
Abstract
Purpose of Review
The goal of this review is to evaluate the management options for achondroplasia, the most common non-lethal skeletal dysplasia. This disease is characterized by short stature and a variety of complications, some of which can be quite severe.
Recent Findings
Despite several attempts to standardize care, there is still no widely accepted consensus. This is in part due to absence of concrete data on the incidence of sudden unexplained death in infants with achondroplasia and the best investigation for ascertaining which individuals could benefit from foramen magnum decompression surgery.
Summary
In this review, we identify the different options of care and management for the various orthopedic, neurologic, and respiratory complications. In parallel, several innovative or drug repositioning therapies are being investigated that would restore bone growth but may also prevent complications. Achondroplasia is the most common non-lethal skeletal dysplasia. It is characterized by short stature and a variety of complications, some of which can be quite severe. Despite several attempts to standardize care, there is still no widely accepted consensus. This is in part due to absence of concrete data on the incidence of sudden unexplained death in infants with achondroplasia and the best investigation for ascertaining which individuals could benefit from foramen magnum decompression surgery. In this review, we identify the different options of care and management for the various orthopedic, neurologic, and respiratory complications. In parallel, several innovative or drug repositioning therapies are being investigated that would restore bone growth but may also prevent complications.
Similar content being viewed by others


Introduction
Achondroplasia is a rare genetic disorder for which no cure is available. This skeletal dysplasia is the most common form of short limb dwarfism and was first reported in 1878 [1]. It is estimated that it affects approximately 250,000 people worldwide [2•, 3, 4]. The incidence is estimated to be 1 in 10,000 to 30,000 live births per year [5], affecting both males and females with equal frequency.
Achondroplasia is characterized by prenatal onset of disproportionate short stature [6, 7]. Most affected children and adults enjoy good general health, but numerous neurological, orthopedic, and otolaryngologic complications can occur in this disorder, and an association with sudden infant death has been reported [8]. The phenotype of achondroplasia is caused by abnormal endochondral bone development, thus mainly affecting the growth of the long bones; the vertebrae; and several bones in the skull, including the temporal, occipital, sphenoid, and ethmoid bones. The trunk is narrow but of normal size. Adult height is 131 ± 5.6 cm in males and 124 ± 5.9 cm in females [7, 9].
Achondroplasia is caused by a single point gain-of-function mutation in the gene coding for fibroblast growth factor receptor 3 (FGFR3) [10]. It follows an autosomal dominant inheritance; though in 80% of the cases, it is a de novo mutation [11] associated with increased paternal age, relative to the general population [12,13,14,15]. In 90% of the cases, the mutation is a Gly380Arg substitution (the most common mutation is 1138G > A transition; a G > C transversion has been reported in 20% of the cases) [11, 16, 17]. Diagnosis can be suspected in utero by ultrasound and/or clinical and radiological features at birth but should be confirmed by molecular testing [18]. To help with diagnosis of skeletal dysplasia including achondroplasia, non-invasive prenatal testing is being developed [19]. The penetrance is 100% and the phenotype is already apparent at birth. Infants with achondroplasia present characteristic features with macrocephaly with frontal bossing, midface hypoplasia, and flat nasal bridge. They have short limbs with predominantly proximal (rhizomelic) shortening of the upper limbs [20], joint laxity, and a trident hand [21]. Intellect is not affected and their lifespan is close to that of the general population. Children with achondroplasia present motor delays notably to acquire standing position and walking due to muscle hypotonia and ligamentous laxity. More than 50% of the patients develop an early obesity that augments the morbidity associated with lumbar lordosis as well as the severity of sleep apnea or orthopedic complications such as genua vara.
Achondroplasia is a short stature condition compatible with good general health and normal life expectancy. Besides the burden of short stature, health issues may arise from complications, due to the particular anatomical features of achondroplasia.
Current Care and Management
Management
Recommendations for management of children with achondroplasia have been proposed and updated over time [2•, 22, 23]. However, these guidelines are mainly based on personal experience of the authors and not on clinical studies.
Treatments aimed at correcting growth have been tried; growth hormone therapy has no significant effect on adult stature [24]. Surgical limb lengthening can improve body proportions and final height up to 20 cm but requires multiple surgeries with high rate of complications and physical burden [25]. The choice of undergoing limb lengthening depends largely on cultural and psychosocial factors.
Current management of achondroplasia consists essentially in prevention and treatment of complications. While there is agreement on complications and their pathophysiology, there is no consensus regarding the type and frequency of surveillance because of the lack of prospective controlled clinical studies. The exact prevalence of each complication is unknown, in some cases overestimated; it is likely that the overall prevalence of complications reaches at least 10%. Figure 1 summarizes the different complications occurring in infants, toddlers and children, or adults. The most difficult task for clinicians faced with parents of a newborn with achondroplasia is to provide the right information and awareness about the condition, without overmedicalizing the infant.

Timelines of the different complications occurring in achondroplasia patients
Monitoring
Growth and development should be monitored according to norms for achondroplasia; special charts exist for height, weight, and occipital frontal circumference (OFC) [6, 22]. OFC is physiologically larger in achondroplasia and should be monitored regularly (monthly in the first year of life) in order to detect any unusual acceleration as a possible sign of hydrocephaly, a rare but potentially severe complication. Gross motor skills are often delayed [26, 27], probably due to the anatomical-biomechanical features of this condition with large head, short limbs, and joint hypermobility [28]. Fine motor and feeding skills are usually not delayed and language delay is variable. Overall, most children reach all normal developmental milestones and have no neurological or intellectual impairment; access to physiotherapy, occupational therapy, and language therapists with experience in achondroplasia may significantly improve timing of autonomy [26].
Complications
The most frequent complications observed in achondroplasia children are ENT issues: upper airways are anatomically smaller in achondroplasia and as a consequence, middle ear effusions and infections occur frequently, sometimes leading to conductive hearing loss and subsequent speech delay; tonsillectomy and adenoidectomy may be necessary to reduce airway obstruction, especially in the case of obstructive sleep apnea [29]. Obstructive sleep apnea is reported in >50% of patients [30], possibly because of a combination of small airways, hypotonia, and midface hypoplasia. Clinical surveillance by an ENT specialist with experience in achondroplasia is recommended particularly between 1 and 4 years of age. Pulmonary complications are rare, but restrictive pulmonary disease may occur, particularly in children living at high altitude [23].
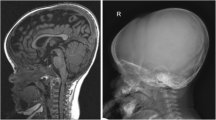
For infants with achondroplasia, their parents, and their doctors, the most frightful complication is sudden infant death. It has been reported to occur in up to 7.5% of achondroplasia infants <1 year [31], but its incidence is probably overestimated; a more recent study reports a sixfold increase in infant mortality in achondroplasia compared to the general population [32]. It is thought to be due to spinal cord compression at the cervicomedullary junction secondary to a stenotic foramen magnum. Sleep-disordered breathing may contribute to increased mortality; however, no clear correlation has been found between central sleep apnea and foramen magnum stenosis. Always in relationship with foramen magnum stenosis, ventriculomegaly is frequent in achondroplasia infants, due to disturbed CSF circulation at the foramen magnum, but true hydrocephaly as a cause of acute intracranial hypertension seems to be much rarer [33, 34]. Some centers perform serial MRI, electrophysiological and sleep studies starting from birth and during the first year of life. However, this surveillance is not without drawbacks: brain and spinal MRI require general anesthesia and somato-sensory evoked potentials may not show changes in presymptomatic phase. In addition, all babies with achondroplasia have a narrow foramen magnum (Fig. 2a), which usually improves with age [35]. No specific marker has been identified so far to distinguish which asymptomatic patients need prophylactic decompression of the foramen magnum, and there is no evidence-based strategy; it is currently recommended that only symptomatic infants (with some clinical neurological signs and/or pathologic polysomnography) undergo brain imaging [36••]. Careful clinical surveillance by a multidisciplinary team experienced in achondroplasia may prove to be as sensible as investigations in detecting critical brainstem compression: increased generalized hypotonia, changes in osteotendinous reflexes (hyperreflexia, clonus, asymmetry), and subtle signs of intracranial hypertension (prominent fontanella, rapid increase of OFC) should be monitored, preferably by the same professional, every 2 months [36 ••]; family doctors should be informed about signs and symptoms to be surveyed in between the visits at the specialized center. Some centers propose sleep studies and even matrasses with detectors of sleep apnea; the utility of these measures has not been proven and the occurrence of sleep apnea appears to be only poorly correlated with foramen magnum stenosis [37, 38].

a MRI image of a four and a half year old boy showing a narrow foramen magnum. This finding is seen in almost all children with achondroplasia and is not by itself an indication to perform neurosurgery. b X-ray showing the typical configuration of the lumbar spine in achondroplasia with shortening in the AP diameter and scalloping if the posterior endplates. More significant shortening of the first lumbar vertebra is also a risk factor for developing a fixed kyphosis and would be an indication for treatment by bracing
Orthopedic complications are possible at all ages. In the first year of life, some degree of kyphosis is always present at the low thoracic and lumbar region (Fig. 2b); it persists until acquirement of standing position, when it reverses to a hyperlordosis. It is recommended to avoid carrying the baby in the first months of life in baby wraps, which do not support the back and favor a kyphotic posture. Later on, the sitting position should not be imposed before the baby has enough muscle strength to sit autonomously. Bracing is not needed in all cases but may be useful to contain kyphosis when the baby is in a sitting position (meals, landau walks). Some children are particularly hypermobile and very delayed in sitting autonomously and standing. In these cases, to avoid a fixed kyphosis, appropriate bracing and/or adapted seats supporting the dorsolumbar spine should be provided. Hypermobility of the knee may also lead to instability and delayed walking but does not usually cause deformity or pain in the first years of life. Physiotherapy may be helpful in gaining correct postures and motoric strategies before free walking is achieved.
In toddlers and children, angular deformities of the lower limbs may develop with walking. Referral to orthopedic surgeons is reserved for patients with a specific problem, such as marked genua vara. External rotation of the hips is frequent and benign and usually resolves when the child starts walking. Obesity is an additional risk factor for orthopedic deformities and joint complications. Starting from when the child has achieved a height of 75 cm, weight/height ratio should be monitored [6] and, if necessary, nutritional counseling be offered.
In adults, the main orthopedic complication is lumbar spinal stenosis between L1 and L4, due to a narrow spinal canal and shorter vertebral arches. It can cause back pain and peripheral nerve compression. Spinal stenosis affects approximately 10% of adults with achondroplasia, and decompression surgery may be necessary in young adults [39].
Daily Living
Living with achondroplasia is associated with all the practical problems of short-limbed short stature in respect to architectural barriers (access to toilets, public desks, automats, public transports, etc). In addition, short arms and limited elbow extension significantly limit autonomy in personal care (dressing, personal hygiene, etc.). Occupational therapy, particularly from school age on, is helpful in finding strategies to overcome these limitations.
Social functioning is normal for most children with achondroplasia. The psychosocial burden of achondroplasia depends on several personal, familial, ethnic, and cultural factors. Some families will have a strong perception of handicap for a child with achondroplasia, while others will not perceive the condition as a problem. Educational guidance for parents of children with achondroplasia should be offered at the achondroplasia multidisciplinary clinic, to prevent overprotection, limited autonomy, and excessive anxiety. Psychological support should be offered to help acceptance of the condition, first to parents of affected infants in order to promote establishment of appropriate affective links and later to children and teenagers facing frustration and rebellion toward diversity. Support groups may also be helpful for patients and families to share experiences and reinforce positive individual resources.
Investigational Therapies
Current care for achondroplasia manages the symptoms and alleviates the consequences of complications. To be effective, a new treatment approach must consider achondroplasia as a complex disease, correcting the short stature and preventing complications. Understanding the pathophysiology of the disease is essential to achieve this and correct the growth of all bones affected by the disease. As stated above, achondroplasia is caused by a mutation in the FGFR3 gene resulting in abnormal endochondral ossification. FGFR3 signaling negatively regulates endochondral bone growth [40] by inhibiting the rate of chondrocyte proliferation and the initiation of chondrocyte hypertrophy [41] through activation of the Stat1 and MAPK pathways [42, 43]. In achondroplasia, the G380R mutation, located in the transmembrane domain of FGFR3, extends the signaling cascade resulting in prolonged bone growth inhibition (Fig. 3a). The dimerization and activation of mutant FGFR3 is ligand-dependent [44,45,46] and stabilizes the ligand/receptor complex by decreasing its internalization [46,47,48].

Schematic representation of FGFR3-mediated inhibition of bone growth in achondroplasia (a) and of the sFGFR3 decoy strategy (b)
Several approaches have been developed to target FGFR3 within the cartilage either by blocking its activation, inhibiting downstream signaling, or increasing its turnover [49 •]. Among them, we have recently evaluated the use of a FGFR3 decoy receptor (sFGFR3) to avoid the FGF ligand from binding to the receptor, thus preventing activation of the intracellular signaling directly downstream of mutant FGFR3 resulting in bone growth activation (Fig. 3b) [45]. The sFGFR3 decoy lacks the transmembrane domain and is thus secreted from the cells, unable to activate the signaling cascade [50]. sFGFR3 was administered subcutaneously for 3 weeks to mice carrying the G380R mutation (Fgfr3 ach/+ mice) and was able to penetrate the growth plate cartilage. At a dose of 2.5 mg/kg twice weekly from age 3 days to 3 weeks, sFGFR3 treatment was effective at restoring normal body, tail, and long bone lengths in treated Fgfr3 ach/+ mice. Interestingly, following treatment, significant reduction in respiratory failure and spinal compression were observed. Treated Fgfr3 ach/+ mice also showed normal rib cage development and decreased vertebrae and skull deformities. No obvious toxicity was observed and treatment did not affect reproduction. One unexpected advantageous side effect was the increase in pelvis size in primiparous-treated Fgfr3 ach/+ females resulting in normal size litters. If this translates into humans, this would be a significant advantage, as during delivery achondroplasia patients must undergo C-section due to small pelvis size. The decoy approach shows promise as a potential treatment for achondroplasia restoring not only the stature but also preventing most of the complications due to the characteristic features of achondroplasia.
A 12 amino acid peptide (called P3) binding with high affinity to the extracellular domain of FGFR3 has been evaluated in mice with thanatophoric dysplasia type II (TDII), a more severe form of FGFR3-related skeletal dysplasia [51]. In vitro, P3 treatment restored chondrocyte proliferation and differentiation by decreasing the MAPK signaling cascade. When administered to neonatal TDII pups, animals survived and treatment partially rescued endochondrial bone growth. Interestingly, this treatment had positive effects on long bones but also rescued neonatal lethality potentially preventing some of the complications caused by spinal and skull deformities.
In 2014, Yamashita et al. evaluated the use of statin to rescue bone growth in achondroplasia and thanatophoric dysplasia type I (TDI) [52••]. The rationale for repositioning this pleiotropic drug is that it appears to stimulate bone growth through anabolic effects on chondrocytes [53,54,55]. While the exact mechanism of action is unknown, it is suspected that statin treatment may accelerate FGFR3 degradation on chondrocytes [52••, 56]. In this study, the authors first develop a cell-based system using induced pluripotent stem cells (iPSCs) generated from dermal fibroblasts of achondroplasia and TDI patients. Following reprogramming of these cells and differentiation into chondrocytes, this cell culture assay displays similarities with patient chondrocytes with decreased chondrogenic potential that was rescued by lovastatin treatment. In vivo, daily intraperitoneal injections of 1 mg/kg rosuvastatin from 3 to 6 weeks of age partially restored limb and skull lengths of Fgfr3 ach/+ mice showing potential therapeutic effects of statins. However, while there are abundant data on the use of statin in humans, little is known about safety in children.
Another repositioning approach has been evaluated recently using meclozine, an over-the-counter antihistamine drug used for motion sickness. The authors initially showed in vitro effects of meclozine on chondrocyte proliferation and differentiation [57]. Transgenic Fgfr3 ach/+ mice orally treated with meclozine showed an improvement in the dwarf phenotype with an increase in bone length [58]. The specific mechanism remains unknown, and the safety profile of chronic administration of meclozine in children has yet to be evaluated.
Small molecule approaches targeting tyrosine kinase domains have been explored for over a decade in anticancer therapies [59]. Their application to skeletal dysplasia appears promising at rescuing the achondroplasia phenotype in vitro and in explant culture systems [60,61,62]. However, the relevance of their in vivo application is unclear because of their absence of selectivity for FGFR3 [60], potentially inhibiting the activities of other FGFR and other tyrosine kinases, inducing multiorgan toxicity [62]. Additional development is needed to further improve the specificity.
Biomarin Pharmaceuticals is currently conducting a phase 2 clinical trial on children with achondroplasia using vosoritide, a C-type natriuretic peptide (CNP) analog [49•]. The CNP approach has been evaluated for nearly two decades now and acts indirectly on FGFR3 signaling though the MAPK signaling pathway not the Stat1 cascade [63, 64]. Skeletal phenotype was rescued in double transgenic mice overexpressing both the mutant FGFR3 receptor and CNP in growth plate chondrocytes [65]. Following chronic infusion of CNP, mice with achondroplasia had longer long bones but also larger foramen magnum, suggesting a potential treatment effect on the occurrence of the complications [66, 67]. Biomarin is now using an analog with a prolonged half-life that proved to be effective at increasing bone length in mice and growth plate width in monkey [68]. The initial phase 2 results are very encouraging demonstrating a favorable safety profile and an increase in 50% of the annualized growth velocity following daily subcutaneous administration of 15 g/kg vosoritide for 12 months compared to the pretreatment growth velocity [69]. The body proportions, calculated as the ratio of upper body length to leg length, which is higher in children with achondroplasia, were not improved by vosoritide treatment. In a prospective observational study, Olney et al. recently showed that CNP plasma levels are elevated in achondroplasia patients suggesting that these patients may have a natural resistance to CNP [70]. Further investigation would be necessary to clarify this as it could interfere with long-term treatment efficacy of vosoritide.
Conclusion
In spite of being the most common form of skeletal dysplasia, there exists no clear consensus on care and management for children and adults with achondroplasia. Developing a routine healthcare is of high importance and would greatly benefit some patients who tend to be overmedicalized or are far removed from specialized centers. A natural history multicenter study has been very recently started to increase the “collaboration among researchers to gather similarly affected patients to answer common clinical research questions”. This type of initiative is essential to improve care and management of these patients. In parallel, the development of biotherapies directly targeting FGFR3 could resolve these issues. However, in addition to rescuing skeletal growth, these therapies should address all of the other (or at least some) clinical aspects of achondroplasia thus preventing the development of complications.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Parrot JM. Sur les malformations achondroplasiques et le dieu Ptah. Bull Anthropol Paris. 1878;1:196.
• Ireland PJ, Pacey V, Zankl A, Edwards P, Johnston LM, Savarirayan R. Optimal management of complications associated with achondroplasia. Appl Clin Genet. 2014;7:117–25. This recent review offers an updated management plan for achondroplasia that included the International Classification of Functioning, Disability, and Health (ICF) model of achondroplasia
Horton WA, Hall JG, Hecht JT. Achondroplasia. Lancet. 2007;370:162–72.
Baujat G, Legeai-Mallet L, Finidori G, Cormier-Daire V, Le Merrer M. Achondroplasia. Best Pract Res Clin Rheumatol. 2008;22:3–18.
Waller DK, Correa A, Vo TM, Wang Y, Hobbs C, Langlois PH, Pearson K, Romitti PA, Shaw GM, Hecht JT. The population-based prevalence of achondroplasia and thanatophoric dysplasia in selected regions of the US. Am J Med Genet A. 2008;146A:2385–9.
Hoover-Fong JE, McGready J, Schulze KJ, Barnes H, Scott CI. Weight for age charts for children with achondroplasia. Am J Med Genet A. 2007;143A:2227–35.
Horton WA, Rotter JI, Rimoin DL, Scott CI, Hall JG. Standard growth curves for achondroplasia. J Pediatr. 1978;93:435–8.
Hecht JT, Francomano CA, Horton WA, Annegers JF. Mortality in achondroplasia. Am J Hum Genet. 1987;41:454–64.
Richette P, Bardin T, Stheneur C. Achondroplasia: from genotype to phenotype. Joint Bone Spine. 2008;75:125–30.
Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet JM, Maroteaux P, Le Merrer M, Munnich A. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371:252–4.
Wilkin DJ, Szabo JK, Cameron R, Henderson S, Bellus GA, Mack ML, Kaitila I, Loughlin J, Munnich A, Sykes B, Bonaventure J, Francomano CA. Mutations in fibroblast growth-factor receptor 3 in sporadic cases of achondroplasia occur exclusively on the paternally derived chromosome. Am J Hum Genet. 1998;63:711–6.
Shinde DN, Elmer DP, Calabrese P, Boulanger J, Arnheim N, Tiemann-Boege I. New evidence for positive selection helps explain the paternal age effect observed in achondroplasia. Hum Mol Genet. 2013;22:4117–26.
Goriely A, Wilkie AO. Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. Am J Hum Genet. 2012;90:175–200.
Orioli IM, Castilla EE, Scarano G, Mastroiacovo P. Effect of paternal age in achondroplasia, thanatophoric dysplasia, and osteogenesis imperfecta. Am J Med Genet. 1995;59:209–17.
Arnheim, N., and Calabrese, P. (2016) Germline stem cell competition, mutation hot spots, genetic disorders, and older fathers. Annu Rev Genomics Hum Genet
Bellus GA, Hefferon TW, Ortiz de Luna RI, Hecht JT, Horton WA, Machado M, Kaitila I, McIntosh I, Francomano CA. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet. 1995;56:368–73.
Xue Y, Sun A, Mekikian PB, Martin J, Rimoin DL, Lachman RS, Wilcox WR. FGFR3 mutation frequency in 324 cases from the international skeletal dysplasia registry. Mol Genet Genomic Med. 2014;2:497–503.
Nahar R, Saxena R, Kohli S, Puri R, Verma IC. Molecular studies of achondroplasia. Indian J Orthop. 2009;43:194–6.
Orhant L, Anselem O, Fradin M, Becker PH, Beugnet C, Deburgrave N, Tafuri G, Letourneur F, Goffinet F, Allach El Khattabi L, Leturcq F, Bienvenu T, Tsatsaris V, Nectoux J. Droplet digital PCR combined with minisequencing, a new approach to analyze fetal DNA from maternal blood: application to the non-invasive prenatal diagnosis of achondroplasia. Prenat Diagn. 2016;36:397–406.
Shelmerdine SC, Brittain H, Arthurs OJ, Calder AD. Achondroplasia: really rhizomelic? Am J Med Genet A. 2016;170:2039–43.
Guzman ER, Day-Salvatore D, Westover T, Rosenberg JC, Beim D, Grabelle H. Prenatal ultrasonographic demonstration of the trident hand in heterozygous achondroplasia. J Ultrasound Med. 1994;13:63–6.
Trotter TL, Hall JG, and American Academy of Pediatrics Committee on, G. Health supervision for children with achondroplasia. Pediatrics. 2005;116:771–83.
Pauli, R. M. (1993) Achondroplasia. In GeneReviews (R) (Pagon, R. A., Adam, M. P., Ardinger, H. H., Wallace, S. E., Amemiya, A., Bean, L. J. H., Bird, T. D., Fong, C. T., Mefford, H. C., Smith, R. J. H., and Stephens, K., eds), Seattle (WA)
Miccoli, M., Bertelloni, S., and Massart, F. (2016) Height outcome of recombinant human growth hormone treatment in achondroplasia children: a meta-analysis. Horm Res Paediatr
Donaldson J, Aftab S, Bradish C. Achondroplasia and limb lengthening: results in a UK cohort and review of the literature. J Orthop. 2015;12:31–4.
Ireland PJ, McGill J, Zankl A, Ware RS, Pacey V, Ault J, Savarirayan R, Sillence D, Thompson EM, Townshend S, Johnston LM. Functional performance in young Australian children with achondroplasia. Dev Med Child Neurol. 2011;53:944–50.
Ireland PJ, Donaghey S, McGill J, Zankl A, Ware RS, Pacey V, Ault J, Savarirayan R, Sillence D, Thompson E, Townshend S, Johnston LM. Development in children with achondroplasia: a prospective clinical cohort study. Dev Med Child Neurol. 2012;54:532–7.
Ireland PJ, Ware RS, Donaghey S, McGill J, Zankl A, Pacey V, Ault J, Savarirayan R, Sillence D, Thompson E, Townshend S, Johnston LM. The effect of height, weight and head circumference on gross motor development in achondroplasia. J Paediatr Child Health. 2013;49:E122–7.
Mogayzel Jr PJ, Carroll JL, Loughlin GM, Hurko O, Francomano CA, Marcus CL. Sleep-disordered breathing in children with achondroplasia. J Pediatr. 1998;132:667–71.
Afsharpaiman S, Sillence DO, Sheikhvatan M, Ault JE, Waters K. Respiratory events and obstructive sleep apnea in children with achondroplasia: investigation and treatment outcomes. Sleep Breath. 2011;15:755–61.
Hecht JT, Horton WA, Reid CS, Pyeritz RE, Chakraborty R. Growth of the foramen magnum in achondroplasia. Am J Med Genet. 1989;32:528–35.
Simmons K, Hashmi SS, Scheuerle A, Canfield M, Hecht JT. Mortality in babies with achondroplasia: revisited. Birth Defects Res A Clin Mol Teratol. 2014;100:247–9.
Erdincler P, Dashti R, Kaynar MY, Canbaz B, Ciplak N, Kuday C. Hydrocephalus and chronically increased intracranial pressure in achondroplasia. Childs Nerv Syst. 1997;13:345–8.
Steinbok P, Hall J, Flodmark O. Hydrocephalus in achondroplasia: the possible role of intracranial venous hypertension. J Neurosurg. 1989;71:42–8.
Mukherjee S, Pringle C, Crocker M. A nine-year review of medicolegal claims in neurosurgery. Ann R Coll Surg Engl. 2014;96:266–70.
•• White KK, Bompadre V, Goldberg MJ, Bober MB, Campbell JW, Cho TJ, Hoover-Fong J, Mackenzie W, Parnell SE, Raggio C, Rapoport DM, Spencer SA, Savarirayan R. Best practices in the evaluation and treatment of foramen magnum stenosis in achondroplasia during infancy. Am J Med Genet A. 2016;170A:42–51. This review highlights our lack of knolwedge regarding sudden unexplained death in achondroplasia, interfering with the establishment of a consensus of care
White KK, Parnell SE, Kifle Y, Blackledge M, Bompadre V. Is there a correlation between sleep disordered breathing and foramen magnum stenosis in children with achondroplasia? Am J Med Genet A. 2016;170A:32–41.
Julliand S, Boule M, Baujat G, Ramirez A, Couloigner V, Beydon N, Zerah M, di Rocco F, Lemerrer M, Cormier-Daire V, Fauroux B. Lung function, diagnosis, and treatment of sleep-disordered breathing in children with achondroplasia. Am J Med Genet A. 2012;158A:1987–93.
Carlisle ES, Ting BL, Abdullah MA, Skolasky RL, Schkrohowsky JG, Yost MT, Rigamonti D, Ain MC. Laminectomy in patients with achondroplasia: the impact of time to surgery on long-term function. Spine (Phila Pa 1976). 2011;36:886–92.
Ornitz DM. FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 2005;16:205–13.
Kozhemyakina E, Lassar AB, Zelzer E. A pathway to bone: signaling molecules and transcription factors involved in chondrocyte development and maturation. Development. 2015;142:817–31.
Murakami S, Balmes G, McKinney S, Zhang Z, Givol D, de Crombrugghe B. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev. 2004;18:290–305.
de Frutos CA, Vega S, Manzanares M, Flores JM, Huertas H, Martinez-Frias ML, Nieto MA. Snail1 is a transcriptional effector of FGFR3 signaling during chondrogenesis and achondroplasias. Dev Cell. 2007;13:872–83.
Monsonego-Ornan E, Adar R, Feferman T, Segev O, Yayon A. The transmembrane mutation G380R in fibroblast growth factor receptor 3 uncouples ligand-mediated receptor activation from down-regulation. Mol Cell Biol. 2000;20:516–22.
Garcia S, Dirat B, Tognacci T, Rochet N, Mouska X, Bonnafous S, Patouraux S, Tran A, Gual P, Le Marchand-Brustel Y, Gennero I, Gouze E. Postnatal soluble FGFR3 therapy rescues achondroplasia symptoms and restores bone growth in mice. Sci Transl Med. 2013;5:203–124.
Monsonego-Ornan E, Adar R, Rom E, Yayon A. FGF receptors ubiquitylation: dependence on tyrosine kinase activity and role in downregulation. FEBS Lett. 2002;528:83–9.
Webster MK, Donoghue DJ. Constitutive activation of fibroblast growth factor receptor 3 by the transmembrane domain point mutation found in achondroplasia. EMBO J. 1996;15:520–7.
Guo C, Degnin CR, Laederich MB, Lunstrum GP, Holden P, Bihlmaier J, Krakow D, Cho YJ, Horton WA. Sprouty 2 disturbs FGFR3 degradation in thanatophoric dysplasia type II: a severe form of human achondroplasia. Cell Signal. 2008;20:1471–7.
• Klag KA, Horton WA. Advances in treatment of achondroplasia and osteoarthritis. Hum Mol Genet. 2016;25:R2–8. This review is of importance as it links achondroplasia and osteoarthritis to elaborate common treatment strategies
Terada M, Shimizu A, Sato N, Miyakaze SI, Katayama H, Kurokawa-Seo M. Fibroblast growth factor receptor 3 lacking the Ig IIIb and transmembrane domains secreted from human squamous cell carcinoma DJM-1 binds to FGFs. Mol Cell Biol Res Commun. 2001;4:365–73.
Jin M, Yu Y, Qi H, Xie Y, Su N, Wang X, Tan Q, Luo F, Zhu Y, Wang Q, Du X, Xian CJ, Liu P, Huang H, Shen Y, Deng CX, Chen D, Chen L. A novel FGFR3-binding peptide inhibits FGFR3 signaling and reverses the lethal phenotype of mice mimicking human thanatophoric dysplasia. Hum Mol Genet. 2012;21:5443–55.
•• Yamashita A, Morioka M, Kishi H, Kimura T, Yahara Y, Okada M, Fujita K, Sawai H, Ikegawa S, Tsumaki N. Statin treatment rescues FGFR3 skeletal dysplasia phenotypes. Nature. 2014;513:507–11. In this paper, the authors very elegantly established induced pluripotent stem cells (iPSCs) using fibroblasts derived from patients. This system can be used to screen drugs that might be used to treat FGFR3 chondrodysplasias
Yudoh K, Karasawa R. Statin prevents chondrocyte aging and degeneration of articular cartilage in osteoarthritis (OA). Aging (Albany NY). 2010;2:990–8.
Simopoulou T, Malizos KN, Poultsides L, Tsezou A. Protective effect of atorvastatin in cultured osteoarthritic chondrocytes. J Orthop Res. 2010;28:110–5.
Baker JF, Walsh PM, Byrne DP, Mulhall KJ. Pravastatin suppresses matrix metalloproteinase expression and activity in human articular chondrocytes stimulated by interleukin-1beta. J Orthop Traumatol. 2012;13:119–23.
Bush JR, Berube NG, Beier F. A new prescription for growth? Statins, cholesterol and cartilage homeostasis. Osteoarthr Cartil. 2015;23:503–6.
Matsushita M, Kitoh H, Ohkawara B, Mishima K, Kaneko H, Ito M, Masuda A, Ishiguro N, Ohno K. Meclozine facilitates proliferation and differentiation of chondrocytes by attenuating abnormally activated FGFR3 signaling in achondroplasia. PLoS One. 2013;8:e81569.
Matsushita M, Hasegawa S, Kitoh H, Mori K, Ohkawara B, Yasoda A, Masuda A, Ishiguro N, Ohno K. Meclozine promotes longitudinal skeletal growth in transgenic mice with achondroplasia carrying a gain-of-function mutation in the FGFR3 gene. Endocrinology. 2015;156:548–54.
Hartmann JT, Haap M, Kopp HG, Lipp HP. Tyrosine kinase inhibitors - a review on pharmacology, metabolism and side effects. Curr Drug Metab. 2009;10:470–81.
Komla-Ebri D, Dambroise E, Kramer I, Benoist-Lasselin C, Kaci N, Le Gall C, Martin L, Busca P, Barbault F, Graus-Porta D, Munnich A, Kneissel M, Di Rocco F, Biosse-Duplan M, Legeai-Mallet L. Tyrosine kinase inhibitor NVP-BGJ398 functionally improves FGFR3-related dwarfism in mouse model. J Clin Invest. 2016;126:1871–84.
Jonquoy A, Mugniery E, Benoist-Lasselin C, Kaci N, Le Corre L, Barbault F, Girard AL, Le Merrer Y, Busca P, Schibler L, Munnich A, Legeai-Mallet L. A novel tyrosine kinase inhibitor restores chondrocyte differentiation and promotes bone growth in a gain-of-function Fgfr3 mouse model. Hum Mol Genet. 2012;21:841–51.
Gudernova I, Vesela I, Balek L, Buchtova M, Dosedelova H, Kunova M, Pivnicka J, Jelinkova I, Roubalova L, Kozubik A, Krejci P. Multikinase activity of fibroblast growth factor receptor (FGFR) inhibitors SU5402, PD173074, AZD1480, AZD4547 and BGJ398 compromises the use of small chemicals targeting FGFR catalytic activity for therapy of short-stature syndromes. Hum Mol Genet. 2016;25:9–23.
Suda M, Ogawa Y, Tanaka K, Tamura N, Yasoda A, Takigawa T, Uehira M, Nishimoto H, Itoh H, Saito Y, Shiota K, Nakao K. Skeletal overgrowth in transgenic mice that overexpress brain natriuretic peptide. Proc Natl Acad Sci U S A. 1998;95:2337–42.
Chusho H, Tamura N, Ogawa Y, Yasoda A, Suda M, Miyazawa T, Nakamura K, Nakao K, Kurihara T, Komatsu Y, Itoh H, Tanaka K, Saito Y, Katsuki M, Nakao K. Dwarfism and early death in mice lacking C-type natriuretic peptide. Proc Natl Acad Sci U S A. 2001;98:4016–21.
Yasoda A, Komatsu Y, Chusho H, Miyazawa T, Ozasa A, Miura M, Kurihara T, Rogi T, Tanaka S, Suda M, Tamura N, Ogawa Y, Nakao K. Overexpression of CNP in chondrocytes rescues achondroplasia through a MAPK-dependent pathway. Nat Med. 2004;10:80–6.
Kake T, Kitamura H, Adachi Y, Yoshioka T, Watanabe T, Matsushita H, Fujii T, Kondo E, Tachibe T, Kawase Y, Jishage K, Yasoda A, Mukoyama M, Nakao K. Chronically elevated plasma C-type natriuretic peptide level stimulates skeletal growth in transgenic mice. Am J Physiol Endocrinol Metab. 2009;297:E1339–48.
Yasoda A, Kitamura H, Fujii T, Kondo E, Murao N, Miura M, Kanamoto N, Komatsu Y, Arai H, Nakao K. Systemic administration of C-type natriuretic peptide as a novel therapeutic strategy for skeletal dysplasias. Endocrinology. 2009;150:3138–44.
Wendt DJ, Dvorak-Ewell M, Bullens S, Lorget F, Bell SM, Peng J, Castillo S, Aoyagi-Scharber M, O’Neill CA, Krejci P, Wilcox WR, Rimoin DL, Bunting S. Neutral endopeptidase-resistant C-type natriuretic peptide variant represents a new therapeutic approach for treatment of fibroblast growth factor receptor 3-related dwarfism. J Pharmacol Exp Ther. 2015;353:132–49.
Noonberg S. BMN 111: vosoritide for achondroplasia. Biomarin R&D Day. 2016:98–135.
Olney RC, Prickett TC, Espiner EA, Mackenzie WG, Duker AL, Ditro C, Zabel B, Hasegawa T, Kitoh H, Aylsworth AS, Bober MB. C-type natriuretic peptide plasma levels are elevated in subjects with achondroplasia, hypochondroplasia, and thanatophoric dysplasia. J Clin Endocrinol Metab. 2015;100:E355–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Sheila Unger and Luisa Bonafé declare no conflicts of interest.
Elvire Gouze has a patent licensed and is the scientific founder of TherAchon.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Rare Bone Disease
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Unger, S., Bonafé, L. & Gouze, E. Current Care and Investigational Therapies in Achondroplasia. Curr Osteoporos Rep 15, 53–60 (2017). https://doi.org/10.1007/s11914-017-0347-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-017-0347-2