
Abstract
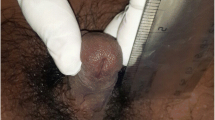
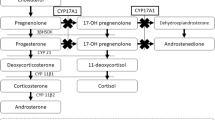
Seventeen alpha-hydroxylase deficiency (17OHD) syndrome is a rare genetic disorder of steroid biosynthesis causing decreased production of glucocorticoids and sex steroids and increased synthesis of mineralocorticoid precursors. There are only 130 cases reported worldwide with documented severe 17OHD. Here, we describe the clinical, hormonal, and molecular genetic characteristics of a Turkish patient with 17OHD, who presented to our clinic due to high blood pressure. A 29-year-old girl with 46,XY genotype was admitted to our nephrology clinic due to uncontrolled hypertension and hypokalemia. The diagnosis was suspected because of primary amenorrhea, absence of sexual maturation, hypertension, and hypokalemia. Endocrine investigation revealed low basal levels of all steroid hormones which require 17-hydroxylation for biosynthesis. Plasma concentrations of ACTH, FSH, and LH were elevated. Imaging did not reveal uterus or adnexial structures. The patient’s hypertension and hypokalemia resolved after glucocorticoid replacement and treatment with potassium-sparing diuretics. 17OHD is a rare form of congenital adrenal hyperplasia in which defects in the biosynthesis of cortisol and sex steroids result in mineralocorticoid excess, hypokalemic hypertension, and sexual abnormalities such as pseudohermaphroditism in males, and sexual infantilism in females. 17OHD should be suspected in patients with hypokalemic hypertension and lack of secondary sexual development so that appropriate therapy can be implemented.

Similar content being viewed by others

References
M. Zachmann, J.A. Vollmin, W. Hamilton et al., Steroid 17, 20-desmolase deficiency: a new cause of male pseudohermaphroditism. Clin. Endocrinol. (Oxf) 1(4), 369–385 (1972)
M.M. Grumbach, I.A. Hughes, F.A. Conte, Disorder of sex differentiation, in Williams Textbook of Endocrinology, 10th edn., ed. by P.R. Larsen, H.M. Kronenberg, S. Melmed, et al. (Saunders, Philadelphia, 2003), pp. 842–1002
A. Bhangoo, J. Aisenberg, A. Chartoffe et al., Novel mutation in cytochrome P450c17 causes complete combined 17alpha-hydroxylase/17, 20-lyase deficiency. J. Pediatr. Endocrinol. Metab. 21(2), 185–190 (2008)
S.L. Siew-Lee Wong, S.G. Shu, C.R. Tsai, Seventeen alpha-hydroxylase deficiency. J. Formos. Med. Assoc. 105(2), 177–181 (2006)
C.L. Benetti-Pinto, D. Vale, H. Garmes et al., 17-hydroxyprogesterone deficiency as a cause of sexual infantilism and arterial hypertension: laboratory and molecular diagnosis–a case report. Gynecol. Endocrinol. 23(2), 94–98 (2007)
C.E. Kater, E.G. Biglieri, Disorders of steroid 17 alphahydroxylase deficiency. Endocrinol. Metab. Clin. North Am. 23, 341–357 (1994)
T. Yanase, E.R. Simpson, M.R. Waterman, 17 alpha-hydroxylase/17, 20-lyase deficiency: from clinical investigation to molecular definition. Endocr. Rev. 12, 91–108 (1991)
S. Kalkan, C. Dizdarer, F. Tuzun et al., Myopathy, hypokalemic hypertension and hypogonadism: a case report of 17 alpha-hydroxylase deficiency syndrome. Gen. Med. J. 16, 71–75 (2006). (In Turkish)
A. German, S. Suraiya, Y. Tenenbaum-Rakover et al., Control of childhood congenital adrenal hyperplasia and sleep activity and quality with morning or evening glucocorticoid therapy. J. Clin. Endocrinol. Metab. 93(12), 4707–4710 (2008)
L. Zhang, H. Wang, T. Hong, Diagnosis and treatment of 17 alpha-hydroxylase deficiency: a case report and literature review. Beijing Da Xue Xue Bao 40(2), 221–222 (2008)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Aydin, Z., Ozturk, S., Gursu, M. et al. Male pseudohermaphroditism as a cause of secondary hypertension: a case report. Endocr 38, 100–103 (2010). https://doi.org/10.1007/s12020-010-9357-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-010-9357-x