
Abstract
In this review, we discuss mechanisms of resistance identified in bacterial agents Staphylococcus aureus and the enterococci towards two priority classes of antibiotics—the fluoroquinolones and the glycopeptides. Members of both classes interact with a number of components in the cells of these bacteria, so the cellular targets are also considered. Fluoroquinolone resistance mechanisms include efflux pumps (MepA, NorA, NorB, NorC, MdeA, LmrS or SdrM in S. aureus and EfmA or EfrAB in the enterococci) for removal of fluoroquinolone from the intracellular environment of bacterial cells and/or protection of the gyrase and topoisomerase IV target sites in Enterococcus faecalis by Qnr-like proteins. Expression of efflux systems is regulated by GntR-like (S. aureus NorG), MarR-like (MgrA, MepR) regulators or a two-component signal transduction system (TCS) (S. aureus ArlSR). Resistance to the glycopeptide antibiotic teicoplanin occurs via efflux regulated by the TcaR regulator in S. aureus. Resistance to vancomycin occurs through modification of the D-Ala-D-Ala target in the cell wall peptidoglycan and removal of high affinity precursors, or by target protection via cell wall thickening. Of the six Van resistance types (VanA-E, VanG), the VanA resistance type is considered in this review, including its regulation by the VanSR TCS. We describe the recent application of biophysical approaches such as the hydrodynamic technique of analytical ultracentrifugation and circular dichroism spectroscopy to identify the possible molecular effector of the VanS receptor that activates expression of the Van resistance genes; both approaches demonstrated that vancomycin interacts with VanS, suggesting that vancomycin itself (or vancomycin with an accessory factor) may be an effector of vancomycin resistance. With 16 and 19 proteins or protein complexes involved in fluoroquinolone and glycopeptide resistances, respectively, and the complexities of bacterial sensing mechanisms that trigger and regulate a wide variety of possible resistance mechanisms, we propose that these antimicrobial resistance mechanisms might be considered complex ‘nanomachines’ that drive survival of bacterial cells in antibiotic environments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The term ‘Antimicrobial Resistance (AMR) Nanomachine’ (or ‘AMR Nanomachine’) has not to our knowledge been coined previously. Yet it may be considered an appropriate term to use for the cascade of molecular mechanisms that drive antimicrobial drug resistances in microorganisms, including drug recognition by intricate microbial sensing and signal transduction machinery and the subsequent efflux and/or deactivation machinery that remove the antimicrobials death threat from microbial cells. Our definition of the AMR nanomachine therefore encompasses the initiation, activity and control of AMR in response to a given antibiotic in bacterial cells. The AMR nanomachine has one overarching goal—one machinery—aimed at the overall process of achieving resistance to an antibiotic (AMR). The individual processes are not able to function independently of each other if AMR is to be achieved—indeed, they are very much interconnected through the antibiotic to which they are reacting, and dependent on each other to achieve the AMR goal. After all, when antibiotic is removed, all these processes are either reduced or cease to function. The AMR nanomachine can be switched on and off by levels of antibiotic present. Together, these machines facilitate the survival of microorganisms in environments containing elevated levels of antimicrobial drugs. Unfortunately, one such environment includes our hospitals and clinics that utilise antimicrobial agents to combat microbial infections. Possession of the resistance ‘nanomachinery’ by pathogenic microorganisms poses a serious threat to our ability to treat serious microbial infections with current therapies. Indeed, resistance exhibited by bacterial pathogens to current antibacterial agents is now recognised to be a major global problem in the fight against infections. Currently 25,000 people per annum die in Europe as a result of infections caused by microorganisms that are untreatable with antimicrobial agents (EARS-Net 2014; Public Health England Report 2015) and it is predicted that there will be 10 million deaths every year globally by 2050 unless action is taken to safeguard the effectiveness of our antibiotics (HM Government (UK) Review 2015). Antibiotic-resistant infections are also estimated to cost the European Union €1.5 billion per year with regard to healthcare expenses and lost productivity; by 2050, costs worldwide are predicted to soar to £66 trillion (Public Health England Report 2015; HM Government (UK) Review 2015).
Major causes of the emergence and development of resistance machines amongst microbial populations are the intense use and misuse of antibiotics (reviewed in Barbosa and Levy 2000). The more that antibiotics are used and distributed in the environment, the greater the generation of multi-antibiotic resistances (e.g. Mladenovic-Antic et al. 2016; Tammer et al. 2016; Barnes et al. 2017; Mascarello et al. 2017; Pitiriga et al. 2017; also see CMO Report 2011; Public Health England and Veterinary Medicines Directorate Report 2015). After all, resistance can be considered a natural phenomenon and, as already mentioned above, a means by which microorganisms protect themselves against exposure to antibiotics in the environment. In the UK human healthcare sector, 531 tonnes of active antibiotics were prescribed in 2013 (Public Health England Report 2013). In spite of high usage, the importance of rational use of antibiotics has been highlighted previously (Aliabadi and Lees 2000). Dosing regimens and durations of antibiotic treatments should be optimised so that they are sufficiently high as to maximise antibacterial effect but as low as possible to reduce the risk of the emergence of resistance (Baquero and Negri 1997; Guillemot et al. 1998; Negri et al. 1994). The use of sub-optimal antibiotic dosages, as well as excessive dosages, increase selection of resistant strains (Odenholt et al. 2003; Baquero et al. 2008; Gullberg et al. 2011); mathematical modelling methods are being explored to investigate optimal doses and durations (e.g. Bonhoeffer et al. 1997; Bergstrom et al. 2004; D’Agata et al. 2008; Geli et al. 2012; Peña-Miller et al. 2014; Paterson et al. 2016).
In an era in which fewer new and novel antibiotics (that might overcome the resistance issue) are being discovered, the Chief Medical Officer for England has called for antimicrobial stewardship measures to be put in place, encompassing the promotion and monitoring of the judicious use of existing antimicrobials to preserve their future effectiveness (CMO Report 2011). Two of the most important classes of antibiotics recognised as critically important to both medicine and agriculture are the fluoroquinolones and the glycopeptides (WHO 2011; OiE 2015). These antibiotic classes are considered of utmost priority with regard to risk management of resistance generation amongst microbial populations (WHO 2011; OiE 2015). This review will focus on these priority classes, describing the fluoroquinolone and glycopeptide resistance machinery found in enterococci and staphylococci (bacteria that are of significance (and common) to both animal husbandry and human medicine practices). It is relevant to mention that most natural variants of resistance determinants arise through point mutations in target sites as well as resistance enzymes and efflux systems, affecting antibiotic binding strengths and catalytic efficiencies (Raquet et al. 1997; Crichlow et al. 1999; Nukaga et al. 2003; Rubtsova et al. 2010; King and Strynadka 2011; Sarovich et al. 2012; Ramirez et al. 2013; Kaitany et al. 2013; June et al. 2014; Shaheen et al. 2015; Mehta et al. 2015). Changes induced by mutations in the sensory/regulatory proteins that control the production of resistance determinants have also been documented (Baptista et al. 1997; DeMarco et al. 2007; Resch et al. 2008; Noguchi et al. 2004; Schmitz et al. 1998). Therefore, a summary of the sensitive target sites in bacterial cells as well as the AMR nanomachinery governing sensing of and resistance to antibiotics (Table 1) is included in the following review of fluoroquinolone and glycopeptide nanomachines.
Fluoroquinolones
The sensitive cellular targets
Fluoroquinolones (and the older generation quinolones that are currently used much less in the clinic) are used to treat infections caused by both Gram-positive and Gram-negative bacteria (Andersson and MacGowan 2003; Andriole 2005; Heeb et al. 2011; Aldred et al. 2014) (and references therein). Research in the early 1990s revealed interactions with either the A subunit of DNA gyrase or a complex of DNA gyrase and DNA (through the A subunit) to inhibit enzyme activity (Hooper and Wolfson 1991). It was later shown that another cellular target for quinolones occurs in a member of the bacterial type II topoisomerases (specifically, topoisomerase IV) as well as the gyrase (Hooper 1999, 2001; Anderson and Osheroff 2001; Drlica et al. 2008, 2009); reviewed in Aldred et al. (2014). The normal roles of these enzymes are to generate double-stranded breaks in the chromosome; DNA gyrase then introduces negative supercoils in DNA in front of the replication fork, whilst topoisomerase IV controls DNA supercoiling and is involved in the decatenation of daughter chromosomes following replication (Aldred et al. 2014; Tomašić and Mašič 2014). Quinolones bind at the interface of enzyme and DNA in the cleavage-ligation active sites and they do so non-covalently (Wohlkonig et al. 2010; Laponogov et al. 2009, 2010; Bax et al. 2010; Aldred et al. 2014). In the case of quinolone-topoisomerase binding, a water-metal ion bridge provides the link between the antibiotic and the enzyme (Fig. 1).

Schematic representation of moxifloxacin binding to topoisomerase IV via a water-metal ion bridge, adapted and redrawn from Aldred et al. (2014) for ciprofloxacin. For clarity, the DNA has been omitted. Moxifloxacin is shown in black; the Mg2+ ion that is chelated by the C3-C4 ketoacid of the antibiotic is shown in pink; the four water molecules coordinated by the Mg2+ ion are shown in blue. The side chains of the conserved acidic Glu88 (Asp in Escherichia coli and Streptococcus pneumoniae) and Ser84 residues of Acinetobacter baumanii topoisomerase IV are shown in orange, together with their hydrogen bonding to the water molecules
Resistance determinants
Fluoroquinolones appear to interact with a wide range of cellular components, possibly facilitating and/or enhancing the generation of a number of mechanisms by which resistance can be mounted. Kaatz et al. (1991, 1993) first described three means by which resistance to fluoroquinolones may be generated in Staphylococcus aureus:
-
(1)
Mutational change in DNA gyrase, evidenced by the isolation of several point mutations in gyrA that confer high-level fluoroquinolone resistance (Sreedharan et al. 1990; Goswitz et al. 1992); mutations in the topoisomerases have similarly subsequently been shown to provide the bases for bacterial resistance generation (Wohlkonig et al. 2010; Aldred et al. 2012, 2013); in general, the more resistant a clinical isolate is, then the more quinolone resistance-associated mutations it contains (Komp Lindgren et al. 2003; reviewed in Jacoby 2005).
-
(2)
The cfx-ofx locus described by Trucksis et al. (1991), which confers lower-level resistance than that generated by gyrA mutations (Table 1); and
-
(3)
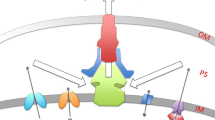
Efflux of (fluoro)quinolones from the cell by efflux pumps. In Gram-positive bacteria, the majority of efflux membrane proteins which include quinolones in their substrate profiles belong to the Major Facilitator Superfamily (MFS) of membrane transporters, e.g. NorA, NorB, NorC, MdeA, LmrS and SdrM in S. aureus (Table 1; Fig. 2) (Ubukata et al. 1989; Kaatz et al. 1991, 1993; Yoshida et al. 1990; Ding et al. 2008; Truong-Boldoc et al. 2006; Huang et al. 2004; Floyd et al. 2010 for LmrS efflux of gatifloxacin; Yamada et al. 2006; and reviewed in Santos Costa et al. 2013 and Correira et al. 2017) and EfmA in Enterococcus faecium (Nishioka et al. 2009). An efflux protein belonging to the Multiple Antibiotic and Toxin Extrusion family (MATE) includes MepA in S. aureus (Kaatz et al. 2005a, 2005b), whilst the ATP-binding cassette family (ABC) includes EfrAB of Enterococcus faecalis (Lee et al. 2003) (Table 1; Fig. 2).

The fluoroquinolone resistance nanomachine. For full details, see text
Amongst Gram-positive bacteria, a further low-level resistance mechanism has been described that is plasmid-borne, Qnr E. faecalis. Qnr proteins resemble DNA mimics and decrease the binding of gyrase and topoisomerase IV to chromosomal DNA. This results in a reduction in the number of available enzyme targets on the bacterial chromosome. Qnr proteins also bind to the gyrase and topoisomerase IV themselves, thereby denying access for quinolones into the cleavage complexes (Fig. 2). These proteins were first discovered in Gram-negative species (reviewed in Tran and Jacoby 2002, Jacoby 2005 and Strahilevitz et al. 2009; Rodriguez-Martinez et al. 2011; and Aldred et al. 2014) but QnrE. faecalis originating from the Gram-positive bacterium Enterococcus faecalis was identified and characterised as a Qnr-like protein that confers intrinsic resistance to fluoroquinolones (Arsene and Leclercq 2007). It is not yet known whether or not Qnr proteins or indeed the cfx-ofx locus constitute separate independent AMR nanomachines from efflux proteins and their regulators.
Resistance regulation determinants
Three systems that regulate expression of the S. aureus Nor MFS multidrug transporters have so far been described (Table 1). MgrA (formerly known as NorR) possesses a helix-turn-helix motif within a region resembling the MarR family of transcriptional regulators (Fig. 2). MgrA positively regulates norA expression (Truong-Bolduc et al. 2003) and negatively regulates the transcription of norB, a gene (tet38) that encodes another more selective transporter Tet38 (Truong-Boldoc et al. 2005) and norC (Truong-Boldoc et al. 2006). Subsequent work established that MgrA is a global regulator affecting approximately 350 genes (Luong et al. 2006) including those involved in autolytic activities and production of alpha-toxin, nuclease and protein A virulence factors (Ingavale et al. 2003; Luong et al. 2003; Truong-Boldoc et al. 2005). MgrA exhibited only weak binding to the norB and tet38 promoter regions and therefore it was proposed that MgrA acts as an indirect regulator of these genes (Truong-Boldoc et al. 2005).
NorG was first identified as a transcriptional regulator of norA (Truong-Boldoc and Hooper 2007). NorG is a member of the GntR-like family of transcriptional regulators (Fig. 2) and it was shown to bind to the promoter regions of norB, norC and abcA as well as norA (Truong-Boldoc and Hooper 2007). It was shown to directly activate norB transcription but repress abcA (an ATP-dependent transporter of the ABC family that confers beta-lactam resistance) (Truong-Boldoc and Hooper 2007).
The third system identified as a regulator of Nor transporter expression in S. aureus is the ArlSR two-component signal transduction system (Fournier et al. 2000; Fournier and Hooper 2000) (Fig. 2; Table 1). Expression from the norA promoter was dependent on the ArlS histidine protein kinase. An arlS mutant which lacks ArlS exhibited increased norA expression (Fournier et al. 2000). Multiple putative binding sites upstream of the transcriptional start point were identified for an 18-kDa DNA-binding protein which could have been ArlR itself or another unidentified protein under ArlSR regulation; the identity of the 18-kDa protein was later shown to be MgrA (Truong-Bolduc et al. 2003; Kaatz et al. 2005b). Finally, the arlS mutant displayed altered growth-phase regulation of NorA confirming the role of the two-component system in norA expression (Fournier et al. 2000). The arlS phenotype also displayed changes in the ability to form biofilms, perform autolysis functions and produce peptidoglycan hydrolase, indicating the importance of the two-component system in multiple cellular functions in addition to quinolone export (Fournier and Hooper 2000).
Amongst the remaining quinolone efflux pumps listed in Table 1, the only other regulator identified so far in S. aureus is MepR (Fig. 2) which is a MarR-like transcriptional repressor of the MepA MATE-type multiple drug resistance pump (Kaatz et al. 2005a; Kaatz et al. 2006). MepR bound upstream of both mepA and its own gene mepR demonstrating autoregulatory activity. Repression of mepA expression by MepR is relieved in the presence of MepA substrates such as benzylalkonium chloride, dequalinium, ethidium bromide and pentamidine. Presumably such relief might also possibly occur using fluoroquinolone substrates, since MepA is an efflux pump with specificity for some fluoroquinolones as well as a wide range of other drugs (Kaatz et al. 2005a; Fabrega et al. 2009; Fernandez and Hancock 2012; Correira et al. 2017).
If, then, the term ‘AMR nanomachine’ can indeed be successfully applied to the cascade of bacterial signal sensing and transduction events required to detect fluoroquinolones and to the subsequent coupling of these detection systems to resistance machinery such as efflux pumps and other protection systems in bacterial cells, then one of the next questions is whether the same term can also be legitimately and appropriately applied to other resistance mechanisms mounted against other families of antibacterial agents. The following section addresses this question by considering the different detection and resistance mechanisms that have evolved for survival in the presence of another important and distinct family of antibacterial agents, the glycopeptides.
Glycopeptides
Glycopeptide antibiotics have been identified as one of the highest priority classes of antimicrobial agents for risk management in clinical and agricultural settings (WHO 2011). Glycopeptide antibiotics are relatively large in size—for example, vancomycin has a molar mass of 1449 Da (Phillips-Jones et al. 2017a)—and therefore, unlike the smaller fluoroquinolones, glycopeptide drugs do not penetrate the membranes of bacterial cells and instead exert their inhibitory effects through interference with crucial bacterial processes outside the cell (Courvalin 2006). Yet bacterial cells must be able to mount resistance to glycopeptides to secure their survival (see below). But unlike the nanomachinery of fluoroquinolone resistance, an arsenal of efflux pumps will of course be redundant against antibiotics that do not enter bacterial cells. Glycopeptide resistance must be exerted by a quite different means. The question is whether the mechanisms for glycopeptide resistance and resistance regulation described below may also be considered parts of a nanomachine.
The sensitive cellular targets
Vancomycin and teicoplanin are two important members of the glycopeptide class which are used to combat serious infections caused by Gram-positive bacteria such as bloodstream infections, infections of the skin, bones and joints, endocarditis and meningitis (Rayner and Munckhof 2005; Kristich et al. 2014; Alvarez et al. 2016). Use of vancomycin in the clinic increased markedly in the 1970s to combat methicillin-resistant Staphylococcus aureus. Indeed, vancomycin has become a drug of last resort to combat infections that are otherwise resistant to other front-line antibiotics. The main reasons why vancomycin is used so reservedly are as follows: (1) the toxicity of the antibiotic and (2) the poor absorption of the drug upon oral administration (Moellering 2006; Levine 2006). Vancomycin administration is carefully monitored to ensure that the concentrations are sufficiently high to be effective against Gram-positive bacterial pathogens (a serum peak level of 25–40 μg/ml and a trough serum concentration of 15–20 μg/ml, the former being equivalent to eight times the minimum inhibitory concentration), but sufficiently low as to minimise the toxic effects of the antibiotic on the patient (Tobin et al. 2002; Levine 2006; Jones 2006; Rybak et al. 2009; van Hal et al. 2013; MacDougall et al. 2016). Vancomycin is typically administered intravenously to adult patients at a starting concentration of 2.5–5.0 mg/ml (Rybak 2006). Recent biophysical investigations of vancomycin in physiologically relevant buffer conditions at both starting and therapeutic concentrations have suggested that the drug adopts two different conformations at these differing concentrations. Using sedimentation equilibrium techniques in the analytical ultracentrifuge, the SEDFIT-MSTAR algorithm and other analyses, it was shown that all the glycopeptide is dimerized at the point of clinical infusion (5 mg/ml) but at the trough serum concentration of 19 μg/ml the drug is mainly monomeric (< 20% dimerized) (Phillips-Jones et al. 2017a). Experiments employing a range of different loading concentrations were consistent with a monomer-dimer equilibrium that is completely reversible and dissociation constants indicative of relatively weak association between monomers (Phillips-Jones et al. 2017a). This is of significance because there is still relatively little understanding about the conformationally relevant form of the antibiotic during its inhibitory activity (see below).
Glycopeptides such as vancomycin inhibit bacterial cell wall biosynthesis by binding to the C-terminal D-Ala-D-Ala residues of the muramyl pentapeptide of peptidoglycan precursor Lipid II (Fig. 3). Vancomycin binding results in inhibition of transpeptidase and transglycosylase activities during peptidoglycan biosynthesis, affecting crosslinking, formation of glycan chains and incorporation of peptidoglycan precursors, resulting in osmotic shock and cell lysis (Nieto and Perkins 1971; Reynolds 1989; Kahne et al. 2005; Jia et al. 2013). It has been established that for many glycopeptides, ligand binding is accompanied by the presence of asymmetric, back-to-back homodimers of the antibiotic formed through sugar-sugar recognition (see Phillips-Jones et al. 2017a for references therein). Experimental evidence shows that dimerization and binding of D-Ala-D-Ala in vitro are generally cooperative phenomena leading to the conclusion that dimerization is important for enhancing antibiotic activity (Mackay et al. 1994). Face-to-face dimers have also been reported (Mackay et al. 1994; Loll et al. 1998), as have higher order dimer-to-dimer, trimers of dimer and hexamer conformations for glycopeptide antibiotics (Loll et al. 2009; Nitanai et al. 2009), though the significance of these higher order conformations regarding inhibitory action and affinity remains to be established. There are examples of glycopeptides that do not dimerize at all; for example, lipophilic monomeric teicoplanin inserts into the membrane through the lipid moiety and it is thought to do so in such a way as to be positioned optimally for inhibitory activity (Beauregard et al. 1995; Sharman et al. 1997).

Schematic representation of vancomycin (top left) adapted and redrawn from Phillips-Jones et al. (2017a) showing the vancosamine - glucose disaccharide (purple) attached to a heptapeptide (green, N-methyl-D-leucine (residue 1); gold, m-chloro-β-hydroxy-D-tyrosine (residue 2); red, asparagine (residue 3); blue, D-phenyl glycine (residue 4); green-grey, p-hydroxy-D-phenylglycine (residue 5); pink, m-chloro- β-hydroxy-D-tyrosine (residue 6) and dark orange, m,m-dihydroxy-L-phenylglycine (residue 7)). Vancomycin binding to its sensitive target sequence (D-Ala-D-Ala) in bacterial peptidoglycan via five hydrogen bonds is shown by the dashed black lines. Hydrogen bond 2 is formed from residue 4 of vancomycin and the N-H group in D-Ala-D-Ala shown in red (middle structure). This hydrogen bond is not formed with the peptidoglycan of vancomycin-resistant bacteria that contain D-Ala-D-Lactate instead of D-Ala-D-Ala (bottom right) resulting in a total of only four hydrogen bonds for vancomycin binding which results in a 1000-fold reduced affinity of the glycopeptide for the peptidoglycan—essentially, resistance to the antibiotic
Resistance determinants
High-level resistance to glycopeptide antibiotics was first reported amongst the enterococci in 1988 (Leclercq et al. 1988; Uttley et al. 1988) and subsequently spread to Staphylococcus aureus including MRSA strains (Sievert et al. 2002). Glycopeptide resistant strains of enterococci and staphylococci have spread across the world at a rapid rate (e.g. Lu et al. 2001; Iverson et al. 2002; Eisner et al. 2005; and reviewed in Schouten et al. 2000; Werner et al. 2008; Périchon and Courvalin 2009).
Resistance to vancomycin occurs by two main mechanisms: (1) target modification—production of low-affinity precursors for peptidoglycan biosynthesis so that instead of D-Ala-D-Ala being incorporated into peptidoglycan monomers, other depsipeptides (D-Ala-D-lactate or D-Ala-D-serine) are synthesised and incorporated instead (Fig. 3). Vancomycin exhibits an approximately 1000-fold reduced binding affinity for D-Ala-D-Lac because of the reduced number of hydrogen bonding sites available (one crucial hydrogen bond is lost (bond 2 in Fig. 3)); and (2) removal of the high affinity precursors usually synthesised in the cell so there are no vancomycin-binding targets available (reviewed in Reynolds and Courvalin 2005; Courvalin 2006; Wright 2011). Amongst the enterococci, there are six types of resistances found (VanA-, VanB-, VanC-, VanD-, VanE- and VanG-type) which execute the above two mechanisms and these have been comprehensively described in Courvalin (2006) and Depardieu et al. (2007). Amongst Staphylococcus aureus isolates, only one of these types (VanA-type) has so far emerged and is thought to have been transferred from enterococci (Sievert et al. 2002; Sievert et al. 2008; Périchon and Courvalin 2009; McCallum et al. 2010). Broadly, the glycopeptide resistance nanomachine comprises (1) enzymes to synthesise the D-Ala-D-Lac or D-Ala-D-Ser depsipeptides, (2) enzymes for hydrolysis of antibiotic-‘susceptible’ peptidoglycan precursors and (3) a regulatory system to control production of these resistance enzymes. In the following sections, discussion is mainly confined to the VanA-type resistance because it is the most common type amongst clinical enterococci and the first (and, to date, only) to have disseminated to staphylococci (Table 1). The reader is referred to the review by Depardieu et al. (2007) for a detailed comparison of the genes/elements involved in each of the six vancomycin resistance types. In the VanA-type resistance, there are nine genes involved in production of transposition ability (orf1 and orf2) (associated with replicative transposition when in the Tn1546 element), regulation of resistance gene expression (the vanS and vanR genes encoding the VanSR two-component regulatory system, described below), vanH and vanA encoding enzymes required for synthesis of D-Ala-D-Lac (dehydrogenase and ligase, respectively), hydrolysis of peptidoglycan precursors by a D,D-dipeptidase (encoded by vanX) and D,D-carboxypeptidase (vanY) and a ninth gene (vanZ) of unknown function (Fig. 4a; Courvalin 2006; Depardieu et al. 2007). This cluster of genes was originally associated with the plasmid-borne Tn1546 transposable element but are also plasmid- and chromosome-borne following horizontal gene transfer to the enterococci (Courvalin 2004; Palmer et al. 2010) and S. aureus (Haaber et al. 2017).

The VanA-type glycopeptide resistance nanomachine. a The VanS-VanR two-component signal transduction system and organisation of the vanA operon. Open arrows represent coding sequences and indicate the direction of transcription. The regulatory and resistance genes are cotranscribed from promoters PR and PH, respectively; b synthesis of peptidoglycan precursors in a VanA-type resistant strain. Ddl, D-Ala:D-Ala ligase; penta, L-Ala-γ-D-Glu-L-Lys-D-Ala-D-Ala; Pentadepsi, L-Ala-γ-D-Glu-L-Lys-D-Ala-D-Lac; Tetra, L-Ala-γ-D-Glu-L-Lys-D-Ala; Tri, L-Ala-γ-D-Glu-L-Lys
S. aureus possesses another mechanism of glycopeptide resistance known as ‘glycopeptide (or vancomycin) intermediate S. aureus’ (GISA or VISA). This type of resistance was first reported in 1997 (Hiramatsu et al. 1997) and strains possessing it characteristically exhibit reduced susceptibility to glycopeptides (Hiramatsu 2001). GISA strains possess thickened peptidoglycan in their cell walls or poorly cross-linked peptidoglycan. Such conditions result in restriction of glycopeptides to the outermost layers of peptidoglycan where they quickly become sequestered by the increased numbers of free D-Ala-D-Ala target binding sites, thereby never reaching the inner layers of peptidoglycan and the crucial sites of active peptidoglycan biosynthesis (Cui et al. 2006) or at least diffuse more slowly to them (Pereira et al. 2007). Alterations and mutations in several genetic loci have been identified as responsible (Table 1) (Howden et al. 2010; Hiramatsu et al. 2014; Hu et al. 2016).
Additional resistance to teicoplanin in S. aureus has also been characterised (Chang et al. 2013, 2014 and refs therein). The transcriptional regulator known as teicoplanin-associated locus regulator (TcaR) belongs to the MarR family of multidrug efflux regulators (Fig. 2) (Grove 2013), involved in teicoplanin and methicillin resistance in staphylococci (Brandenberger et al. 2000).
Resistance regulation determinants
Here we consider the A-type resistance to vancomycin only, as it is common to both enterococci and staphylococci. For regulators of other glycopeptide resistances, the reader is referred to Table 1 and Depardieu et al. (2007) and Hong et al. (2008).
The A-type vanHAXYZ resistance genes are regulated by the VanSR two-component signal transduction system (Arthur et al. 1992). VanS is the membrane-bound sensor kinase component involved in signal sensing and VanR is the partner response regulator component responsible for activating resistance gene expression at the PH promoter (Fig. 4b) (Arthur et al. 1992; Wright et al. 1993; Holman et al. 1994; Courvalin 2006). Expression of equivalent genes of other resistance types (Types B-E and G) is also under VanSR control (Depardieu et al. 2007). Expression of the vanSR genes themselves is initiated from the distinct PR promoter which is under autoregulatory control (Arthur et al. 1997). In the presence of glycopeptides, VanR is phosphorylated by VanS~P. VanR~P binds to the PR and PH promoters, promoting transcription of the vanHAXYZ genes and its own synthesis (Arthur et al. 1997). However, in the absence of glycopeptides (or VanS), VanR~P is still generated due to the activities of low molecular weight phosphodonors such as acetyl phosphate and/or cross-talking histidine kinases resulting in constitutive low-level activation of the PR and PH promoters (Fig. 4b). In the absence of glycopeptides, it is suggested that VanS serves as a phosphatase, removing phosphate from VanR through its phosphatase activity and reducing resistance gene expression in the absence of inducer. Conversely, when inducer is present, VanS transitions from phosphatase to kinase mode, resulting in increased VanR phosphorylation and elevated levels of VanR~P for induction of the vanHAXYZ resistance genes (Arthur et al. 1997, 1999).
The precise nature of the activating ligand for VanS has been the subject of intense interest for many years (see the comprehensive review by Hong et al. 2008). Using a variety of approaches such as reporter genes, VanX activity assays, measurements of induction of Lac-containing precursors or through detection of induced vancomycin resistance of pretreated cultures, all the evidence pointed towards vancomycin or teicoplanin as inducers of VanA-type resistance (Ulijasz et al. 1996; Arthur et al. 1999; Lai and Kirsch 1996; Mani et al. 1998; Grissom-Arnold et al. 1997; Baptista et al. 1996; Allen and Hobbs 1995; Handwerger and Kolokathis 1990; reviewed in Hong et al. 2008). However, because the structurally unrelated antibiotic moenomycin also induces VanA resistance, it was thought that the molecular effector for VanS must be a cell wall intermediate such as Lipid II which would accumulate in cells exposed to both moenomycin and vancomycin cell wall-active antibiotics (Lai and Kirsch 1996; Mani et al. 1998; Grissom-Arnold et al. 1997; Baptista et al. 1996; Allen and Hobbs 1995; Handwerger and Kolokathis 1990).
Two biophysical approaches—hydrodynamic methods in an analytical ultracentrifuge and circular dichroism spectroscopy—recently established that the purified intact VanS membrane protein from E. faecium B4147 interacts directly with vancomycin and teicoplanin (Phillips-Jones et al. 2017a, b) (Fig. 5). Hydrodynamic experiments in buffers containing 20% glycerol (to maintain VanS solubility) revealed that vancomycin elicits an increase in VanS sedimentation coefficient, s, of over 33% with the appearance of additional higher s components suggesting higher oligomeric forms of the receptor in the presence of the antibiotic (Fig. 5a) (Phillips-Jones et al. 2017b). These results demonstrate that VanS interacts with vancomycin. Circular dichroism measurements confirmed this finding; difference spectra obtained in the near-UV region (which interrogates changes in the tertiary structural environments of aromatic residues) for VanS alone and for VanS + 5-fold vancomycin were clearly different especially in the 280–300-nm region contributed by tyrosine and tryptophan residues (Fig. 5b) (Phillips-Jones et al. 2017b). CD-based titration experiments in the presence of detergent using increasing concentrations of vancomycin and teicoplanin revealed Kd values in the regions of 70 μM, and 30 and 170 μM, respectively (Hughes et al. 2017). Such Kd values are indicative of relatively weak binding. Weak binding may be a feasible explanation for signal transduction processes that are rapid and reversible, though it has not yet been demonstrated that the weak binding by vancomycin demonstrated in Phillips-Jones et al. (2017b) is accompanied by increased levels of VanR phosphorylation by VanS. Alternatively, the weak binding measured in these studies may reflect the absence of an essential binding accessory factor in this in vitro system, or it could reflect the absence of the natural membrane environment required for full VanS function. The latter possibility seems reasonable as VanS remained monomeric throughout all experimental conditions tested, in the presence or absence of detergent, including those associated with ligand binding which are known to induce dimerization in other sensor kinases (Phillips-Jones et al. 2017a, b). But the demonstration in these studies of vancomycin binding to VanS, albeit weakly, provides the first evidence in the clinical enterococci for the involvement of vancomycin as a molecular effector of VanA-type VanS activation. Indeed, Hughes et al. (2017) tested components of Lipid II to determine whether they too demonstrated interactions with VanS and no spectral changes were found (Hughes et al. 2017). Studies of distantly related VanS sensors in actinomycetes and VanB-type enterococci have previously provided evidence that the antibiotic itself or the antibiotic bound to the D-Ala-D-Ala substrate serves as the inducing effectors (Koteva et al. 2010; Kwun et al. 2013).

a Sedimentation coefficient concentration distribution, c(s) vs s20,b, (where s20,b is the sedimentation coefficient at 20.0 oC in buffer b) profile for intact VanS (5.4 μM) (black line) in HGN buffer (containing 20% glycerol) pH ~ 7.9, I = 0.1, at 20.0 °C. The rotor speed was 40,000 rpm. The profile for 12.8 μM vancomycin is shown (grey line). VanS and vancomycin is shown by the red line under the same conditions. b (leftmost): VanS (9 μM) difference CD spectrum (solid black line); VanS in the presence of 5-fold vancomycin (45 μM) difference spectrum (dashed black line); (rightmost): vancomycin (45 μM) difference spectrum. Reactions contained 10 mM HEPES, 20% (v/v) glycerol, 100 mM NaCl and 0.05% n-dodecyl-β-D-maltoside, pH 7.9. Unsmoothed data shown. Both panels reproduced with permission from Phillips-Jones et al. (2017b)
Conclusion
Based on the above considerations of two quite different sets of resistance mechanisms, namely fluoroquinolone and glycopeptide resistances, it is clear that the enterococci and staphylococci expend significant levels of energy into ensuring survival in an antibiotic environment. There are 16 possible proteins and/or distinct complexes that have arisen amongst different strains for resistance to fluoroquinolones—though not all are likely to be present in any one individual cell (Table 1). For VanA-type glycopeptide and teicoplanin resistances, there are up to 19 proteins or complexes involved (Table 1). Therefore, in the same way that the term ‘antibiotic resistome’ has been used for all antibiotic resistance genes and their precursors in bacteria (Wright 2007), the term ‘antimicrobial resistance (AMR) nanomachines’ proposed here seems appropriate (for both fluoroquinolone and glycopeptide resistances) to reflect the large number of gene products (encoded by resistome genes) that are responsible for coordinating the sensing of these antibiotics, for transferring signal information, and for the expression of a wide variety of resistance options such as efflux pumps and efflux regulatory machinery, target protection and target modification.
Biophysical investigations have played an important role in identifying how some of the AMR nanomachines work. In this review we have focused on the use of hydrodynamic studies and circular dichroism spectroscopy to investigate the interactions between glycopeptides and the sensory receptor VanS. Such techniques are likely to prove useful for investigating the interactions between other nanomachine components in the future.
References
Aldred KJ, Kerns RJ, Osheroff N (2014) Mechanism of quinolone action and resistance. Biochemistry 53:1565–1574
Aldred KJ, McPherson SA, Turnbough CL Jr, Kerns RJ, Osheroff N (2013) Topoisomerase IV-quinolone interactions are mediated through a water-metal ion bridge: mechanistic basis of quinolone resistance. Nucleic Acids Res 41:4628–4639
Aldred KJ, McPherson SA, Wang P, Kerns RJ, Graves DE, Turnbough CL Jr, Osheroff N (2012) Drug interactions with Bacillus anthracis topoisomerase IV: biochemical basis for quinolone action and resistance. Biochemistry 51:370–381
Aliabadi FS, Lees P (2000) Antibiotic treatment for animals: effect on bacterial population and dosage regimen optimisation. Int J Antimicrob Agents 14:307–313
Allen NE, Hobbs JN (1995) Induction of vancomycin resistance in Enterococcus faecium by non-glycopeptide antibiotics. FEMS Microbiol Lett 132:107–114
Alvarez R, Lopez Cortes LE, Molina J et al (2016) Optimizing the clinical use of vancomycin. Antimicrob Agents Chemother 60:2601–2609
Anderson VE, Osheroff N (2001) Type II topoisomerases as targets for quinolone antibacterials: turning Dr Jekyll into Mr Hyde. Curr Pharm Des 7:337–353
Andersson MI, MacGowan AP (2003) Development of the quinolones. J Antimicrob Chemother 51(Suppl. S1):1–11
Andriole VT (2005) The quinolones: past, present and future. Clin Infect Dis 41(Suppl 2):S113–S119
Arsene S, Leclercq R (2007) Role of a qnr-like gene in the intrinsic resistance of Enterococcus faecalis to fluoroquinolones. Antimicrob Agents Chemother 51:3254–3258
Arthur M, Depardieu F, Courvalin P (1999) Regulated interactions between partner and non-partner sensors and response regulators that control glycopeptide resistance gene expression in enterococci. Microbiology 145:1849–1858
Arthur M, Depardieu F, Gerbaud G, Galimand M, Leclercq R, Courvalin P (1997) The VanS sensor negatively controls VanR-mediated transcriptional activation of glycopeptide resistance genes of Tn1546 and related elements in the absence of induction. J Bacteriol 179:97–106
Arthur M, Molinas C, Courvalin P (1992) The VanS-VanR two-component regulatory system controls synthesis of depsipeptide peptidoglycan precursors in Enterococcus faecium BM4147. J Bacteriol 174:2582–2591
Baptista M, Depardieu F, Courvalin P, Arthur M (1996) Specificity of induction of glycopeptide resistance genes in Enterococcus faecalis. Antimicrob Agents Chemother 40:2291–2295
Baptista M, Depardieu F, Reynolds P, Courvalin P, Arthur M (1997) Mutations leading to increased levels of resistance to glycopeptide antibiotics in VanB-type enterococci. Mol Microbiol 25:93–105
Baquero F, Martinez J-L, Cantón R (2008) Antibiotics and antibiotic resistance in water environments. Curr Opin Biotechnol 19:260–265
Baquero F, Negri MC (1997) Strategies to minimize the development of antibiotic resistance. J Chemother 9(Suppl 3):29–37
Barbosa TM, Levy SB (2000) The impact of antibiotic use on resistance development and persistence. Drug Resist Updat 3:303–311
Barnes SL, Rock C, Harris AD et al (2017) The impact of reducing antibiotics on the transmission of multidrug-resistant organisms. Infect Control Hosp Epidemiol 38:663–669
Bax BD, Chan PF, Egglestone DS, Fosberry A, Gentry DR, Gorrec F, Giordano I, Hann MM, Hennessy A, Hibbs M, Huang J, Jones E, Jones J, Brown KK, Lewis CJ, May EW, Saunders MR, Singh O, Spitzfaden CE, Shen C, Shillings A, Theobald AJ, Wohlkonig A, Pearson ND, Gwynn MN (2010) Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 466:935–940
Beauregard DA, Williams DH, Gwynn MN, Knowles DJC (1995) Dimerization and membrane anchors in extracellular targeting of vancomycin group antibiotics. Antimicrob Agents Chemother 39:781–785
Bergstrom CT, Lo M, Lipsitch M (2004) Ecological theory suggests that antimicrobial cycling will not reduce antimicrobial resistance in hospitals. Proc Natl Acad Sci U S A 101:13285–13290
Bonhoeffer S, Lipsitch M, Levin BR (1997) Evaluating treatment protocols to prevent antibiotic resistance. Proc Natl Acad Sci U S A 94:12106–12111
Brandenberger M, Tschierske M, Giachino P, Wada A, Berger-Bachi B (2000) Inactivation of a novel three-cistronic operon tcaR-tcaA-tcaB increases teicoplanin resistance in Staphylococcus aureus. Biochim Biophys Acta 1523:135–139
Chang Y-M, Chen CK-M, Ko T-P, Chang-Chien MW, Wang AH (2013) Structural analysis of the antibiotic-recognition mechanism of MarR proteins. Acta Crystallogr D Biol Crystallogr 69:1138–1149
Chang Y-M, Ho C-H, Chen CK-M, Maestre-Reyna M, Chang-Chien MW, Wang AH-J (2014) TcaR-ssDNA complex crystal structure reveals new DNA binding mechanism of the MarR family of proteins. Nucleic Acids Res 42:5314–5321
CMO Report (2011) Annual Report of the Chief Medical Officer Volume 2: infections and the rise of antimicrobial resistance. Department of Health, UK
Correira S, Poeta P, Hebraud M, Capelo JL, Igrejas G (2017) Mechanisms of quinolone action and resistance: where do we stand? J Med Microbiol 66:551–559
Courvalin P (2004) Genetics of glycopeptide resistance in Gram-positive pathogens. Int J Med Microbiol 294:479–486
Courvalin P (2006) Vancomycin resistance in Gram-positive cocci. Clin Infect Dis 42:S25–S34
Crichlow GV, Kuzin AP, Nukaga M, Mayama K, Sawai T, Knox JR (1999) Structure of the extended-spectrum class C beta-lactamase of Enterobacter cloacae GC1, a natural mutant with a tandem tripeptide insertion. Biochemist 38:10256–10261
Cui L, Iwamoto A, Lian J-Q, Neoh H-M, Maruyama T, Horikawa Y, Hiramatsu K (2006) Novel mechanism of antibiotic resistance originating in vancomycin-intermediate Staphylococcus aureus. Antimicrob Agents Chemother 50:428–438
D’Agata EMC, Dupont-Rouzeyrol M, Magal P, Olivier D, Ruan S (2008) The impact of different antibiotic regimens on the emergence of antimicrobial-resistant bacteria. PLoS One 3:e4036
DeMarco CE, Cushing LA, Frempong-Manso E, Seo SM, Jaravaza TA, Kaatz GW (2007) Efflux-related resistance to norfloxacin, dyes and biocides in bloodstream isolates of Staphylococcus aureus. Antimicrob Agents Chemother 51:3235–3239
Depardieu F, Podglajen I, Leclercq R, Collatz E, Courvalin P (2007) Modes and modulations of antibiotic resistance gene expression. Clin Microbiol Rev 20:79–114
Ding Y, Onodera Y, Lee JC, Hooper DC (2008) NorB, an efflux pump in Staphylococcus aureus strain MW2, contributes to bacterial fitness in abscesses. J Bacteriol 190:7123–7129
Drlica K, Hiasa H, Kerns R, Malik M, Mustaev A, Zhao X (2009) Quinolones: action and resistance updated. Curr Top Med Chem 9:981–998
Drlica K, Malik M, Kerns RJ, Zhao X (2008) Quinolone-mediated bacterial death. Antimicrob Agents Chemother 52:385–392
EARS-Net Annual Report (2014) European Centre for Disease Prevention and Control Report. Antimicrobial resistance surveillance in Europe: Annual Report of the European Antimicrobial Resistance Surveillance Network (EARS-Net)
Eisner A, Feierl G, Gorkiewicz G, Dieber F, Kessler HH, Marth E, Köfer J (2005) High prevalence of VanA-type vancomycin-resistant enterococci in Austrian poultry. Appl Environ Microbiol 71:6407–6409
Fabrega A, Madurga S, Giratt E, Vila J (2009) Mechanism of action of and resistance to quinolones. Microb Biotechnol 2:40–61
Fernandez L, Hancock RE (2012) Adaptive and mutational resistance: role of porins and efflux pumps in drug resistance. Clin Microbiol Rev 25:661–681
Floyd JL, Smith KP, Kumar SH, Floyd JT, Varela MF (2010) LmrS is a multidrug efflux pump of the major facilitator superfamily from Staphylococcus aureus. Antimicrob Agents Chemother 54:5406–5412
Fournier B, Aras R, Hooper DC (2000) Expression of the multidrug resistance transporter NorA from Staphylococcus aureus is modified by a two-component regulatory system. J Bacteriol 182:664–671
Fournier B, Hooper DC (2000) A new two-component regulatory system involved in adhesion, autolysis and extracellular proteolytic activity of Staphylococcus aureus. J Bacteriol 182:3955–3964
Geli P, Laxminarayan R, Dunne M, Smith DL (2012) ‘One-size-fits-all’? Optimizing treatment duration for bacterial infections. PLoS One 7:e29838
Goswitz JJ, Willard KE, Fasching CE, Peterson LR (1992) Detection of gyrA mutations associated with ciprofloxacin resistance in methicillin-resistant Staphylococcus aureus, analysis by polymerase chain reaction and automated direct DNA sequencing. Antimicrob Agents Chemother 36:1166–1169
Grissom-Arnold J, Alborn WE, Nicas TI, Jaskunas SR (1997) Induction of VanA vancomycin resistance genes in Enterococcus faecalis: use of a promoter fusion to evaluate glycopeptide and non-glycopeptide induction signals. Microb Drug Resist 3:53–64
Grove A (2013) MarR family transcription factors. Curr Biol 23:R142–R143
Guillemot D, Carbon C, Balkau B, Geslin P, Lecoeur H, Vauzelle-Kervroedan F, Bouvenot G, Eschwege E (1998) Low dosage and long treatment duration of beta-lactam: risk factors for carriage of penicillin-resistant Streptococcus pneumoniae. JAMA 279:365–370
Gullberg E et al (2011) Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog 7:1–9
Haaber J, Penadés JR, Ingmer H (2017) Transfer of antibiotic resistance in Staphylococcus aureus. Trends Microbiol 25:893–905
Handwerger S, Kolokathis A (1990) Induction of vancomycin resistance in Enterococcus faecium by inhibition of transglycosylation. FEMS Microbiol Lett 58:167–170
Heeb S, Fletcher MP, Chhabra SR, Diggle SP, Williams P, Cámara M (2011) Quinolones: from antibiotics to autoinducers. FEMS Microbiol Rev 35:247–274
Hiramatsu K (2001) Vancomycin-resistant Staphylococcus aureus: a new model of antibiotic resistance. Lancet Infect Dis 1:147–155
Hiramatsu K, Hanaki H, Ino T, Yabuta K, Oguri T, Tenover F (1997) Methicillin-resistance Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother 40:135–136
Hiramatsu K, Kayayama Y, Matsuo M, Aiba Y, Saito M, Hishinuma T, Iwamoto A (2014) Vancomycin-intermediate resistance in Staphylococcus aureus. J Global Antmicrob Resist 2:213–224
HM Government (UK) Report (2015) Rapid diagnostics: stopping unnecessary use of antibiotics. Review on Antimicrobial Resistance. Chaired by Jim O’Neill
Holman TR, Wu Z, Wanner BL, Walsh CT (1994) Identification of the DNA-binding site for thephosphorylated VanR protein required for vancomycin resistance in Enterococcus faecium. Biochemistry 33:4625–4631
Hong H-J, Hutchings MI, Buttner MJ (2008) Vancomycin resistance VanS/VanR two-component systems. Chapter 14 in. Bacterial signal transduction: networks & drug targets (ed. R. Utsumi) Landes Bioscience & Springer Science+Business Media
Hooper DC (1999) Mode of action of fluoroquinolones. Drugs 58(Suppl 2):6–10
Hooper DC (2001) Mechanisms of action of antimicrobials: focus on fluoroquinolones. Clin Infect Dis 32(Suppl 1):S9–S15
Hooper DC, Wolfson JS (1991) Mode of action of the new quinolones: new data. Eur J Microbiol Infect Dis 10:223–231
Howden BP, Davies JK, Johnson PDR, Stinear TP, Grayson ML (2010) Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin intermediate and heterogenous vancomycin-intermediate strains: resistance mechanisms, laboratory detection and clinical implications. Clin Microbiol Rev 23:99–139
Hu Q, Peng H, Rao X (2016) Molecular events for promotion of vancomycin resistance in vancomycin intermediate Staphylococcus aureus. Front Microbiol 7:1601–1618
Huang J, O’Toole PW, Shen W, Amrine-Madsen H, Jiang X, Lobo N, Palmer LM, Voelker L, Fan F, Gwynn MN, McDevitt D (2004) Novel chromosomally encoded multidrug efflux transporter MdeA in Staphylococcus aureus. Antimicrob Agents Chemother 48:909–917
Hughes CS, Longo E, Phillips-Jones MK, Hussain R (2017) Characterisation of the selective binding of antibiotics vancomycin and teicoplanin by the VanS receptor regulating type A vancomycin resistance in the enterococci. Biochi. Biophys Acta 1861:1951–1959
Ingavale SS, van Wamel W, Cheung AL (2003) Characterization of RAT, an autolysis regulator in Staphylococcus aureus. Mol Microbiol 48:1451–1466
Iverson A, Kühn I, Franklin A, Mölby R (2002) High prevalence of vancomycin-resistant enterococci in Swedish sewage. Appl Environ Microbiol 68:2838–2842
Jacoby GA (2005) Mechanisms of resistance to quinolones. Clin Infect Dis 41:S120–S126
Jia Z, O’Mara ML, Zuegg J, Cooper MA, Mark AE (2013) Vancomycin: ligand recognition, dimerization and super-complex formation. FEBS J 280:1294–1307
Jones RN (2006) Microbiological features of vancomycin in the 21st century: minimum inhibitory concentration creep, bacteriocidal/static activity and applied breakpoints to predict clinical outcomes or detect resistant strains. Clin Infect Dis 42:S13–S24
June CM, Vallier BC, Bonomo RA, Leonard DA, Powers RA (2014) Structural origins of oxacillinase specificity in class D beta-lactamases. Antimicrob Agents Chemother 58:333–341
Kaatz GW, DeMarco CE, Seo SM (2006) MepR, a repressor of the Staphylococcus aureus MATE family multidrug efflux pump MepA, is a substrate-responsive regulatory protein. Antimicrob Agents Chemother 50:1276–1281
Kaatz GW, McAleese F, Seo SM (2005) Multidrug resistance in Staphylococcus aureus due to overexpression of a novel multidrug and toxin extrusion (MATE) transport protein. Antimicrob Agents Chemother 49:1857–1864
Kaatz GW, Seo SM, Ruble CA (1991) Mechanisms of fluoroquinolone resistance in Staphylococcus aureus. J Infect Dis 163:1080–1086
Kaatz GW, Seo SM, Ruble CA (1993) Efflux-mediated fluoroquinolone resistance in Staphylococcus aureus. Antimicrob Agents Chemother 37:1086–1094
Kaatz GW, Thyagarajan RV, Seo SM (2005) Effect of promoter region mutations and mgrA overexpression on transcription of norA, which encodes a Staphylococcus aureus multidrug efflux transporter. Antimicrob Agents Chemother 49:161–169
Kahne D, Leimkuhler C, Lu W, Walsh C (2005) Glycopeptide and lipoglycopeptide antibiotics. Chem Rev 105:425–448
Kaitany K-C, Klinger NV, June CM, Ramey ME, Bonomo RA, Powers RA, Leonard DA (2013) Structures of the Class D carbapenemases OXA23 and OXA146: mechanistic basis of activity against carbapenems, extended-spectrum cephalosporins and aztreonam. Antimicrob Agents Chemother 57:4848–4855
King D, Strynadka N (2011) Crystal structure of New Dehli metallo-beta-lactamase reveals molecular basis for antibiotic resistance. Protein Sci 20:1484–1491
Komp Lindgren P, Karlsson A, Hughes D (2003) Mutation rate and evolution of fluoroquinolone resistance in Escherichia coli isolates from patients with urinary tract infections. Antimicrob Agents Chemother 47:3222–3232
Koteva K, Hong H-J, Wang XD, Nazi I, Hughes D, Naldrett MJ, Buttner MJ, Wright GD (2010) A vancomycin photoprobe identifies the histidine kinase VanSSC as a vancomycin receptor. Nat Chem Biol 6:327–329
Kristich CJ, Rice LB, Arias CA (2014) Enterococcal infection—treatment and antibiotic resistance. In: Enterococci: from commensals to leading causes of drug-resistant infection. In: Gilmore M.S, Clewell D B, Ike Y, Shankar N (Eds). Boston, Massachusetts Eye and Ear Infirmary, p 123–184. https://www.ncbi.nlm.nih.gov/books/NBK190424/pdf/Bookshelf_NBK190424.pdf. Date of access: 2 Feb 2017
Kwun MJ, Novotna G, Hesketh AR, Hill L, Hong H-J (2013) In vivo studies suggest that induction of VanS-dependent vancomycin resistance requires binding of the drug to D-Ala-D-Ala termini in the peptidoglycan cell wall. Antimicrob Agents Chemother 57:4470–4480
Lai MH, Kirsch DR (1996) Induction signals for vancomycin resistance encoded by the vanA gene cluster in Enterococcus faecium. Antimicrob Agents Chemother 40:1645–1648
Laponogov I, Pan XS, Veselkov DA, McAuley KE, Fisher LM, Sanderson MR (2010) Structural basis of gate-DNA breakage and resealing by type II topoisomerases. PLoS One 5:e11338
Laponogov I, Sohi MK, Veselkov DA, Pan XS, Sawhney R, Thompson AW, McAuley KE, Fisher LM, Sanderson MR (2009) Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nat Struct Mol Biol 16:667–669
Leclercq R, Derlot E, Duval J, Courvalin P (1988) Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N Engl J Med 319:157–161
Lee E-W, Huda N, Kuroda T, Mizushima T, Tsuchiya T (2003) EfrAB, an ABC multidrug efflux pump in Enterococcus faecalis. Antimicrob Agents Chemother 47:3733–3738
Levine DP (2006) Vancomycin: a history. Clin Infect Dis 42:S5–12
Loll PJ, Derhovanessian A, Shapovalov MV, Kaplan J, Yang L, Axelsen PH (2009) Vancomycin forms ligand-mediated supramolecular complexes. J Mol Biol 385:200–211
Loll PJ, Miller R, Weeks CM, Axelsen PH (1998) A ligand-mediated dimerization mode for vancomycin. Chem Biol 5:293–298
Lu J-J, Perng C-L, Ho M-F, Chiueh T-S, Lee W-H (2001) High prevalence of VanB2 vancomycin-resistant Enterococcus faecium in Taiwan. J Clin Microbiol 39:2140–2145
Luong TT, Dunman PM, Murphy E, Projan SJ, Lee CY (2006) Transcription profiling of the mgrA regulon in Staphylococcus aureus. J Bacteriol 188:1899–1910
Luong TT, Newell SW, Lee CY (2003) Mgr, a novel global regulator in Staphylococcus aureus. J Bacteriol 185:3703–3710
MacDougall J, Ahern J, Civalier M, Pierce K, Cohen R (2016) Identification of risk factors for initial elevated vancomycin trough concentrations. J Pharm Technol 32:29–33
Mackay JP, Gerhard U, Beauregard DA, Williams DH, Westwell MS, Searle MS (1994) Glycopeptide antibiotic activity and the possible role of dimerization: a model for biological signalling. J Am Chem Soc 116:4581–4590
Mani N, Sancheti P, Jiang ZD, McNaney C, Decenzoi M, Knight B, Stankis M, Kuranda M, Rothstein DM (1998) Screening systems for detecting inhibitors of cell wall transglycosylation in Enterococcus. J Antibiot 51:471–479
Mascarello M, Simonetti O, Knezevich A et al (2017) Correlation between antibiotic consumption and resistance of bloodstream bacteria in a University Hospital in North Eastern Italy, 2008-2014. Infection 45:459–467
McCallum N, Berger-Bächi B, Senn MM (2010) Regulation of antibiotic resistance in Staphylococcus aureus. Int J Med Microbiol 300:118–129
Mehta SC, Rice K, Palzkill T (2015) Natural variants of the KPC-2 carbapenemase have evolved increased catalytic efficiency for ceftazidime hydrolysis at the cost of enzyme stability. PLOS Pathog 11:e1004949
Mladenovic-Antic S, Kocic B, Velickovic-Radovanovic R et al (2016) Correlation between antimicrobial consumption and antimicrobial resistance of Pseudomonas aeruginosa in a hospital setting: a 10 year study. J Clin Pharmacol Ther 41:532–537
Moellering RC (2006) Vancomycin: a 50-year reassessment. Clin Infect Dis 42:S3–S4
Negri M-A, Morosini M, Loza E, Baquero F (1994) In vitro selective antibiotic concentrations of β-lactams for penicillin-resistant Streptococcus pneumoniae populations. Antimicrob Agents Chemother 38:122–125
Nieto M, Perkins HR (1971) The specificity of combination between ristocetins and peptides related to bacterial cell wall mucopeptide precursors. Biochem J 124:845–852
Nishioka T, Ogawa W, Kuroda T, Katsu T, Tsuchiya T (2009) Gene cloning and characterization of EfmA, a multidrug efflux pump, from Enterococcus faecium. Biol Pharm Bull 32:483–488
Nitanai Y, Kikuchi T, Kakoi K, Hanamaki S, Fujisawa I, Aoki K (2009) Crystal structures of the complexes between vancomycin and cell wall precursor analogues. J Mol Biol 385:1422–1432
Noguchi N, Okada H, Narui K, Sasatsu M (2004) Comparison of the nucleotide sequence and expression of norA genes and microbial susceptibility in 21 strains of Staphylococcus aureus. Microb Drug Resist Mech Epidem Dis 10:197–203
Nukaga M, Mayama K, Hujer AM, Bonomo RA, Knox JR (2003) Ultrahigh resolution structure of a class A beta-lactamase: on the mechanism and specificity of the extended-spectrum SHV-2 enzyme. J Mol Biol 328:289–301
Odenholt I, Gustafsson I, Löwdin E, Cars O (2003) Suboptimal antibiotic dosage as a risk factor for selection of penicillin-resistant Streptococcus pneumoniae: in vitro kinetic model. Antimicrob Agents Chemother 47:518–523
OiE (2015) OiE list of antimicrobial agents of Veterinary Importance
Palmer KL, Kos VN, Gilmore MS (2010) Horizontal gene transfer and the genomics of enterococcal antibiotic resistance. Curr Opin Microbiol 13:632–639
Paterson IK, Hoyle A, Ochoa G, Baker-Austin C, Taylor NGH (2016) Optimising antibiotic usage to treat bacterial infections. Sci Rep 6:37853
Peña-Miller R, Fuentes-Hernandez A, Reding C, Gudelj I, Beardmore R (2014) Testing the optimality properties of a dual antibiotic treatment in a two-locus, two-allele model. J R Soc Interface 11:20131935
Pereira PM, Filipe SR, Tomasz A, Pinho MC (2007) Fluorescence ratio imaging microscopy shows decreased access of vancomycin to cell wall synthetic sites in vancomycin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 51:3627–3633
Périchon B, Courvalin P (2009) VanA-type vancomycin-resistant Staphylococcus aureus. Antimicr Agents Chemother 53:4580–4587
Phillips-Jones MK, Lithgo R, Dinu V, Gillis RB, Harding JE, Adams GG, Harding SE (2017a) Full hydrodynamic reversibility of the weak dimerization of vancomycin and elucidation of its interaction with VanS monomers at clinical concentration. Sci Rep 7:12697
Phillips-Jones MK, Channell G, Kelsall CJ, Hughes CS, Ashcroft AE, Patching SG, Dinu V, Gillis RB, Adams GA, Harding SE (2017b) Hydrodynamics of the VanA-type VanS histidine kinase: an extended solution conformation and first evidence for interactions with vancomycin. Sci Rep 7:46180
Pitiriga V, Vrioni G, Saroglou G et al (2017) The impact of antibiotic stewardship programs in combating quinolone resistance: a systematic review and recommendations for more efficient interventions. Adv Ther 34:854–865
Public Health England and Veterinary Medicines Directorate Report (2015) UK One Health Report: Joint report on human and animal antibiotic use, sales and resistance, 2013. PHE publications gateway number 2015160
Public Health England Report (2013) UK One Health Report: Joint report on human and animal antibiotic use, sales and resistance, 2013 published by Public Health England
Public Health England Report (2015). Health matters: antimicrobial resistance, 2015 published by Public Health England
Ramirez MS, Nikolaidis N, Tolmasky ME (2013) Rise and dissemination of aminoglycoside resistance: the aac(6′)-Ib paradigm. Front Microbiol 4:121
Raquet X, Lamotte-Brasseur J, Bouillenne F, Frère J-M (1997) A disulphide bridge near the active site of carbapenem-hydrolysing class A beta-lactamases might explain their unusual substrate profile. Proteins Struct Funct Genet 27:47–58
Rayner C, Munckhof WJ (2005) Antibiotics currently used in the treatment of infections caused by Staphylococcus aureus. Intern Med J 35:S3–S16
Resch M, Striegl H, Henssler EM, Sevvana M, Egerer-Sieber C, Schiltz E, Hillen W, Muller YA (2008) A protein functional leap: how a single mutation reverses the function of the transcription regulator TetR. Nucleic Acid Res 36:4390–4401
Reynolds PE (1989) Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur J Clin Microbiol Infect Dis 8:943–950
Reynolds PE, Courvalin P (2005) Vancomycin resistance in enterococci due to synthesis of precursors terminating in D-Alanyl-D-Serine. Antimicrob Agents Chemother 49:21–25
Rodriguez-Martinez JM, Cano ME, Velasco C, Martinez-Martinez L, Pascual A (2011) Plasmid-mediated quinolone resistance: an update. J Infect Chemother 17:149–182
Rubtsova M, Ulyashova MM, Bachmann TT, Schmid RD, Egorov AM (2010) Multiparametric determination of genes and their point mutations for identification of beta-lactamases. Biochem Mosc 75:1628–1649
Rybak MJ (2006) The pharmacokinetic and pharmacodynamics properties of vancomycin. Clin Infect Dis 42:S35–S39
Rybak M, Lomaestro B, Rotschafer JC, Moellering R, Craig W, Billeter M, Dalovisio JR, Levine DP (2009) Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America and the Society of Infectious Diseases Pharmacists. Am J Health Syst Pharm 66:82–98
Santos Costa S, Viveiros M, Amaral L, Couto I (2013) Multidrug efflux pumps in Staphylococcus aureus. Open Microbiol J 7(Suppl. 1-M5):59–71
Sarovich DS, Price EP, Von Schulze AT, Cook JM, Mayo M, Watson LM, Richardson L, Seymour ML, Tuanyok A, Engelthaler DM, Pearson T, Peacock SJ, Currie BJ, Keim P, Wagner DM (2012) Characterisation of ceftazidime resistance mechanisms in clinical isolates of Burkholderia pseudomallei from Australia. PLoS One 7:e30789
Schmitz FJ, Hertel B, Hofmann B, Scheuring S, Verhoef J, Fluit AC, Heinz HP, Köhrer K, Jones ME (1998) Relationship between mutations in the coding and promoter regions of the norA genes in 42 unrelated clinical isolates of Staphylococcus aureus and the MICs of norfloxacin for these strains. J Antimicrob Chemother 42:561–563
Schouten MA, Hoogkamp-Korstanje JAA, Meis JFG, Voss A, the European VRE Study Group (2000) Prevalence of vancomycin-resistant enterococci in Europe. Eur J Clin Microbiol Infect Dis 19:816–822
Shaheen A, Ismat F, Iqbal M, Haque A, De Zorzi R, Mirza O, Walz T, Rahman M (2015) Characterisation of putative multidrug resistance transporters of the major facilitator superfamily expressed in Salmonella typhi. J Infect Chemother 21:357–362
Sharman GJ, Try AC, Dancer RJ, Cho YR, Staroske T, Bardsley B, Maguire AJ, Cooper MA, O’Brien DP, Williams DH (1997) The roles of dimerization and membrane anchoring in activity of glycopeptide antibiotics against vancomycin-resistant bacteria. J Am Chem Soc 119:12041–12047
Sievert DM, Boulton ML, Stolman G, Johnson D, Stobierski MG, Downes FP, Somsel PA, Rudrik JT, Brown W, Hafeez W, Lundstrom T, Flanagan E, Johnson R, Mitchell J, Chang S (2002) Staphylococcus aureus resistant to vancomycin. MMWR Morb Mortal Wkly Rep 51:565–567
Sievert DM, Rudrik JT, Patel JB, McDonald C, Wilkins MJ, Hageman JC (2008) Vancomycin-resistant Staphylococcus aureus in the United States, 2002-2006. Clin Infect Dis 46:668–674
Sreedharan S, Oram M, Jensen B, Peterson LR, Fisher LM (1990) DNA gyrase gyrA mutations in ciprofloxacin-resistant strains of Staphylococcus aureus: close similarity with quinolone resistance mutations in Escherichia coli. J Bacteriol 172:7260–7262
Strahilevitz J, Jacoby GA, Hooper DC, Robicsek A (2009) Plasmid-mediated quinolone resistance: a multifaceted threat. Clin Microbiol Rev 22:664–689
Tammer I, Geginat G, Lange S et al (2016) Antibiotic consumption and the development of antibiotic resistance in surgical units. Zentralbl Chir 141:53–61
Tobin CM, Darville JM, Thomson AH, Sweeney G, Wilson JF, MacGowan AP, White LO (2002) Vancomycin therapeutic drug monitoring: is there a consensus view? The results of a UK national external quality assessment scheme (UK NEQAS) for antibiotic assays questionnaire. J Antimicrob Chemother 50:713–718
Tomašić T, Mašič LP (2014) Prospects for developing new antibacterials targeting bacterial type IIA topoisomerases. Curr Top Med Chem 14:130–151
Tran JH, Jacoby GA (2002) Mechanism of plasmid-mediated quinolone resistance. Proc Natl Acad Sci U S A 99:5638–5642
Trucksis M, Wolfson JS, Hooper DC (1991) A novel locus conferring fluoroquinolone resistance in Staphylococcus aureus. J Bacteriol 173:5854–5860
Truong-Boldoc QC, Dunman PM, Strahilevitz J, Projan SJ, Hooper DC (2005) MgrA is a multiple regulator of two new efflux pumps in Staphylococcus aureus. J Bacteriol 187:2395–2405
Truong-Boldoc QC, Hooper DC (2007) The transcriptional regulators NorG and MgrA modulate resistance to both quinolones and β-lactams in Staphylococcus aureus. J Bacteriol 189:2996–3005
Truong-Boldoc QC, Strahilevitz J, Hooper DC (2006) NorC, a new efflux pump regulated by MgrA of Staphylococcus aureus. Antimicrob Agents Chemother 50:1104–1107
Truong-Bolduc QC, Zhang X, Hooper DC (2003) Characterisation of NorR protein, a multifunctional regulator of norA expression in Staphylococcus aureus. J Bacteriol 185:3127–3138
Ubukata K, Itoh-Yamashita N, Konno M (1989) Cloning and expression of the norA gene for fluoroquinolone resistance in Staphylococcus aureus. Antimicrob Agents Chemother 33:1535–1539
Ulijasz AT, Grenader A, Weisblum B (1996) A vancomycin-inducible lacZ reporter system in Bacillus subtilis: induction by antibiotics that inhibit cell wall synthesis and by lysozyme. J Bacteriol 178:6305–6309
Uttley AHC, Collins CH, Naidoo J, George RC (1988) Vancomycin-resistant enterococci. Lancet i:57-58
Van Hal SJ, Paterson DL, Lodise TP (2013) Systematic review and meta-analysis of vancomycin-induced nephrotoxicity associated with dosing schedules that maintain troughs between 15 and 20 milligrams per litre. Antimicrob Agents Chemother 57:734–744
Werner G, Coque TM, Hammerum AM, Hope R, Hryniewicz W, Johnson A, Klare I, Kristinsson KG, Leclercq R, Lester CH, Lillie M, Novais C, Olsson-Liljequist B, Peixe LV, Sadowy E, Simonsen GS, Top J, Vuopio-Varkila J, Willems RJ, Witte W, Woodford N (2008) Emergence and spread of vancomycin resistance among enterococci in Europe. Eur Secur 13:1–11
WHO (2011) Critically important antimicrobials for human medicine. 3rd revision. World Health Organisation Advisory Group on Integrated Surveillance of Antimicrobial Resistance (AGISAR)
Wohlkonig A, Chan PF, Fosberry AP, Homes P, Huang J, Kranz M, Leydon VR, Miles TJ, Pearson ND, Perera RL, Shillings AJ, Gwynn MN, Bax BD (2010) Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat Struct Mol Biol 17:1152–1153
Wright GD (2007) The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol 5:175–186
Wright GD (2011) Molecular mechanisms of antibiotic resistance. Chem Commun 47:4055–4061
Wright GD, Holman TR, Walsh CT (1993) Purification and characterisation of VanR and the cytosolic domain of VanS: a two-component regulatory system required for vancomycin resistance in Enterococcus faecium BM4147. Biochemistry 32:5057–5063
Yamada Y, Hideka K, Shiota S, Kuroda T, Tsuchiya T (2006) Gene cloning and characterization of SdrM, a chromosomally-encoded multidrug efflux pump, from Staphylococcus aureus. Biol Pharm Bull 29:554–556
Yoshida H, Bogaki M, Nakamura S, Ubukata K, Konno M (1990) Nucleotide sequence and characterisation of the Staphylococcus aureus norA gene, which confers resistance to quinolones. J Bacteriol 172:6942–6949
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Mary K. Phillips-Jones declares that she has no conflict of interest. Stephen E. Harding declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
This paper was submitted to the Special Issue ‘Multiscale structural biology: Biophysical principles and mechanisms underlying the action of bio-nanomachines’ in Honour of Professor Fumio Arisaka on the occasion of his 70th birthday.
This article is part of a Special Issue on ‘Biomolecules to Bio-nanomachines—Fumio Arisaka 70th Birthday’ edited by Damien Hall, Junichi Takagi and Haruki Nakamura.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Phillips-Jones, M.K., Harding, S.E. Antimicrobial resistance (AMR) nanomachines—mechanisms for fluoroquinolone and glycopeptide recognition, efflux and/or deactivation. Biophys Rev 10, 347–362 (2018). https://doi.org/10.1007/s12551-018-0404-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-018-0404-9