitt J, Benavente R, Alsheimer M (2010) Mammalian sperm head formation involves different polarization of two novel LINC complexes. PLoS One 5:e12072. https://doi.org/10.1371/journal.pone.0012072
Gob E, Meyer-Natus E, Benavente R, Alsheimer M (2011) Expression of individual mammalian Sun1 isoforms depends on the cell type. Commun Integr Biol 4:440–442. https://doi.org/10.4161/cib.4.4.15369
Goodchild RE, Dauer WT (2005) The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J Cell Biol 168:855–862. https://doi.org/10.1083/jcb.200411026
Gorlich D, Kutay U (1999) Transport between the cell nucleus and the cytoplasm. Annu Rev Cell Dev Biol 15:607–660. https://doi.org/10.1146/annurev.cellbio.15.1.607
Gotic I et al (2010) Lamina-associated polypeptide 2alpha loss impairs heart function and stress response in mice. Circ Res 106:346–353. https://doi.org/10.1161/CIRCRESAHA.109.205724
Grossman E, Medalia O, Zwerger M (2012) Functional architecture of the nuclear pore complex. Annu Rev Biophys 41:557–584. https://doi.org/10.1146/annurev-biophys-050511-102328
Hagan I, Yanagida M (1995) The product of the spindle formation gene sad1+ associates with the fission yeast spindle pole body and is essential for viability. J Cell Biol 129:1033–1047
Haque F et al (2006) SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol Cell Biol 26:3738–3751. https://doi.org/10.1128/MCB.26.10.3738-3751.2006
Haque F, Mazzeo D, Patel JT, Smallwood DT, Ellis JA, Shanahan CM, Shackleton S (2010) Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J Biol Chem 285:3487–3498. https://doi.org/10.1074/jbc.M109.071910
Harris CA, Andryuk PJ, Cline S, Chan HK, Natarajan A, Siekierka JJ, Goldstein G (1994) Three distinct human thymopoietins are derived from alternatively spliced mRNAs. Proc Natl Acad Sci U S A 91:6283–6287
Haskell GT et al (2017) Whole exome sequencing identifies truncating variants in nuclear envelope genes in patients with cardiovascular disease. Circ Cardiovasc Genet 10. https://doi.org/10.1161/CIRCGENETICS.116.001443
Haywood AF et al (2013) Recurrent missense mutations in TMEM43 (ARVD5) due to founder effects cause arrhythmogenic cardiomyopathies in the UK and Canada. Eur Heart J 34:1002–1011. https://doi.org/10.1093/eurheartj/ehs383
Hellemans J et al (2004) Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat Genet 36:1213–1218. https://doi.org/10.1038/ng1453
Hennen J, Saunders CA, Mueller JD, Luxton GWG (2018) Fluorescence fluctuation spectroscopy reveals differential SUN protein oligomerization in living cells. Mol Biol Cell 29:1003–1011. https://doi.org/10.1091/mbc.E17-04-0233
Hershberger RE, Hedges DJ, Morales A (2013) Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 10:531–547. https://doi.org/10.1038/nrcardio.2013.105
Ho CY, Jaalouk DE, Vartiainen MK, Lammerding J (2013) Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature 497:507–511. https://doi.org/10.1038/nature12105
Hodgkinson KA et al (2013) The natural history of a genetic subtype of arrhythmogenic right ventricular cardiomyopathy caused by a p.S358L mutation in TMEM43. Clin Genet 83:321–331. https://doi.org/10.1111/j.1399-0004.2012.01919.x
Hodgkinson KA et al (2016) Long-term clinical outcome of arrhythmogenic right ventricular cardiomyopathy in individuals with a p.S358L mutation in TMEM43 following implantable cardioverter defibrillator therapy. Circ Arrhythm Electrophysiol 9. https://doi.org/10.1161/CIRCEP.115.003589
Hoger TH, Zatloukal K, Waizenegger I, Krohne G (1990) Characterization of a second highly conserved B-type lamin present in cells previously thought to contain only a single B-type lamin. Chromosoma 100:67–69
Holaska JM (2008) Emerin and the nuclear lamina in muscle and cardiac disease. Circ Res 103:16–23. https://doi.org/10.1161/CIRCRESAHA.108.172197
Holaska JM, Wilson KL, Mansharamani M (2002) The nuclear envelope, lamins and nuclear assembly. Curr Opin Cell Biol 14:357–364
Holt I et al (2016) Specific localization of nesprin-1-alpha2, the short isoform of nesprin-1 with a KASH domain, in developing, fetal and regenerating muscle, using a new monoclonal antibody. BMC Cell Biol 17:26. https://doi.org/10.1186/s12860-016-0105-9
Honda T, Kanai Y, Ohno S, Ando H, Honda M, Niwano S, Ishii M (2016) Fetal arrhythmogenic right ventricular cardiomyopathy with double mutations in TMEM43. Pediatr Int 58:409–411. https://doi.org/10.1111/ped.12832
Huber MD, Guan T, Gerace L (2009) Overlapping functions of nuclear envelope proteins NET25 (Lem2) and emerin in regulation of extracellular signal-regulated kinase signaling in myoblast differentiation. Mol Cell Biol 29:5718–5728. https://doi.org/10.1128/MCB.00270-09
Ishimura A, Ng JK, Taira M, Young SG, Osada S (2006) Man1, an inner nuclear membrane protein, regulates vascular remodeling by modulating transforming growth factor beta signaling. Development 133:3919–3928. https://doi.org/10.1242/dev.02538
Ishimura A, Chida S, Osada S (2008) Man1, an inner nuclear membrane protein, regulates left-right axis formation by controlling nodal signaling in a node-independent manner. Dev Dyn 237:3565–3576. https://doi.org/10.1002/dvdy.21663
Jaalouk DE, Lammerding J (2009) Mechanotransduction gone awry. Nat Rev Mol Cell Biol 10:63–73. https://doi.org/10.1038/nrm2597
Jahed Z, Fadavi D, Vu UT, Asgari E, Luxton GWG, Mofrad MRK (2018) Molecular insights into the mechanisms of SUN1 oligomerization in the nuclear envelope. Biophys J 114:1190–1203. https://doi.org/10.1016/j.bpj.2018.01.015
Jahn D et al (2012) A truncated lamin A in the Lmna -/- mouse line: implications for the understanding of laminopathies. Nucleus 3:463–474. https://doi.org/10.4161/nucl.21676
Jiang C et al (2017) TMEM43/LUMA is a key signaling component mediating EGFR-induced NF-kappaB activation and tumor progression. Oncogene 36:2813–2823. https://doi.org/10.1038/onc.2016.430
Jungwirth M, Dear ML, Brown P, Holbrook K, Goodchild R (2010) Relative tissue expression of homologous torsinB correlates with the neuronal specific importance of DYT1 dystonia-associated torsinA. Hum Mol Genet 19:888–900. https://doi.org/10.1093/hmg/ddp557
Kayman-Kurekci G et al (2014) Mutation in TOR1AIP1 encoding LAP1B in a form of muscular dystrophy: a novel gene related to nuclear envelopathies. Neuromuscul Disord 24:624–633. https://doi.org/10.1016/j.nmd.2014.04.007
Ketema M, Wilhelmsen K, Kuikman I, Janssen H, Hodzic D, Sonnenberg A (2007) Requirements for the localization of nesprin-3 at the nuclear envelope and its interaction with plectin. J Cell Sci 120:3384–3394. https://doi.org/10.1242/jcs.014191
Ketema M, Kreft M, Secades P, Janssen H, Sonnenberg A (2013) Nesprin-3 connects plectin and vimentin to the nuclear envelope of Sertoli cells but is not required for Sertoli cell function in spermatogenesis. Mol Biol Cell 24:2454–2466. https://doi.org/10.1091/mbc.E13-02-0100
Khatau SB et al (2012) The distinct roles of the nucleus and nucleus-cytoskeleton connections in three-dimensional cell migration. Sci Rep 2:488. https://doi.org/10.1038/srep00488
Kim DI, Roux KJ (2016) Filling the void: proximity-based labeling of proteins in living cells. Trends Cell Biol 26:804–817. https://doi.org/10.1016/j.tcb.2016.09.004
Kim CE, Perez A, Perkins G, Ellisman MH, Dauer WT (2010) A molecular mechanism underlying the neural-specific defect in torsinA mutant mice. Proc Natl Acad Sci U S A 107:9861–9866. https://doi.org/10.1073/pnas.0912877107
Kim DI, Jensen SC, Noble KA, Kc B, Roux KH, Motamedchaboki K, Roux KJ (2016) An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell 27:1188–1196. https://doi.org/10.1091/mbc.E15-12-0844
Kubben N et al (2011) Post-natal myogenic and adipogenic developmental: defects and metabolic impairment upon loss of A-type lamins. Nucleus 2:195–207. https://doi.org/10.4161/nucl.2.3.15731
Lammerding J et al (2004) Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 113:370–378. https://doi.org/10.1172/JCI19670
Lammerding J, Hsiao J, Schulze PC, Kozlov S, Stewart CL, Lee RT (2005) Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J Cell Biol 170:781–791. https://doi.org/10.1083/jcb.200502148
Lee KK, Haraguchi T, Lee RS, Koujin T, Hiraoka Y, Wilson KL (2001) Distinct functional domains in emerin bind lamin A and DNA-bridging protein BAF. J Cell Sci 114:4567–4573
Lei K et al (2009) SUN1 and SUN2 play critical but partially redundant roles in anchoring nuclei in skeletal muscle cells in mice. Proc Natl Acad Sci U S A 106:10207–10212. https://doi.org/10.1073/pnas.0812037106
Liang WC, Mitsuhashi H, Keduka E, Nonaka I, Noguchi S, Nishino I, Hayashi YK (2011) TMEM43 mutations in Emery-Dreifuss muscular dystrophy-related myopathy. Ann Neurol 69:1005–1013. https://doi.org/10.1002/ana.22338
Lin F, Worman HJ (1995) Structural organization of the human gene (LMNB1) encoding nuclear lamin B1. Genomics 27:230–236. https://doi.org/10.1006/geno.1995.1036
Lin F et al (2000) MAN1, an inner nuclear membrane protein that shares the LEM domain with lamina-associated polypeptide 2 and emerin. J Biol Chem 275:4840–4847
Lin F, Morrison JM, Wu W, Worman HJ (2005) MAN1, an integral protein of the inner nuclear membrane, binds Smad2 and Smad3 and antagonizes transforming growth factor-beta signaling. Hum Mol Genet 14:437–445. https://doi.org/10.1093/hmg/ddi040
Liu J, Lee KK, Segura-Totten M, Neufeld E, Wilson KL, Gruenbaum Y (2003) MAN1 and emerin have overlapping function(s) essential for chromosome segregation and cell division in Caenorhabditis elegans. Proc Natl Acad Sci U S A 100:4598–4603. https://doi.org/10.1073/pnas.0730821100
Lombardi ML, Lammerding J (2011) Keeping the LINC: the importance of nucleocytoskeletal coupling in intracellular force transmission and cellular function. Biochem Soc Trans 39:1729–1734. https://doi.org/10.1042/BST20110686
Lu D, Lian H, Zhang X, Shao H, Huang L, Qin C, Zhang L (2010) LMNA E82K mutation activates FAS and mitochondrial pathways of apoptosis in heart tissue specific transgenic mice. PLoS One 5:e15167. https://doi.org/10.1371/journal.pone.0015167
Malone CJ, Fixsen WD, Horvitz HR, Han M (1999) UNC-84 localizes to the nuclear envelope and is required for nuclear migration and anchoring during C. elegans development. Development 126:3171–3181
Manilal S, Nguyen TM, Sewry CA, Morris GE (1996) The Emery-Dreifuss muscular dystrophy protein, emerin, is a nuclear membrane protein. Hum Mol Genet 5:801–808
Margalit A, Neufeld E, Feinstein N, Wilson KL, Podbilewicz B, Gruenbaum Y (2007) Barrier to autointegration factor blocks premature cell fusion and maintains adult muscle integrity in C. elegans. J Cell Biol 178:661–673. https://doi.org/10.1083/jcb.200704049
Mattout-Drubezki A, Gruenbaum Y (2003) Dynamic interactions of nuclear lamina proteins with chromatin and transcriptional machinery. Cell Mol Life Sci 60:2053–2063. https://doi.org/10.1007/s00018-003-3038-3
McFadden DG, Barbosa AC, Richardson JA, Schneider MD, Srivastava D, Olson EN (2005) The Hand1 and Hand2 transcription factors regulate expansion of the embryonic cardiac ventricles in a gene dosage-dependent manner. Development 132:189–201. https://doi.org/10.1242/dev.01562
McKeon FD, Kirschner MW, Caput D (1986) Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature 319:463–468. https://doi.org/10.1038/319463a0
McNally EM, Mestroni L (2017) Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res 121:731–748. https://doi.org/10.1161/CIRCRESAHA.116.309396
Meinke P, Schirmer EC (2016) The increasing relevance of nuclear envelope myopathies. Curr Opin Neurol 29:651–661. https://doi.org/10.1097/WCO.0000000000000359
Meinke P et al (2014) Muscular dystrophy-associated SUN1 and SUN2 variants disrupt nuclear-cytoskeletal connections and myonuclear organization. PLoS Genet 10:e1004605. https://doi.org/10.1371/journal.pgen.1004605
Mejat A, Misteli T (2010) LINC complexes in health and disease. Nucleus 1:40–52. https://doi.org/10.4161/nucl.1.1.10530
Melcon G et al (2006) Loss of emerin at the nuclear envelope disrupts the Rb1/E2F and MyoD pathways during muscle regeneration. Hum Mol Genet 15:637–651. https://doi.org/10.1093/hmg/ddi479
Mendez-Lopez I, Worman HJ (2012) Inner nuclear membrane proteins: impact on human disease. Chromosoma 121:153–167. https://doi.org/10.1007/s00412-012-0360-2
Merner ND et al (2008) Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet 82:809–821. https://doi.org/10.1016/j.ajhg.2008.01.010
Mewborn SK et al (2010) Altered chromosomal positioning, compaction, and gene expression with a lamin A/C gene mutation. PLoS One 5:e14342. https://doi.org/10.1371/journal.pone.0014342
Milting H et al (2015) The TMEM43 Newfoundland mutation p.S358L causing ARVC-5 was imported from Europe and increases the stiffness of the cell nucleus. Eur Heart J 36:872–881. https://doi.org/10.1093/eurheartj/ehu077
Mislow JM, Holaska JM, Kim MS, Lee KK, Segura-Totten M, Wilson KL, McNally EM (2002a) Nesprin-1alpha self-associates and binds directly to emerin and lamin A in vitro. FEBS Lett 525:135–140
Mislow JM, Kim MS, Davis DB, McNally EM (2002b) Myne-1, a spectrin repeat transmembrane protein of the myocyte inner nuclear membrane, interacts with lamin A/C. J Cell Sci 115:61–70
Montes de Oca R, Lee KK, Wilson KL (2005) Binding of barrier to autointegration factor (BAF) to histone H3 and selected linker histones including H1.1. J Biol Chem 280:42252–42262. https://doi.org/10.1074/jbc.M509917200
Montes de Oca R, Shoemaker CJ, Gucek M, Cole RN, Wilson KL (2009) Barrier-to-autointegration factor proteome reveals chromatin-regulatory partners. PLoS One 4:e7050. https://doi.org/10.1371/journal.pone.0007050
Montes de Oca R, Andreassen PR, Wilson KL (2011) Barrier-to-autointegration factor influences specific histone modifications. Nucleus 2:580–590. https://doi.org/10.4161/nucl.2.6.17960
Morgan JT et al (2011) Nesprin-3 regulates endothelial cell morphology, perinuclear cytoskeletal architecture, and flow-induced polarization. Mol Biol Cell 22:4324–4334. https://doi.org/10.1091/mbc.E11-04-0287
Mounkes LC, Kozlov SV, Rottman JN, Stewart CL (2005) Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum Mol Genet 14:2167–2180. https://doi.org/10.1093/hmg/ddi221
Muchir A et al (2000) Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet 9:1453–1459
Muchir A, Pavlidis P, Bonne G, Hayashi YK, Worman HJ (2007a) Activation of MAPK in hearts of EMD null mice: similarities between mouse models of X-linked and autosomal dominant Emery Dreifuss muscular dystrophy. Hum Mol Genet 16:1884–1895. https://doi.org/10.1093/hmg/ddm137
Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G, Worman HJ (2007b) Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J Clin Invest 117:1282–1293. https://doi.org/10.1172/JCI29042
Muchir A, Shan J, Bonne G, Lehnart SE, Worman HJ (2009) Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum Mol Genet 18:241–247. https://doi.org/10.1093/hmg/ddn343
Muchir A, Wu W, Choi JC, Iwata S, Morrow J, Homma S, Worman HJ (2012) Abnormal p38alpha mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum Mol Genet 21:4325–4333. https://doi.org/10.1093/hmg/dds265
Naetar N et al (2008) Loss of nucleoplasmic LAP2alpha-lamin A complexes causes erythroid and epidermal progenitor hyperproliferation. Nat Cell Biol 10:1341–1348. https://doi.org/10.1038/ncb1793
Nagano A et al (1996) Emerin deficiency at the nuclear membrane in patients with Emery-Dreifuss muscular dystrophy. Nat Genet 12:254–259. https://doi.org/10.1038/ng0396-254
Nigro V et al (1995) SSCP detection of novel mutations in patients with Emery-Dreifuss muscular dystrophy: definition of a small C-terminal region required for emerin function. Hum Mol Genet 4:2003–2004
Nikolova V et al (2004) Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest 113:357–369. https://doi.org/10.1172/JCI19448
Nikolova-Krstevski V et al (2011) Nesprin-1 and actin contribute to nuclear and cytoskeletal defects in lamin A/C-deficient cardiomyopathy. J Mol Cell Cardiol 50:479–486. https://doi.org/10.1016/j.yjmcc.2010.12.001
Nishioka Y, Imaizumi H, Imada J, Katahira J, Matsuura N, Hieda M (2016) SUN1 splice variants, SUN1_888, SUN1_785, and predominant SUN1_916, variably function in directional cell migration. Nucleus 7:572–584. https://doi.org/10.1080/19491034.2016.1260802
Osada S, Ohmori SY, Taira M (2003) XMAN1, an inner nuclear membrane protein, antagonizes BMP signaling by interacting with Smad1 in Xenopus embryos. Development 130:1783–1794
Ozawa R et al (2006) Emerin-lacking mice show minimal motor and cardiac dysfunctions with nuclear-associated vacuoles. Am J Pathol 168:907–917. https://doi.org/10.2353/ajpath.2006.050564
Padiath QS et al (2006) Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet 38:1114–1123. https://doi.org/10.1038/ng1872
Padmakumar VC, Abraham S, Braune S, Noegel AA, Tunggal B, Karakesisoglou I, Korenbaum E (2004) Enaptin, a giant actin-binding protein, is an element of the nuclear membrane and the actin cytoskeleton. Exp Cell Res 295:330–339. https://doi.org/10.1016/j.yexcr.2004.01.014
Padmakumar VC et al (2005) The inner nuclear membrane protein Sun1 mediates the anchorage of Nesprin-2 to the nuclear envelope. J Cell Sci 118:3419–3430. https://doi.org/10.1242/jcs.02471
Pan D, Estevez-Salmeron LD, Stroschein SL, Zhu X, He J, Zhou S, Luo K (2005) The integral inner nuclear membrane protein MAN1 physically interacts with the R-Smad proteins to repress signaling by the transforming growth factor-{beta} superfamily of cytokines. J Biol Chem 280:15992–16001. https://doi.org/10.1074/jbc.M411234200
Perovanovic J et al (2016) Laminopathies disrupt epigenomic developmental programs and cell fate. Sci Transl Med 8:335ra358. https://doi.org/10.1126/scitranslmed.aad4991
Petrie RJ, Koo H, Yamada KM (2014) Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix. Science 345:1062–1065. https://doi.org/10.1126/science.1256965
Pickersgill H, Kalverda B, de Wit E, Talhout W, Fornerod M, van Steensel B (2006) Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat Genet 38:1005–1014. https://doi.org/10.1038/ng1852
Postel R, Ketema M, Kuikman I, de Pereda JM, Sonnenberg A (2011) Nesprin-3 augments peripheral nuclear localization of intermediate filaments in zebrafish. J Cell Sci 124:755–764. https://doi.org/10.1242/jcs.081174
Potter C et al (2017) Multiple isoforms of Nesprin1 are integral components of ciliary rootlets. Curr Biol 27:2014–2022 e6. https://doi.org/10.1016/j.cub.2017.05.066
Prokocimer M et al (2009) Nuclear lamins: key regulators of nuclear structure and activities. J Cell Mol Med 13:1059–1085. https://doi.org/10.1111/j.1582-4934.2008.00676.x
Puckelwartz MJ et al (2009) Disruption of nesprin-1 produces an Emery Dreifuss muscular dystrophy-like phenotype in mice. Hum Mol Genet 18:607–620. https://doi.org/10.1093/hmg/ddn386
Puckelwartz MJ et al (2010) Nesprin-1 mutations in human and murine cardiomyopathy. J Mol Cell Cardiol 48:600–608. https://doi.org/10.1016/j.yjmcc.2009.11.006
Quijano-Roy S et al (2008) De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann Neurol 64:177–186. https://doi.org/10.1002/ana.21417
Raffaele Di Barletta M et al (2000) Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery-Dreifuss muscular dystrophy. Am J Hum Genet 66:1407–1412. https://doi.org/10.1086/302869
Rajgor D, Mellad JA, Autore F, Zhang Q, Shanahan CM (2012) Multiple novel nesprin-1 and nesprin-2 variants act as versatile tissue-specific intracellular scaffolds. PLoS One 7:e40098. https://doi.org/10.1371/journal.pone.0040098
Rajkumar R, Sembrat JC, McDonough B, Seidman CE, Ahmad F (2012) Functional effects of the TMEM43 Ser358Leu mutation in the pathogenesis of arrhythmogenic right ventricular cardiomyopathy. BMC Med Genet 13:21. https://doi.org/10.1186/1471-2350-13-21
Raju GP, Dimova N, Klein PS, Huang HC (2003) SANE, a novel LEM domain protein, regulates bone morphogenetic protein signaling through interaction with Smad1. J Biol Chem 278:428–437. https://doi.org/10.1074/jbc.M210505200
Randles KN et al (2010) Nesprins, but not sun proteins, switch isoforms at the nuclear envelope during muscle development. Dev Dyn 239:998–1009. https://doi.org/10.1002/dvdy.22229
Razafsky D, Hodzic D (2015) A variant of Nesprin1 giant devoid of KASH domain underlies the molecular etiology of autosomal recessive cerebellar ataxia type I. Neurobiol Dis 78:57–67. https://doi.org/10.1016/j.nbd.2015.03.027
Rebelo S, da Cruz ESEF, da Cruz ESOA (2015) Genetic mutations strengthen functional association of LAP1 with DYT1 dystonia and muscular dystrophy. Mutat Res Rev Mutat Res 766:42–47. https://doi.org/10.1016/j.mrrev.2015.07.004
Reil M, Dabauvalle MC (2013) Essential roles of LEM-domain protein MAN1 during organogenesis in Xenopus laevis and overlapping functions of emerin. Eur J Cell Biol 92:280–294. https://doi.org/10.1016/j.ejcb.2013.10.008
Renou L et al (2008) Heart-hand syndrome of Slovenian type: a new kind of laminopathy. J Med Genet 45:666–671. https://doi.org/10.1136/jmg.2008.060020
Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY (2013) Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339:1328–1331. https://doi.org/10.1126/science.1230593
Rober RA, Weber K, Osborn M (1989) Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. Development 105:365–378
Robson MI et al (2016) Tissue-specific gene repositioning by muscle nuclear membrane proteins enhances repression of critical developmental genes during myogenesis. Mol Cell 62:834–847. https://doi.org/10.1016/j.molcel.2016.04.035
Rockman HA et al (1991) Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci U S A 88:8277–8281
Ronaldson-Bouchard K et al (2018) Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556:239–243. https://doi.org/10.1038/s41586-018-0016-3
Rothballer A, Schwartz TU, Kutay U (2013) LINCing complex functions at the nuclear envelope: what the molecular architecture of the LINC complex can reveal about its function. Nucleus 4:29–36. https://doi.org/10.4161/nucl.23387
Roux KJ, Kim DI, Raida M, Burke B (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196:801–810. https://doi.org/10.1083/jcb.201112098
Rowat AC, Lammerding J, Ipsen JH (2006) Mechanical properties of the cell nucleus and the effect of emerin deficiency. Biophys J 91:4649–4664. https://doi.org/10.1529/biophysj.106.086454
Santos M et al (2014) Identification of a novel human LAP1 isoform that is regulated by protein phosphorylation. PLoS One 9:e113732. https://doi.org/10.1371/journal.pone.0113732
Schirmer EC, Florens L, Guan T, Yates JR 3rd, Gerace L (2003) Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science 301:1380–1382. https://doi.org/10.1126/science.1088176
Senior A, Gerace L (1988) Integral membrane proteins specific to the inner nuclear membrane and associated with the nuclear lamina. J Cell Biol 107:2029–2036
Shackleton S et al (2000) LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet 24:153–156. https://doi.org/10.1038/72807
Shanahan CM, Weissberg PL, Metcalfe JC (1993) Isolation of gene markers of differentiated and proliferating vascular smooth muscle cells. Circ Res 73:193–204
Shimojima M et al (2017) Emerin plays a crucial role in nuclear invagination and in the nuclear calcium transient. Sci Rep 7:44312. https://doi.org/10.1038/srep44312
Shin JY et al (2013) Lamina-associated polypeptide-1 interacts with the muscular dystrophy protein emerin and is essential for skeletal muscle maintenance. Dev Cell 26:591–603. https://doi.org/10.1016/j.devcel.2013.08.012
Shin JY et al (2014) Depletion of lamina-associated polypeptide 1 from cardiomyocytes causes cardiac dysfunction in mice. Nucleus 5:260–459. https://doi.org/10.4161/nucl.29227
Siragam V et al (2014) TMEM43 mutation p.S358L alters intercalated disc protein expression and reduces conduction velocity in arrhythmogenic right ventricular cardiomyopathy. PLoS One 9:e109128. https://doi.org/10.1371/journal.pone.0109128
Siu CW et al (2012) Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging (Albany NY) 4:803–822. https://doi.org/10.18632/aging.100503
Solovei I et al (2013) LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152:584–598. https://doi.org/10.1016/j.cell.2013.01.009
Sosa BA, Rothballer A, Kutay U, Schwartz TU (2012) LINC complexes form by binding of three KASH peptides to domain interfaces of trimeric SUN proteins. Cell 149:1035–1047. https://doi.org/10.1016/j.cell.2012.03.046
Sosa BA, Kutay U, Schwartz TU (2013) Structural insights into LINC complexes. Curr Opin Struct Biol 23:285–291. https://doi.org/10.1016/j.sbi.2013.03.005
Starr DA, Fridolfsson HN (2010) Interactions between nuclei and the cytoskeleton are mediated by SUN-KASH nuclear-envelope bridges. Annu Rev Cell Dev Biol 26:421–444. https://doi.org/10.1146/annurev-cellbio-100109-104037
Starr DA, Han M (2002) Role of ANC-1 in tethering nuclei to the actin cytoskeleton. Science 298:406–409. https://doi.org/10.1126/science.1075119
Stroud MJ, Banerjee I, Veevers J, Chen J (2014) Linker of nucleoskeleton and cytoskeleton complex proteins in cardiac structure, function, and disease. Circ Res 114:538–548. https://doi.org/10.1161/CIRCRESAHA.114.301236
Stroud MJ, Feng W, Zhang J, Veevers J, Fang X, Gerace L, Chen J (2017) Nesprin 1alpha2 is essential for mouse postnatal viability and nuclear positioning in skeletal muscle. J Cell Biol 216:1915–1924. https://doi.org/10.1083/jcb.201612128
Stroud MJ et al (2018) Luma is not essential for murine cardiac development and function. Cardiovasc Res 114:378–388. https://doi.org/10.1093/cvr/cvx205
Stubenvoll A, Rice M, Wietelmann A, Wheeler M, Braun T (2015) Attenuation of Wnt/beta-catenin activity reverses enhanced generation of cardiomyocytes and cardiac defects caused by the loss of emerin. Hum Mol Genet 24:802–813. https://doi.org/10.1093/hmg/ddu498
Stuurman N, Heins S, Aebi U (1998) Nuclear lamins: their structure, assembly, and interactions. J Struct Biol 122:42–66. https://doi.org/10.1006/jsbi.1998.3987
Sullivan T et al (1999) Loss of A-type Lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 147:913–920
Swift J et al (2013) Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341:1240104. https://doi.org/10.1126/science.1240104
Tapia O, Fong LG, Huber MD, Young SG, Gerace L (2015) Nuclear envelope protein Lem2 is required for mouse development and regulates MAP and AKT kinases. PLoS One 10:e0116196. https://doi.org/10.1371/journal.pone.0116196
Taylor MR et al (2005) Thymopoietin (lamina-associated polypeptide 2) gene mutation associated with dilated cardiomyopathy. Hum Mutat 26:566–574. https://doi.org/10.1002/humu.20250
Towbin BD et al (2012) Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell 150:934–947. https://doi.org/10.1016/j.cell.2012.06.051
van Steensel B, Belmont AS (2017) Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169:780–791. https://doi.org/10.1016/j.cell.2017.04.022
Vergnes L, Peterfy M, Bergo MO, Young SG, Reue K (2004) Lamin B1 is required for mouse development and nuclear integrity. Proc Natl Acad Sci U S A 101:10428–10433. https://doi.org/10.1073/pnas.0401424101
Vohanka S et al (2001) A mutation in the X-linked Emery-Dreifuss muscular dystrophy gene in a patient affected with conduction cardiomyopathy. Neuromuscul Disord 11:411–413
Voit T et al (1988) Emery-Dreifuss muscular dystrophy: disease spectrum and differential diagnosis. Neuropediatrics 19:62–71. https://doi.org/10.1055/s-2008-1052404
Vytopil M et al (2003) Mutation analysis of the lamin A/C gene (LMNA) among patients with different cardiomuscular phenotypes. J Med Genet 40:e132
Wagner N, Krohne G (2007) LEM-domain proteins: new insights into lamin-interacting proteins. Int Rev Cytol 261:1–46. https://doi.org/10.1016/S0074-7696(07)61001-8
Wang Y, Herron AJ, Worman HJ (2006) Pathology and nuclear abnormalities in hearts of transgenic mice expressing M371K lamin A encoded by an LMNA mutation causing Emery-Dreifuss muscular dystrophy. Hum Mol Genet 15:2479–2489. https://doi.org/10.1093/hmg/ddl170
Wang W et al (2012) Structural insights into SUN-KASH complexes across the nuclear envelope. Cell Res 22:1440–1452. https://doi.org/10.1038/cr.2012.126
Warren DT, Zhang Q, Weissberg PL, Shanahan CM (2005) Nesprins: intracellular scaffolds that maintain cell architecture and coordinate cell function? Expert Rev Mol Med 7:1–15. https://doi.org/10.1017/S1462399405009294
Wilhelmsen K et al (2005) Nesprin-3, a novel outer nuclear membrane protein, associates with the cytoskeletal linker protein plectin. J Cell Biol 171:799–810. https://doi.org/10.1083/jcb.200506083
Wilson KL, Foisner R (2010) Lamin-binding Proteins. Cold Spring Harb Perspect Biol 2:a000554. https://doi.org/10.1101/cshperspect.a000554
Worman HJ (2012) Nuclear lamins and laminopathies. J Pathol 226:316–325. https://doi.org/10.1002/path.2999
Worman HJ, Courvalin JC (2000) The inner nuclear membrane. J Membr Biol 177:1–11
Worman HJ, Fong LG, Muchir A, Young SG (2009) Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest 119:1825–1836. https://doi.org/10.1172/JCI37679
Worman HJ, Ostlund C, Wang Y (2010) Diseases of the nuclear envelope. Cold Spring Harb Perspect Biol 2:a000760. https://doi.org/10.1101/cshperspect.a000760
Wu W, Shan J, Bonne G, Worman HJ, Muchir A (2010) Pharmacological inhibition of c-Jun N-terminal kinase signaling prevents cardiomyopathy caused by mutation in LMNA gene. Biochim Biophys Acta 1802:632–638. https://doi.org/10.1016/j.bbadis.2010.04.001
Wu W, Muchir A, Shan J, Bonne G, Worman HJ (2011) Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation 123:53–61. https://doi.org/10.1161/CIRCULATIONAHA.110.970673
Xing XW, Li LY, Liu G, Fu JJ, Tan XJ, Lu GX (2004) Identification of a novel gene SRG4 expressed at specific stages of mouse spermatogenesis. Acta Biochim Biophys Sin Shanghai 36:351–359
Yamada T, Kobayashi T (1996) A novel emerin mutation in a Japanese patient with Emery-Dreifuss muscular dystrophy. Hum Genet 97:693–694
Zhang Q et al (2001) Nesprins: a novel family of spectrin-repeat-containing proteins that localize to the nuclear membrane in multiple tissues. J Cell Sci 114:4485–4498
Zhang Q, Ragnauth C, Greener MJ, Shanahan CM, Roberts RG (2002) The nesprins are giant actin-binding proteins, orthologous to Drosophila melanogaster muscle protein MSP-300. Genomics 80:473–481
Zhang Q et al (2005) Nesprin-2 is a multi-isomeric protein that binds lamin and emerin at the nuclear envelope and forms a subcellular network in skeletal muscle. J Cell Sci 118:673–687. https://doi.org/10.1242/jcs.01642
Zhang Q et al (2007a) Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet 16:2816–2833. https://doi.org/10.1093/hmg/ddm238
Zhang X et al (2007b) Syne-1 and Syne-2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development 134:901–908. https://doi.org/10.1242/dev.02783
Zhang J et al (2010) Nesprin 1 is critical for nuclear positioning and anchorage. Hum Mol Genet 19:329–341. https://doi.org/10.1093/hmg/ddp499
Zhang Z et al (2015) Normalization of Naxos plakoglobin levels restores cardiac function in mice. J Clin Invest 125:1708–1712. https://doi.org/10.1172/JCI80335
Zhen YY, Libotte T, Munck M, Noegel AA, Korenbaum E (2002) NUANCE, a giant protein connecting the nucleus and actin cytoskeleton. J Cell Sci 115:3207–3222
Zhou Z et al (2012) Structure of Sad1-UNC84 homology (SUN) domain defines features of molecular bridge in nuclear envelope. J Biol Chem 287:5317–5326. https://doi.org/10.1074/jbc.M111.304543
Zhou C et al (2017) Novel nesprin-1 mutations associated with dilated cardiomyopathy cause nuclear envelope disruption and defects in myogenesis. Hum Mol Genet 26:2258–2276. https://doi.org/10.1093/hmg/ddx116
Zimmermann WH et al (2006) Engineered heart tissue grafts improve systolic and diastolic function in infarcted rat hearts. Nat Med 12:452–458. https://doi.org/10.1038/nm1394
Acknowledgements
We would like to thank Lucy Coker for helping to create the scientific illustration in Figure 1.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by British Heart Foundation Intermediate Fellowship: FS/17/57/32934.
Conflict of interest
Matthew J. Stroud declares that he has no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by the author.
Additional information
This article is part of a Special Issue on ‘Heart Failure Due to Non-Myofibrillar Defects’ edited by Elisabeth Ehler and Katja Gehmlich.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Stroud, M.J. Linker of nucleoskeleton and cytoskeleton complex proteins in cardiomyopathy. Biophys Rev 10, 1033–1051 (2018). https://doi.org/10.1007/s12551-018-0431-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-018-0431-6