
Abstract
Myotonia congenita is a genetic disease caused by mutations in the CLCN1 gene, which encodes for the major chloride skeletal channel ClC-1, involved in the normal repolarization of muscle action potentials and consequent relaxation of the muscle after contraction. Two allelic forms are recognized, depending on the phenotype and the inheritance pattern: the autosomal dominant Thomsen disease with milder symptoms and the autosomal recessive Becker disorder with a severe phenotype. Before the recent advances of molecular testing, the diagnosis and genetic counseling of families was a challenge due to the large number of mutations in the CLCN1 gene, found both in homozygous or in heterozygous state. Here, we studied a consanguineous family in which three members presented a variable phenotype of myotonia, associated to a combination of three different mutations in the CLCN1 gene. A pathogenic splicing site mutation which causes the skipping of exon 17 was present in homozygosis in one very severely affected son. This mutation was present in compound heterozygosis in the consanguineous parents, but interestingly it was associated to a different second variant in the other allele: c.1453 A > G in the mother and c.1842 G > C in the father. Both displayed variable, but less severe phenotypes than their homozygous son. These results highlight the importance of analyzing the combination of different variants in the same gene in particular in families with patients displaying different phenotypes. This approach may improve the diagnosis, prognosis, and genetic counseling of the involved families.
Similar content being viewed by others
Introduction
Myotonia congenita is an inherited disease caused by mutations in the major chloride skeletal channel ClC-1, coded by the CLCN1 gene (Steinmeyer et al. 1991). ClC-1 channels are found spanning the cell membrane forming pores that act as gates in the control of the flow of extracellular chloride ions into the cells. Such ions regulate and stabilize cells’ electrical charge, which is extremely important for correct muscle contraction.
The dysfunction of this channel is related to a muscle membrane hyperexcitability and a delay in muscle fiber relaxation. Clinically, the patients show muscle stiffness following voluntary contraction (Lehmann-Horn and Jurkat-Rott 1999). Due to hyperexcitability of the skeletal muscle membrane, a delayed muscle relaxation occurs, causing repetitive electrical discharges of affected muscle fibers (Wu et al. 2002). The myotonia decreases after repetitive movements. Muscular hypertrophy is an important clinical signal, and patients can present an athletic-like phenotype. Muscle atrophy and transient or permanent weakness can be observed in some patients.
The worldwide prevalence of myotonia congenita has been estimated in approximately 1:100,000 cases (OMIM #160800; OMIM #255700).
The clinical picture in congenital myotonia is highly variable among affected patients and even among patients of the same family. The phenotypic spectrum ranges from mild myotonia identified only on electromyographic examination to severe and disabling myotonia with transient weakness and myopathy.
Two allelic forms are recognized, depending on the phenotype and the inheritance pattern: the autosomal dominant Thomsen disease with milder symptoms and the autosomal recessive Becker disorder with a severe phenotype. Before the recent advances of molecular testing, the diagnosis and genetic counseling of families were a challenge because of the identification of a large number of mutations in the CLCN1 gene, found both in homozygous or in heterozygous state (Colding-Jørgensen 2004).
In this report, we studied a consanguineous family whose members present a variable phenotype of myotonia, associated to a different combination of mutations in the two alleles of the CLCN1 gene. A pathogenic splicing site mutation in the CLCN1 gene which causes exon 17 skipping was present in homozygosis in one severely affected son. This mutation was present in compound heterozygosis with other two different variants in the CLCN1 gene in each one of the consanguineous parents, both with variable but less severe phenotype.
This report highlights the importance of studying the combination of different variants aiming to elucidate their participation in the phenotype of the patients, in the prognosis and genetic counseling of the involved families.
Materials and Methods
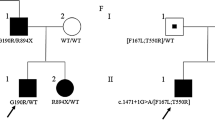
The CLCN1 gene was sequenced in seven family members, as shown in Fig. 1. DNA samples were extracted from whole peripheral blood by standard procedures and amplified by PCR using primers for the 23 exons of the CLCN1 gene and amplicons were submitted to Sanger sequencing analysis.

Pedigree of the family and all the tested individuals, with the respective CLCLN1 gene genotype identified
In one case, II-2, a custom next-generation sequencing panel containing more than 90 genes related to monogenic neuromuscular disorders was done in order to search for additional possible variants in CLCN1 and other candidate genes. The Nextera Rapid Capture Custom Enrichment Kit (Illumina) was used for library preparation and capture. Sequencing was performed on a HiSeq 2500 equipment (Illumina). The fastq files were aligned against the GRCh37/hg19 human genome reference using BWA-MEM, with a post-processing step using Picard Tools 1.81 to convert sam files to bam files and to mark PCR duplicates. Genotypes were called using the Genome Analysis Toolkit (GATK) UnifiedGenotyper, version 3.7, following the GATK best practices. The variants were annotated using ANNOVAR. Variants were filtered according to allelic number from our center cohort, population frequencies, pathogenicity, and previous reports.
Muscle biopsy was performed in the patient (II-2), after informed consent, and a biceps muscle fragment was frozen in liquid nitrogen immediately after removal and stored at − 70 °C until use. Routine histological and histochemical procedures were done, with staining for HE, modified Gomori trichrome, NADH, ATPase 9.4, 4.3 and acid and alkaline phosphatase (Dubowitz et al. 2013) (Fig. 2).

Muscle biopsy analysis of patient II-2. a Western blot analysis for dystrophin, dysferlin, and the 94-kDa band of calpain 3 in a control (C) and the patient (II-2). b Histological analysis showing staining for HE, ATPase 9.4, and NADH, showing no significant alterations. c |Immunohistochemical analysis for dystrophin (rod domains antibody), γ-SG, and α2-laminin, showing normal staining of muscle membrane in both control and the patient
Immunofluorescence analysis was done using antibodies for dystrophin (primary antibody: VP-D508, Vector, 1:20—anti-mouse-CY3, C2181, Sigma, 1:200), γ-sarcoglycan (primary antibody: SC28280, Santa Cruz, 1:100—anti-mouse-Alexa 594, A-21442, Invitrogen, 1:1000), and α2-laminin (primary antibody: SC59854, Santa Cruz, 1:100—anti-mouse-Alexa 594, A-21471, Invitrogen, 1:1000). Images were captured with a Zeiss Axio Imager 2 Research Microscope (Zeiss) at ×200 magnification. In addition, western blot analysis was done using antibodies for dystrophin (VP-D508, Vector, 1:100), dysferlin (VP-D503, Vector, 1:50), and calpain-3 (VP-D304, Vector, 1:100). The membrane was incubated with the Mouse IgG (H + L) Secondary Antibody (Invitrogen), 1:1000, and revealed using the Novex™ ECL Chemiluminescent Substrate Reagent Kit (Invitrogen). Images were obtained using photo-documenter (ImageQuant TL, GE Healthcare Life Sciences).
Results
Clinical Report
The female index case II-2 was first evaluated in 1994 at the age of 33. At that time, she complained of diffuse muscle pain, intense fatigue, frequent falls, difficulty in climbing stairs, and myotonia. She presented marked calf hypertrophy. Her son, III-2, on the occasion, was a 14-month-old baby who presented a delayed neuromotor development and seizures that were related to perinatal anoxia. Patient II-2 returned for re-evaluation on November 2011, presenting the same complaints of muscle pain and fatigue, and reporting a stable clinical picture. Physiotherapist evaluation revealed muscle weakness of proximal predominance in the upper limbs and lower limbs, cervical and paravertebral weakness without facial impairment, difficulty in climbing, diffuse muscle hypertrophy, and hyperlordosis. On respiratory assessment, mild ventilatory disturbance of a restrictive pattern (FVC 80% predicted) was observed. Serum CK level was normal. Patient III-2 was also evaluated on November 2011 at the age of 18 years. He presented significant intellectual disability, and seizures which were controlled with medications. He also presented muscle pain and urinary incontinence and difficulty walking and climbing stairs. He had an echocardiogram with mild mitral valve prolapse. Physical evaluation revealed muscle weakness in the upper and lower limbs, more important than that observed in the mother, facial weakness (not detected in his mother), and important cervical weakness. Mild myotonia and subtle Gowers maneuver were also observed. He had hard but not enlarged calves, lumbar hyperlordosis, and thoracic kyphosis. Respiratory assessment was suggestive of mild ventilatory disorder. Serum CK levels were within normal range. Her husband (II-3) referred a mild muscle weakness since the age of 40, mainly in the upper limbs (arms and hands), with difficulty in lifting weight and shaking hands, without any other limitations. He had no complaints of myotonia in the hands, mentioning only weakness. At first, the weakness was mild and did not bring limitations. Her oldest son (III-1), now 32 years old, remains asymptomatic until now.
This family lived in a distant state of Brazil, and in addition to the two evaluations done in our Center in São Paulo, their clinical status was updated through frequent telephone contacts. In a recent call (2020), patient II-2 reported that both she and her son III-2 had worsened their respiratory condition and a non-invasive ventilation was recommended. She also informed that the two have cardiac arrhythmia. Her husband, now aged 63 years, referred that he is gradually getting worse, mainly in the last year. He continues to deny pain or cramps and refers to muscle locking only in stressful situations. He has noticed a progressive and significant calves’ hypertrophy. Today he climbs stairs with difficulty, holding on to the railing. The main complaint is overall muscle weakness. The maternal grandmother (I-1) and paternal grandmother (I-3) died recently at the age of 75 and 77 due to cardiac problems, and the last one was affected by Alzheimer disease.
The patient II-2 muscle biopsy showed very mild myopathic alterations, including moderate variation on fiber size, but no internally located nuclei, nor endomysial or perimysial connective tissue proliferation. Muscle immunofluorescence and western blot analyses revealed a normal quantity and distribution of proteins related to neuromuscular disorders (dystrophin, dysferlin, calpain 3, γ-SG, and α2-laminin) (Fig. 2).
Molecular Analysis of the CLCN1 Gene
Sanger sequencing of the whole CLCN1 gene identified at least three different variants segregating in the family:
-
a)
c.2172 + 1 G > T in intron 17—splicing site recessive pathogenic mutation found in I-3, II-2, II-3, and III-1, in heterozygous state, and in III-2 in the homozygous state.
-
b)
c.1453 A > G p.(Met485Val) in exon 13—recessive pathogenic mutation found in I-1 and II-2 in the heterozygous state
-
c)
c.1842 G > C p.(Lys614Asn) in exon 16—variant of uncertain significance (VUS) found in II-3 and III-1 in the heterozygous state.
The distribution of these mutations is shown in Fig. 1.
We did not find any significant pathogenic variant in other analyzed NMD genes in the patient II-2 NGS study.
Discussion
More than 300 mutations in the CLCN1 gene have been described in patients with myotonia congenita based on the Human Gene Mutation Database (HGMD) and Leiden Open Variation Database (LOVD). The number of mutations causing the autosomal recessive form of the disorder (Becker disease) is expressively higher than the amount of identified autosomal dominant form (Thomsen disease) (Orsini et al. 2020). Likewise, Becker disease patients are more severely affected than Thomsen disease patients (Cherian et al. 2008).
Clinical phenotypic differences are mainly explained by morphological and functional alterations on the ClC-1 channel. The ClC-1 channel is a homodimer presenting a “double-barreled” structure with two conduction pathways as physically independent “fast gates” and “slow gates” (Pusch 2002) Becker phenotype is caused by mutations at the fast gates level, and they can modify the structure or function of the two protein subunits, reducing the transport of chloride ions into skeletal muscle cells. Regarding the AD Thomsen disease, mutations are mainly found at the slow gates level in one subunit of the homodimer, so that the altered protein gains a dominant negative effect, affecting the dimerization and impairing the unaltered subunit functioning (Imbrici et al. 2015). However, some mutations have been found in both diseases, so that genotype-phenotype correlation and distinction between the two forms is not always easy and clear.
By sequencing the CLCN1 gene in this consanguineous family, we unexpectedly found an interesting combination of different mutations, associated to different phenotypes in the family.
The mutation c.2172 + 1 G > T is classified as pathogenic. This mutation was described in the homozygous state in patients affected by Becker disease. It is located at a donor splicing site in intron 17 of the CLCN1 gene, and mutation at this point leads to exon 17 skipping (Chen et al. 2004). The variant is present with very low frequency in the gnomAD population database (8.6 × 10–6). The presence of the mutation in patient I-3, with no phenotype, also adds more evidence to the recessive character of this mutation. As patient III-2 is homozygous for this mutation, his severely affected myotonic state might have contributed to his perinatal anoxia episode, and the further consequences on his phenotype.
Both II-2 and II-3, who are first-degree cousins, are heterozygous for this same mutation, and would not be expected to be clinically affected since carriers of this mutation have been described as asymptomatic (Chen et al. 2004). However, patient II-2 was clearly affected, and patient II-3 also showed a milder muscle commitment. Surprisingly, the molecular analysis showed that both were also carriers of another different variant in the other allele of the CLCN1 gene. Patient II-2 was a compound heterozygous, harboring the c.2172 + 1 G > T pathogenic mutation associated in trans with the c.1453 A > G p.(Met485Val) variant in the second allele. The p.(Met485Val) missense variant has been reported in patients with MC, in either homozygous or compound heterozygous state (Meyer-Kleine et al. 1995; Kubisch et al. 1998; Fialho et al. 2007; Dupré et al. 2009; Mazón et al. 2012; Brugnoni et al. 2013; Skálová et al. 2013; Tan et al. 2014; Hoche et al. 2014). Expression functional studies in Xenopus oocytes highlighted the great reduction of the single channel conductance caused by this mutation (Wollnik et al. 1997). The total frequency of the variant on the gnomAD frequency database is 3.8 × 10–4 in the heterozygous state. Based on that evidences, the p.(Met485Val) variant is classified as pathogenic. Therefore, the clinical phenotype of patient II-2 could be explained by the presence of two pathogenic mutations in the CLCN1 gene. On the other hand, patient I-1, who carried only this mutation, was apparently asymptomatic, and died at the age of 75 years due to a sudden cardiac arrest. According to the information given by patient II-2, she always presented enlarged calves, which could be a discrete manifestation of this mutation in heterozygous state.
An observation that drew our attention was that one of the patients mentioned by Chen et al. (2004) was receiving diazepam, and the other suffered of depression. Patient II-2 is also under benzodiazepine therapy. This medication is commonly used for its effects in muscle relaxation, but taking into account the psychological state of the patients, it would be interesting to elucidate the possible neurological effects of CLCN1 mutations.
The mildly affected patient II-3 was a compound heterozygous for the c.2172 + 1 G > T pathogenic mutation associated to the variant c.1842 G > C, p.(Lys614Asn) in the second allele. This VUS, a variant of uncertain significance, was previously reported in a family with myotonia congenita, although the mutation did not segregate with the disease. Three patients in this family harbored the p.(Met128Val) variant in the CLCN1 gene, so that this mutation is considered responsible for the autosomal dominant inheritance pattern in the family (Colding-Jørgensen et al. 2003). Pathogenicity prediction by in silico analysis classifies the p.(Lys614Asn) as probably damaging, since this amino acid is conserved in mammals and this change might cause structural alterations on the secondary protein structure. Pathogenicity of this mutation could be reinforced because a different mutation at the same position was identified in compound heterozygosis in a MC patient (Trip et al. 2008). The total allele frequency of p.(Lys614Asn) in gnomAD is 1.5 × 10–3, and there is an entry of one unaffected homozygous female. Thus, we cannot conclude on the pathogenicity of this variant. However, since patient II-3 presented some clinical signals after the age of 40, and the older son (III-1) is asymptomatic at the age of 32 years, we speculated whether the combination of c.2172 + 1 G > T and p.(Lys614Asn) is leading to a more significant impairment of the function of the channel, possibly with an aging effect, although p.(Lys614Asn) alone might not be damaging enough to cause the disease. This possibility could have an important implication in the prognosis of the young individual, since disease manifestation could occur later.
In conclusion, with the improvement of molecular technology, screening for mutations will identify more and more combined variants in genes altered in AR or AD phenotypes, which may have important implication for the diagnosis, prognosis, and genetic counseling of the involved families.
Availability of Data and Material
All data generated and/or analyzed during the study are available upon request.
References
Brugnoni R, Kapetis D, Imbrici P et al (2013) A large cohort of myotonia congenita probands: novel mutations and a high-frequency mutation region in exons 4 and 5 of the CLCN1 gene. J Hum Genet 58:581–587. https://doi.org/10.1038/jhg.2013.58
Chen L, Schaerer M, Lu ZH et al (2004) Exon 17 skipping inCLCN1 leads to recessive myotonia congenita. Muscle Nerve 29:670–676. https://doi.org/10.1002/mus.20005
Cherian A, Baheti N, Kuruvilla A (2008) Muscle channelopathies and electrophysiological approach. Ann Indian Acad Neurol 11:20. https://doi.org/10.4103/0972-2327.40221
Colding-Jørgensen E, DunØ M, Schwartz M, Vissing J (2003) Decrement of compound muscle action potential is related to mutation type in myotonia congenita. Muscle Nerve 27:449–455. https://doi.org/10.1002/mus.10347
Colding-Jørgensen E (2004) Thomsens sygdom (myotonia congenita) [Thomsen disease (myotonia congenita)]. Ugeskr Laeger 166:3179–3184
Dubowitz V, Sewry CA, Oldfors A (2013) Muscle biopsy: a practical approach, 4th Edtion. Elsevier
Dupré N, Chrestian N, Bouchard J-P et al (2009) Clinical, electrophysiologic, and genetic study of non-dystrophic myotonia in French-Canadians. Neuromuscul Disord 19:330–334. https://doi.org/10.1016/j.nmd.2008.01.007
Fialho D, Schorge S, Pucovska U et al (2007) Chloride channel myotonia: exon 8 hot-spot for dominant-negative interactions. Brain 130:3265–3274. https://doi.org/10.1093/brain/awm248
Hoche F, Seidel K, Barbosa-Sicard E et al (2014) Novel N-terminal truncating CLCN1 mutation in severe becker disease. Muscle Nerve 50:866–867. https://doi.org/10.1002/mus.24312
Imbrici P, Altamura C, Pessia M et al (2015) ClC-1 chloride channels: state-of-the-art research and future challenges. Front Cell Neurosci 09. https://doi.org/10.3389/fncel.2015.00156
Kubisch C, Schmidt-Rose T, Fontaine B et al (1998) ClC-1 chloride channel mutations in myotonia congenita: variable penetrance of mutations shifting the voltage dependence. Hum Mol Genet 7:1753–1760. https://doi.org/10.1093/hmg/7.11.1753
Lehmann-Horn F, Jurkat-Rott K (1999) Voltage-gated ion channels and hereditary disease. Physiol Rev 79:1317–1372. https://doi.org/10.1152/physrev.1999.79.4.1317
Mazón MJ, Barros F, De la Peña P et al (2012) Screening for mutations in Spanish families with myotonia. Functional analysis of novel mutations in CLCN1 gene. Neuromuscul Disord 22:231–243. https://doi.org/10.1016/j.nmd.2011.10.013
Meyer-Kleine C, Steinmeyer K, Ricker K et al (1995) Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCNI) leading to myotonia. Am J Hum Genet 57:1325
Orsini C, Petillo R, D’ Ambrosio P et al (2020) CLCN1 molecular characterization in 19 South-Italian patients with dominant and recessive type of myotonia congenita. Front Neurol 11. https://doi.org/10.3389/fneur.2020.00063
Pusch M (2002) Myotonia caused by mutations in the muscle chloride channel geneCLCN1. Hum Mutat 19:423–434. https://doi.org/10.1002/humu.10063
Skálová D, Zídková J, Voháňka S et al (2013) CLCN1 mutations in Czech patients with myotonia congenita, in silico analysis of novel and known mutations in the human dimeric skeletal muscle chloride channel. PLoS ONE 8. https://doi.org/10.1371/journal.pone.0082549
Steinmeyer K, Klocke R, Ortland C et al (1991) Inactivation of muscle chloride channel by transposon insertion in myotonic mice. Nature 354:304–308. https://doi.org/10.1038/354304a0
Tan SV, Z’Graggen WJ, Boërio D et al (2014) Chloride channels in myotonia congenita assessed by velocity recovery cycles. Muscle Nerve 49:845–857. https://doi.org/10.1002/mus.24069
Trip J, Drost G, Verbove DJ et al (2008) In tandem analysis of CLCN1 and SCN4A greatly enhances mutation detection in families with non-dystrophic myotonia. Eur J Hum Genet 16:921–929. https://doi.org/10.1038/ejhg.2008.39
Wollnik B, Kubisch C, Steinmeyer K, Pusch M (1997) Identification of functionally important regions of the muscular chloride channel ClC-1 by analysis of recessive and dominant myotonic mutations. Hum Mol Genet 6:805–811. https://doi.org/10.1093/hmg/6.5.805
Wu F-F, Ryan A, Devaney J et al (2002) Novel CLCN1 mutations with unique clinical and electrophysiological consequences. Brain 125:2392–2407. https://doi.org/10.1093/brain/awf246
Acknowledgments
We would like to thank Fundação de Amparo à Pesquisa do Estado de São Paulo—Centro de Pesquisa, Inovação e Difusão (FAPESP-CEPID), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Instituto Nacional de Ciência e Tecnologia (INCT), FINEP, for the financial support. We would also like to thank the technical support of the following researchers: Adriano S. Senkevics, Dinorah Zilbersztajn-Gotlieb, Lydia U. Yamamoto, Viviane P. Muniz, and Leticia Nogueira. We are also very grateful to Dr. Paula Onofre-Oliveira for the English correction and improvement.
Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo—Centro de Pesquisa, Inovação e Difusão (FAPESP-CEPID), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Instituto Nacional de Ciência e Tecnologia (INCT), FINEP.
Author information
Authors and Affiliations
Contributions
Lucas Santos Souza conceived, designed, and performed the analysis and elaborated, wrote, and reviewed the manuscript. Priscila Calyjur performed the analysis. Antonio Fernando Ribeiro Jr performed the muscle protein analysis. Juliana Gurgel-Giannetti performed the medical and/or physical evaluation of the patients, and contributed with the manuscript revision. Rita Cassia Mingroni Pavanello performed the medical and/or physical evaluation of the patients. Mayana Zatz performed the genetic counseling and contributed with the manuscript revision. Mariz Vainzof conceived, designed, and performed the analysis and elaborated, wrote, and reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethics Approval
This work is in accordance and was approved by the ethics committee of the Biosciences Institute of the University of Sao Paulo, and the DNA samples are deposited in the biobank repository of the Human Genome and Stem Cells Research Center of IB-USP.
Consent to Participate
All subjects participating in this study signed an appropriated informed consent.
Consent for Publication
All subjects participating in this study signed an appropriated informed consent for publication.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Souza, L.S., Calyjur, P., Ribeiro, A.F. et al. Association of Three Different Mutations in the CLCN1 Gene Modulating the Phenotype in a Consanguineous Family with Myotonia Congenita. J Mol Neurosci 71, 2275–2280 (2021). https://doi.org/10.1007/s12031-020-01785-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-020-01785-4