
Abstract
Purpose of Review
Richter syndrome (RS) is an uncommon but aggressive evolution of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). RS is an unmet clinical need in the field of CLL. Recent advances in understanding the biology of this condition provide the rationale for testing new therapeutic concepts in order to improve the outcome of patients developing RS, which is so far poor. In this review, we summarize disease characteristics and available therapeutic options for RS.
Recent Findings
Current regimens with novel agents in monotherapy have shown little impact on survival. Nevertheless, the better reported outcome for RS has been achieved with the combination of chemo-immunotherapy with a novel agent, confirming the synergistic effect of the approaches. Still, the frailty of this population may impose a less toxic management leaving most patients with no reasonable therapeutic option.
Summary
Treatment options for RS need to be further expanded. Preclinical models in current development may allow to explore actionable pathways and identify new drug targeted combinations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The transformation of CLL or SLL into an aggressive lymphoma was firstly described in 1928 as a “reticular cell sarcoma” by Maurice Richter [1] and then nominated in his honor as Richter syndrome. As described within the WHO Classification, RS may present as two different pathologic entities: the diffuse large B cell lymphoma (DLBCL) variant or, rarely, the Hodgkin lymphoma (HL) variant [2].
Some clinical clues of transformation (development of new B symptoms, asymmetric arise of bulky lymph nodes with/without associated organ dysfunction from invasive or obstructive neoplastic growth, and/or sudden rise of lactate dehydrogenase (LDH) levels) should promptly rise suspicion of this life-threatening complication in a patient with CLL. This rare evolution is estimated to occur in 0.5–1% of patients with CLL/SLL per year [3]. Different genetic characteristics explaining the aggressiveness and chemorefractoriness of RS have been identified, including TP53, NOTCH1, MYC, and CDKN2A mutations or disruptions [4,5,6]. Only a fraction of RS (~ 20% with DLBCL morphology and ~ 70% with HL morphology) harbors distinct IGHV-D-J rearrangements compared to the preceding CLL, representing de novo lymphomas developing in a CLL patient [6, 7].
The DLBCL variant of RS is associated with a dismal prognosis with a median survival of < 1 year [6, 8, 9•]. Treatment options commonly used in this setting are based on regimens of de novo DLBCL. However, the limited efficacy obtained with conventional treatments led to the consolidation strategy of stem cell transplantation (SCT) in selected patients. The HL variant shows in contrast better survivals when treated with HL regimens. Several studies evaluating the role of novel agents are ongoing in RS, showing promising benefits when combined with conventional chemo-immunotherapy.
Morphology and RS Subtypes
DLBCL Variant
The DLBCL variant is described in approximately 90% of RS. The morphology of the DLBCL variant of RS is characterized by confluent sheets of large neoplastic B lymphocytes resembling either centroblasts (60–80% of cases) or immunoblasts (20–40% of cases) [1, 6, 10]. CLL transformation should be differentiated from CLL progression, which can be associated with the expansion of the proliferation centers in the lymph nodes with confluent and enriched proliferating cells [1]. These forms of “aggressive” CLL or “accelerated” CLL have an outcome intermediate between typical CLL and classic RS. Though not clearly defined by the current WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues, some morphologic criteria have been proposed to correctly distinguish RS from an “accelerated CLL”: (i) tumor of large B cells with nuclear size equal or larger than macrophage nuclei or more than twice a normal lymphocyte and (ii) diffuse growth pattern of such large cells (not just presence of small foci) [11, 12]. The immune phenotype of tumor cells invariably express CD20, while CD5 expression is maintained only in a fraction (~ 30%) of cases, and CD23 expression is even more rare (~ 15% of cases) [7]. PD-1 expression, described only on the paraimmunoblasts of proliferation centers, is common in DLBCL variants, whereas it is only rarely found in de novo DLBCL specimens.
Based on the analysis of the rearrangement of IGHV-D-J genes, most (~ 80%) of the DLBCL variants of RS are clonally related to the preceding CLL phase, thus representing true transformations [5, 7]. This information profoundly impacts on prognosis, with clonally related cases having a median survival of approximately 12 months, while clonally unrelated RS show a similar survival to DLBCL de novo cases (nearly 65 months).
HL Variant
The presence of classical Reed-Sternberg cells harboring a CD30-positive/CD15-positive/CD20-negative phenotype in a proper polymorphous background of small T cells, epithelioid histiocytes, eosinophils, and plasma cells defines the HL variant of RS [10]. This variant accounts for only 5–10% of RS. Only a fraction (~ 30%) of the HL variant of RS are clonally related to CLL [10], while most cases (65–75%) are EBV positive with distinct immunoglobulin rearrangements compared to the paired CLL, thus representing de novo, EBV-driven lymphomas arising in a CLL patient [10].
Epidemiology, Genetics, and Risk Factors
Prevalence of DLBCL variant RS is highly variable (1–23%) and depends on a number of factors: (i) whether the analysis is restricted to biopsy-proven cases or also includes patients with clinically suspected transformation; (ii) the diagnostic aggressiveness in case of rapidly progressive lymphadenopathy; and (iii) in the set of clinical trials, the selection of patients who fit the eligibility criteria for trial participation, and in which the therapy used may have influenced the risk of transformation. The prevalence of RS in a large cohort (n = 2975) of prospectively monitored patients with advanced CLL enrolled in trials of the GCLLSG was 3% [13•]. Transformation can occur early after the diagnosis of CLL, with a reported median time to transformation of 1.8–1.9 years for DLBCL [3, 14] and 4.6–7.5 years for HL [15, 16], with a fraction of patients never being treated for CLL before the transformation.
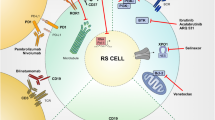
The genetics of the DLBCL variant is different from that of a de novo DLBCL, lacking molecular lesions in signaling pathways and B cell differentiation programs, while sharing with other transformed lymphomas (i.e., transformed follicular lymphoma) lesions affecting general regulators of tumor suppression, cell cycle, and proliferation (Fig. 1) [4,5,6]. Somatic mutations of TP53, NOTCH1, MYC, and CDKN2A account for the aggressive phenotype of the DLBCL variant, which combines chemoresistance and rapid disease kinetics (Fig. 1) [4,5,6].

Transformed lymphoma vulnerabilities and targets for treatment. A representation of the complex process and molecular pathogenesis of transformed lymphomas, resulting from a number of epigenetic and genetic lesions occurring in the tumor cell population (reported in black). Non-genetic mechanisms as pathway activation and changes in immune checkpoints profile are also involved in transformation. Communication between the tumoral cells (in blue) and T cells is established by direct contact, chemokine/cytokine receptors, adhesion molecules and ligand-receptor interactions. Environmental or auto-/self-antigens and homotypic IG interactions trigger BCR activation, which stimulate underlying CLL proliferation. Immune inhibitory molecules (PD-L1 among others) facilitate tumor cells to evade immune-response and maintain tolerance. Recurrently mutated genes in RS affect DNA repair, B cell receptor, and chromatin modification. Potential and established targeted treatments in RS are reported in red. TAM, tumor associate macrophage; BCR, B cell receptor
TP53 is the most frequently disrupted gene in the DLBCL variant, acquiring at the time of transformation either mutation or deletion in ~ 60% of cases. Being a master regulator of the DNA-damage-response pathway which leads to cell apoptosis if activated (i.e., as in response to the antiproliferative effect of chemotherapies), TP53 loss may explain the chemorefractory phenotype generally shown by RS. CDKN2A, which can be found deleted in 30% of cases, is a negative regulator of cell cycle transition from G1 phase to S phase [5, 6]. Cell cycle deregulation by CDKN2A may explain the rapidly progressive behavior of DLBCL variant. MYC genetic alterations sustain ~ 40% of DLBCL variant [5, 17]. MYC is involved in a transcription regulating network which is generally deregulated by somatic structural alterations of this gene in ~ 30% of cases [4,5,6, 14]. The usage of the subset 8 configuration in the B cell receptor (BCR), which has been reported to have an association to NOTCH1 somatic mutations and which shows an unlimited propensity to autonomous BCR signaling and to respond to multiple auto-antigens and immune stimuli from the microenvironment, may explain the particular aggressiveness of CLL harboring subset 8 BCR and their increased propensity to transform into RS [10, 18]. In one study, the 5-year transformation rate of patients with CLL and subset 8 usage has been reported at nearly 70% [10]. The mutational status of NOTCH1 is the only validated risk factor for transformation, with a significantly higher cumulative probability of patients with CLL developing DLBCL variant (45%) compared to CLL without NOTCH1 mutations (4%) [19,20,21].
EBV infection has been suggested as a pathogenetic trigger of DLBCL variant RS. The observation that the overwhelming majority (85–100%) of DLBCL transformed from CLL does not carry EBV infection in the malignant cells, however, does not favor this hypothesis [6].
The role of the exposure to a prior CLL treatment as a risk factor for transformation is controversial, and a proportion of patients who develop RS show no prior therapy for the underlying CLL. In this setting, better outcomes have been reported (median overall survival 35 months for treatment-naïve CLL patients vs 4 months for previously treated in one report [22•]; 46 months vs 7 months, respectively, in one report [9•]). The effect of novel agents on RS development is progressively being reported. Clonal evolution leading to transformation seems to be similar in the novel agent treatment setting to that of chemo-immunotherapy (CIT), with frequent associations of MYC, CDKN2A, TP53, and NOTCH1 disruption [23••, 24••].
While novel agents do not seem to increase the proportion of RS, transformation rates of 5–16% in high-risk and heavily pretreated patients in study population on novel agents have been reported. In this setting, RS occurs typically within the first 18 months of treatment, with a median OS of approximately 6 months after transformation [25].
Early recognition of RS transformation helps to avoid the exposure of patients to multiple lines of therapy that, being targeted to CLL progression, are of little efficacy for the transformed clone. This concept prompts the need for a close monitoring of CLL patients harboring risk factors of RS development.
Diagnosis
Patients with known CLL developing physical deterioration, fever in the absence of infection, rapid and discordant growth of localized lymph nodes, and/or sudden and excessive rise in lactate dehydrogenase (LDH) levels should be suspected for Richter transformation. Likewise, in the case of extra-nodal masses developing in patients with a known CLL, RS might be included in the differential diagnosis. Nonetheless, the specificity of these clinical findings for transformation is only 50–60%, with the remaining cases showing either progressive or “accelerated” CLL, or even a solid cancer [26].
Histologic documentation, with an open biopsy considered as gold standard for RS diagnosis is mandatory to diagnose RS. Samples obtained with fine needle biopsy or aspiration may not be representative of the pathologic architecture of the tumor, with possible false-positive diagnosis (i.e., fine needle biopsy of an enlarged proliferation center, which may be occasionally observed in lymph nodes of progressive or “accelerated” CLL) [27]. Since RS is often restricted to one single lesion at transformation, any biopsy aimed at exploring whether RS has occurred should be directed at the index lesion.
The 18FDG PET/CT has an established role in supporting the choice of whether to perform a biopsy and may tailor the biopsy to the likely transformed site since sites affected by RS are expected to have SUVs overlapping with those of de novo DLBCL [26, 28, 29]. The high negative predictive value (97%) of 18FDG PET/CT when a standard uptake value (SUV) cutoff of < 5 is chosen supports a non-biopsy approach suggesting that a transformation is not likely. Conversely, because of the limited positive predictive value (53%) of 18FDG PET/CT with an SUV ≥ 5, a biopsy should be directed at the index lesion (i.e., the lesion showing the most avid 18FDG uptake, the lesion with the largest diameter by imaging, and/or the lesion showing the most rapid kinetics of progression) [26, 28, 29].
A sensitivity of 91% and a specificity of 95% have been recently reported when using an SUV cutoff of 10, with positive and negative predictive values of 60.6% and 99.2%, respectively [30]. The same report shows a better proportion of correctly classified patients as RS with 94.6% when choosing an SUV cutoff of 10, compared to 73.5% when using an SUV cutoff of 5. Moreover, a better correlation with outcomes has been reported, with median OS of 6.9 months for patients with lesions with an SUV value ≥ 10 compared to 56.9 months for patients with lesions with an SUV < 10. In the largest series of PET/CT prospectively performed in patients following kinase inhibitor discontinuation [31], a SUV threshold ≥ 10 showed low positive (63%) and negative (50%) predictive values. Considering that, the threshold of 10 did not turn out to be a useful noninvasive method to rule out RS post-kinase inhibitor therapy. In the same study, 5 of 8 of the biopsy-confirmed RS showed a SUV ranging from 5 to 9, whereas only 3 of 8 RS had a SUV ≥ 10, further reinforcing the notion that a lower threshold (i.e., SUV 5) should also be used in the setting of kinase inhibitor failure to rule our RS [31].
Prognosis of RS
The prognosis of DLBCL variant of RS is poor. Based on the number of presenting risk factors (Zubrod performance status > 1, LDH levels above normal values, platelet count ≤ 100 × 109/L, tumor size ≥ 5 cm, and > 2 prior lines of therapy), a validated RS prognostic score segregated 4 risk groups: low risk showing a median survival of 13–45 months (0–1 risk factors); low intermediate risk with a median survival of 11–32 months (2 risk factors); high intermediate risk showing a median survival of 4 months (3 risk factors); and high risk with a median survival of 1–4 months (4–5 risk factors) [32]. The most influent prognostic factor is the clonal relationship between the transformed DLBCL and the underlying CLL. Indeed, patients with a clonally unrelated DLBCL show a longer median survival (5 years) compared with patients with a clonally related DLBCL transformation (8–16 months) [6, 22]. RS after ibrutinib or venetoclax shows an even more aggressive behavior. Outcomes are generally poor for patients with a refractory disease, which overall accounts for more than 80% of cases [32,33,34,35,36,37].
Treatment Options of the DLBCL Variant of RS
Chemo-immunotherapy Approach
Regimens indicated for aggressive B cell non-Hodgkin lymphomas have been proposed to treat patients developing the DLBCL variant of RS. The choice of treatment for patients presenting RS needs to be evaluated in view of their history and comorbidities.
The reported response rate of 8 courses of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) is 67% (complete response, CR 7%), with a median progression-free survival (PFS) of 10 months and a median overall survival (OS) of 21 months. Hematotoxicity is reported in 65% of patients, while infections are the most common severe non-hematologic toxicity in 28% of patients [38]. In a retrospective series of 48 patients treated with R-CHOP, the overall response rate (ORR) was 37%, with a median OS of 35 months [22•].
Ofatumumab (O), an anti-CD20 monoclonal antibody with greater complement-mediated cytotoxicity than rituximab, combined with CHOP showed an ORR of 46% (CR 27%, PR 19%) with a median PFS of 6 months and a median OS of 11 months. Adverse events under CHOP-O were infections and hematologic toxicities (thrombocytopenia, febrile neutropenia, sepsis) [39, 40].
R-EPOCH (rituximab, etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin) is used in high-grade B cell lymphoma with rearrangements of MYC and BCL2 and/or BCL6 (double-hit and triple-hit lymphomas). Since MYC is frequently rearranged in the DLBCL variant of RS, R-EPOCH has been investigated in this disease as first-line therapy showing a 20% response rate, a median PFS of 3 months, and a median OS of 6 months [41]. Characteristics of underlying CLL influenced outcomes of R-EPOCH, with worse PFS and OS in deletion 17p and complex karyotype patients. Main adverse events were due to hematologic toxicities (febrile neutropenia and infections) [41].
The hyper-CVAD regimen, a fractioned cyclophosphamide, vincristine, doxorubicin, and dexamethasone regimen, alone or in alternating combination with methotrexate and ara-C, resulted in response rates of ~ 40% with poor median OS. These aggressive regimens were invariably complicated by severe hematotoxicity in all cases, translating into a high severe infection rate of 50% and a treatment-related mortality of ~ 20% [42] despite the prophylaxis with granulocyte-macrophage colony stimulating factor (GM-CSF) [43].
Platinum-containing regimens with the combination of oxaliplatin, fludarabine, ara-C, and rituximab have been explored within the OFAR 1 and OFAR 2 trials. The ORR of OFAR 1 trial was 50% (CR 6–20%), though with short duration of response (mean PFS of 3 months and mean OS of 6–8 months) and severe myelosuppression [44]. The OFAR 2 trial, designed with the aim of improving clinical outcomes and decreasing toxicities with modification of oxaliplatin and cytarabine doses, did not show actual improvement of toxicity rates with 80% of patients developing grade 3–4 neutropenia/thrombocytopenia and 20% grade 3–4 infections. The ORR was 39% (CR 6.5%); the median PFS was 3 months; the median OS was 7 months; and at 2 years, only 19.7% of patients with RS were alive [45].
Radioimmunotherapy
No responses have been documented in 7 RS patients treated with radio-immunotherapy in a single institution trial investigating 90Y ibritumomab tiutexan, with 100% of progression at a median time of 40 days [46, 47].
Role of Stem Cell Transplantation Consolidation
Due to the unsatisfactory durability of response after chemo-immunotherapy, SCT has been explored as post-remission therapy in RS fit patients. However, only 10–15% of patients with RS can access SCT generally due to their frailty (age, performance status) and donor availability [48].
The efficacy of SCT in RS is granted by dose intensity delivered by high-dose cytotoxic therapy and, in the case of allogeneic SCT, graft-versus-leukemia activity. Indeed, in patients undergoing autologous SCT, no clear plateau in relapse-free survival is described, but only a fraction of relapses seems related to RS, while the remainder are due to CLL. This data suggests that autologous SCT may be efficacious on the eradication of the RS component but not on the underlying CLL component. The plateaus of relapse-free survival among RS patients treated with reduced intensity conditioning (RIC) allogeneic SCT support the presence of a graft-versus-leukemia effect in RS [48].
The benefit of receiving SCT is reported as a longer median survival (5 years vs < 1 year for patients not receiving SCT) [48]. At 3 years, the survival after allogeneic SCT was 36% and 59% after autologous SCT, with a respective relapse-free survival of 27% and of 45%. The non-relapse mortality at 3 years was 26% after allogeneic SCT and 12% after autologous SCT [48].
The main factor influencing the post-transplant outcome is disease status at SCT. Indeed, patients who undergo SCT with chemotherapy-sensitive RS had a superior survival compared to those who undergo transplantation with active and progressive disease. The major benefit of SCT was obtained in young (< 60 years) patients. Among patients receiving allogeneic SCT, those conditioned with a reduced intensity regimen had the longest survival [48].
A recent analysis from the German CLL Study Group showed a median OS of 17 months in 3 patients undergoing allogeneic SCT for RS [13•].
A meta-analysis assessing the efficacy of allogeneic SCT for RS patients reported a relapse rate of 28% with a non-relapse mortality of 24% which is in line with previous reports on lymphoid malignancies [49].
Overall, these data suggest that both autologous SCT and reduced intensity conditioning allogeneic SCT can be effective in young patients with a chemosensitive RS. For patients suitable to transplant but lacking a donor, autologous stem cell transplantation may be an alternative option.
HL Variant
In the setting of the HL variant RS, the response rate of ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine) is 40–60%, with a median overall survival of 4 years. Indeed, the standard of care for de novo HL is the most frequently used regimen for patients with the HL variant of RS. [50,51,52,53]. ABVD is associated with the risk of serious pulmonary toxic effects due to the bleomycin exposure [53]. Applying the results from the advanced stage HL trials, bleomycin can be omitted after two cycles of ABVD if interim PET shows negative Deauville score (score 1–3). Escalation to BEACOPP in fit and younger patients might be considered in case of a positive interim PET. For older and unfit patients, the addition of radiotherapy could be an option [54•]. Stem cell transplantation is less used for consolidation in this setting, because of the longer survival observed compared to the DLBCL variant.
Novel Agents in RS
Transformed lymphomas show common molecular signatures presenting deregulation of tumor suppression, cell cycle and proliferation pathways [7, 55]. Recent studies have revealed the molecular pathogenesis of transformed lymphomas including RS, showing a complex process, resulting from a number of epigenetic and genetic lesions occurring in the tumor cell population. Non-genetic mechanisms as pathway activation and changes in immune checkpoints are also involved in transformation (Fig. 1). This novel knowledge encouraged clinical investigations on a variety of targeted therapeutic strategies (Table 1), also prompted by the unsatisfactory response rates obtained with conventional chemo-immunotherapy associated to a short response duration without a SCT for consolidation, which cannot be proposed to the majority of RS patients because of the constrains imposed by a combination of age, poor performance status, lack donor availability and refractoriness to induction treatments.
The nucleo-cytoplasmic transport of proteins is often misregulated in cancer and depends on the activity of export proteins, including XPO1 which transports tumor suppressor proteins. An increased activity of nuclear exportation, with a related inhibition of the physiologic tumor-suppressor processes, is often observed in tumoral diseases. Selinexor is a selective inhibitor of nuclear export aiming at retaining tumor suppressor proteins in the nucleus, thus activating them in tumor cells. In a phase I study, selinexor showed signal of activity in 33% of the patients with the DLBCL variant of RS [56]. Few grade 3–4 adverse events were reported (5%) [56]. The phase 2 study (NCT02138786) has been terminated early, due to enrollment challenges.
Bruton’s tyrosine kinase (BTK), a component of the B cell receptor (BCR) signaling pathway, is a strong regulator of cell proliferation and survival in B cell malignancies. Targeted BTK inhibition is described to act in CLL with growth inhibition and cell death by blocking BCR-induced BTK activation [57, 58]. This activity is maintained in patients with high-risk disease (i.e., CLL with TP53 disruption). In a study of four patients with RS, responses in three patients, including one CR and two partial responses (PRs), were reported [59]. Other case studies reported responses in patients with DLBCL variant RS on ibrutinib [60, 61], with PFS of up to 16 months [61].
Acalabrutinib is a second-generation oral BTK inhibitor which selectively and irreversibly binds cysteine residues on BTK [62]. In the ACE-CL-001 phase I/II trial, the overall response rate to acalabrutinib, a highly selective BTK inhibitor, was 38% among DLBCL variant RS, the median PFS was 3 months and the median duration of response 5 months [62].
Constitutive AKT phosphorylation is significantly increased in high-risk CLL patients harboring TP53 and NOTCH1 mutations in comparison to wild-type patients. Furthermore, pAKT immunofluorescence showed increased expression and frequency in RS patients in comparison to both CLL and de novo DLBCL patients. Genetic over-activation of AKT in the murine Eμ-TCL1 CLL mouse model resulted in the transformation into high-grade lymphoma with phenotypic features of RS. Collectively, the data provide evidence that activation of AKT causes transformation of CLL into aggressive lymphoma [63••]. The PI3K inhibitor idelalisib showed some activity in patients with RS [64•]. These data prompt the investigation of PI3K inhibitors in this setting.
Since most of the DLBCL variant of RS show TP53 disruption, novel drugs for this condition need to act independently of TP53. Venetoclax is a specific inhibitor of BCL2 that acts in a TP53-independent way and is effective in high-risk CLL [65]. Venetoclax is a specific inhibitor of BCL2 that acts with a TP53-independent mechanism and is effective in high-risk CLL. In the M12-175 (NCT01328626) phase I study, a limited number of DLBCL variant RS were treated with escalating doses of venetoclax, achieving a response rate of 43% (no CRs) [65]. The venetoclax-R-EPOCH combination was assessed in a phase 2 study in RS (NCT03054896) [66••]. Of the 21 evaluable patients who have started combination therapy, 16 responded (ORR 59%); 48% had CR, all of whom also showed undetectable bone marrow minimal residual disease (MRD) for the underlying CLL. With a median follow-up of 9 months, the reported median PFS and OS are both 16 months. Toxicities from intensive CIT and venetoclax were described including grade 3–4 neutropenia (58%), anemia (50%), thrombocytopenia (50%), and febrile neutropenia (38%). No tumor lysis syndrome (TLS) occurred with daily venetoclax ramp-up after 1 lead in cycle of R-EPOCH [66••].
The DLBCL variant of RS frequently occurs upon an exhausted immune system, due to immune checkpoint deregulation. This scenario includes expression of high levels of checkpoint inhibitory molecules (i.e., PD-1) on RS tumor cells. Pembrolizumab, an antibody that targets the PD-1 receptor, provided signals of activity in DLBCL variant RS (NCT02332980) [67]. Objective responses were observed in 44% (4/9) DLBCL variant RS patients. All responses were observed in patients with transformation after prior therapy with ibrutinib. The median OS of this cohort was 10.7 months but was not reached in DLBCL variant RS previously exposed to ibrutinib.
Synergistic antitumor effects between ibrutinib and inhibition of the PD-1 and PD-L1 pathway have been reported in preclinical studies [68]. The inhibition of interleukin 2-inducible T cell kinase, which plays a part in T cell proliferation and differentiation, might explain the role of ibrutinib in the modulation of the immune system. A phase 1/2a study was designed to assess the safety and efficacy of ibrutinib in combination with nivolumab in patients with relapsed or refractory hematological malignancies including high-risk CLL/SLL, follicular lymphoma, DLBCL, and RS [69••]. Overall response was seen in the Richter’s transformation cohort (13 [65%] of 20 patients), with two CRs and 11 PRs. The median duration of response was 6.9 months for the RS cohort.
Preliminary data on the administration of CAR-T cells in the setting of RS report discouraging responses (one disease progression, one evolution to PBL), but further studies are warranted [70, 71]. Whether the condition of the T cell pool can influence the proliferation of CAR-T cells has not been reported. Quantitative and qualitative impairment of immune system is observed in patients with CLL, including alterations of the innate immune system (i.e., defective function of neutrophils, natural killer (NK) cells, and decreased complement activity) and of the adaptive immune response (i.e., deficits in cell-mediated immunity with hypogammaglobulinemia, down-regulation of T cell function and defects in antibody dependent cellular cytotoxicity) [72]. In addition to impaired cytotoxicity and expansion, CAR-T cell exhaustion can lead to the failure of CAR-T cell therapy [73].
Results from a pilot study aiming at assessing the efficacy of concurrent ibrutinib through leukapheresis, lymphodepletion, and CD19 CAR-T cells infusion in heavily pretreated high-risk R/R CLL patients who had failed ibrutinib showed high response rates in all patients (4 patients with DLBCL variant CLL) with an ORR of 83%. Tolerability was acceptable, with most patients well tolerating the combination of ibrutinib and CD19 CAR-T cells, but caution is warranted in patients with CRS while receiving ibrutinib after CAR-T cell immunotherapy [74•].
Conclusions and Future Perspectives
The recent genetic tools helped in understanding the molecular basis of RS and led to depict RS as a complex entity based on clonal and nonclonal evolutionary patterns, which impact on outcomes. Relapsed/refractory patients with CLL on novel agents are a new prognostic group with a potential adverse outcome when eventually experimenting transformation. Occurring mostly in elderly patients with different comorbidities, RS can have a limited treatment due to potential toxicities in this fragile population. Even if improved outcomes have been reported over the last 20 years (particularly after the introduction of rituximab), the outcome of RS patients is still poor.
The development of new preclinical models mimicking human RS may help in identifying new treatment targets and elaborating strategies for patients developing this aggressive disease. Early intervention policies for the high-risk CLL population might be explored. The trend of the increased use of novel agents versus standard CIT should likely prevent the selection of high-risk chemoresistant clones and the accumulation of genomic instability due to treatment toxicity.
An international and common effort in developing preclinical models, prognosticators, biobanks and databases should be pursued to improve outcomes in patients with RS.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Richter MN. Generalized reticular cell sarcoma of lymph nodes associated with lymphatic leukemia. Am J Pathol. 1928;4:285–92.
Müller-Hermelink HK, Montserrat E, Catovsky D, Campo E, Harris NL, Stein H. Chronic lymphocytic leukemia/small lymphocytic lymphoma. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW, editors. World Health Organization Classification of Tumours, Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC; 2008. p. 180–2.
Parikh SA, Rabe KG, Call TG, Zent CS, Habermann TM, Ding W, et al. Diffuse large B-cell lymphoma (Richter syndrome) in patients with chronic lymphocytic leukaemia (CLL): a cohort study of newly diagnosed patients. Br J Haematol. 2013;162:774–82.
Chigrinova E, Rinaldi A, Kwee I, Rossi D, Rancoita PM, Strefford JC, et al. Two main genetic pathways lead to the transformation of chronic lymphocytic leukemia to Richter syndrome. Blood. 2013;122:2673–782.
Fabbri G, Khiabanian H, Holmes AB, Wang J, Messina M, Mullighan CG, et al. Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J Exp Med. 2013;210:2273–88.
Rossi D, Spina V, Deambrogi C, Rasi S, Laurenti L, Stamatopoulos K, et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011;117:3391–401.
Mao Z, Quintanilla-Martinez L, Raffeld M, Richter M, Krugmann J, Burek C, et al. IgVH mutational status and clonality analysis of Richter’s transformation. Am J Surg Pathol. 2007;31:1605–14.
Tsimberidou AM, O’Brien S, Khouri I, Giles FJ, Kantarjian HM, Champlin R, et al. Clinical outcomes and prognostic factors in patients with Richter’s syndrome treated with chemotherapy or chemoimmunotherapy with or without stem-cell transplantation. J Clin Oncol. 2006;24:2343–51.
. Wang Y, Tschautscher MA, Rabe KG, et al. Clinical characteristics and outcomes of Richter transformation: experience of 204 patients from a single center. Haematologica. 2020;105:765–73 A large single-institution cohort study defining the main characteristics of RS.
Rossi D, Spina V, Cerri M, Rasi S, Deambrogi C, De Paoli L, et al. Stereotyped B-cell receptor is an independent risk factor of chronic lymphocytic leukemia transformation to Richter syndrome. Clin Cancer Res. 2009;15:4415–22.
Soilleux EJ, Wotherspoon A, Eyre TA, Clifford R, Cabes M, Schuh AH. Diagnostic dilemmas of high-grade transformation (Richter's syndrome) of chronic lymphocytic leukaemia: results of the phase II National Cancer Research Institute CHOP-OR clinical trial specialist haemato-pathology central review. Histopathology. 2016;69:1066–76.
Giné E, Martinez A, Villamor N, López-Guillermo A, Camos M, Martinez D, et al. Expanded and highly active proliferation centers identify a histological subtype of chronic lymphocytic leukemia ("accelerated" chronic lymphocytic leukemia) with aggressive clinical behavior. Haematologica. 2010;95:1526–33.
Al-Sawaf O, Robrecht S, Bahlo J, et al. Richter transformation in chronic lymphocytic leukemia (CLL)-a pooled analysis of German CLL study group (GCLLSG) front line treatment trials. Leukemia. 2020. https://doi.org/10.1038/s41375-020-0797-xA large cohort of patients enrolled in clinical studies by the GCLLSG.
Rossi D, Berra E, Cerri M, Deambrogi C, Barbieri C, Franceschetti S, et al. Aberrant somatic hypermutation in transformation of follicular lymphoma and chronic lymphocytic leukemia to diffuse large B-cell lymphoma. Haematologica. 2006;91:1405–9.
Parikh SA, Habermann TM, Chaffee KG, Call TG, Ding W, Leis JF, et al. Hodgkin transformation of chronic lymphocytic leukemia: incidence, outcomes, and comparison to de novo Hodgkin lymphoma. Am J Hematol. 2015;90(4):334–8.
Mauro FR, Galieni P, Tedeschi A, Laurenti L, del Poeta G, Reda G, et al. Factors predicting survival in chronic lymphocytic leukemia patients developing Richter syndrome transformation into Hodgkin lymphoma. Am J Hematol. 2017;92:529–35.
De Paoli L, Cerri M, Monti S, Rasi S, Spina V, Bruscaggin A, et al. MGA, a suppressor of MYC, is recurrently inactivated in high risk chronic lymphocytic leukemia. Leuk Lymphoma. 2013;54:1087–90.
Gounari M, Ntoufa S, Apollonio B, Papakonstantinou N, Ponzoni M, Chu CC, et al. Excessive antigen reactivity may underlie the clinical aggressiveness of chronic lymphocytic leukemia stereotyped subset #8. Blood. 2015;125(23):3580–7.
Rossi D, Rasi S, Fabbri G, Spina V, Fangazio M, Forconi F, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood. 2012;119:521–9.
Rossi D, Rasi S, Spina V, Fangazio M, Monti S, Greco M, et al. Different impact of NOTCH1and SF3B1 mutations on the risk of chronic lymphocytic leukemia transformation to Richter syndrome. Br J Haematol. 2012;158:426–9.
Villamor N, Conde L, Martínez-Trillos A, Cazorla M, Navarro A, Beà S, et al. NOTCH1 mutations identify a genetic subgroup of chronic lymphocytic leukemia patients with high risk of transformation and poor outcome. Leukemia. 2013;27:1100–6.
Abrisqueta P, Delgado J, Alcoceba M, et al. Clinical outcome and prognostic factors of patients with Richter syndrome: real-world study of the Spanish chronic lymphocytic leukemia study group (GELLC). Br J Haematol. 2020. https://doi.org/10.1111/bjh.16748A recent report of the characteristics of RS patients from the Spanish CLL Study Group (GELLC).
Kadri S, Lee J, Fitzpatrick C, et al. Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv. 2017;1:715–27 A recent molecular study reporting genomic profiles of serial samples from 9 patients with ibrutinib-relapsed disease, including 6 with Richter transformation. RS cells are described as clonal descendants of circulating leukemia cells progressively undergoing evolution and drifts. Interestingly, Authors report that transformed lymphoma cells in tissue may acquire a different BTK mutation from that in the CLL leukemia cells.
Vaisitti T, Braggio E, Allan JN, et al. Novel Richter syndrome xenograft models to study genetic architecture, biology, and therapy responses. Cancer res. 2018;78:3413–20 In this paper are described the first two patient-derived xenograft models of Richter syndrome which represent ex vivo models to study biology of the disease and to test novel therapeutic strategies.
Woyach JA, Ruppert AS, Guinn D, Lehman A, Blachly JS, Lozanski A, et al. BTKC481S-mediated resistance to Ibrutinib in chronic lymphocytic leukemia. J Clin Oncol. 2017;35:1437–43.
Bruzzi JF, Macapinlac H, Tsimberidou AM, Truong MT, Keating MJ, Marom EM, et al. Detection of Richter's transformation of chronic lymphocytic leukemia by PET/CT. J Nucl Med. 2006;47:1267–73.
Gascoyne RD. XIV. The pathology of transformation of indolent B cell lymphomas. Hematol Oncol. 2015;33 Suppl 1:75–79.
Falchi L, Keating MJ, Marom EM, Truong MT, Schlette EJ, Sargent RL, et al. Correlation between FDG/PET, histology, characteristics, and survival in 332 patients with chronic lymphoid leukemia. Blood. 2014;123(18):2783–90.
Mauro FR, Chauvie S, Paoloni F, Biggi A, Cimino G, Rago A, et al. Diagnostic and prognostic role of PET/CT in patients with chronic lymphocytic leukemia and progressive disease. Leukemia. 2015;29(6):1360–5.
Michallet AS, Sesques P, Rabe KG, Itti E, Tordot J, Tychyj-Pinel C, et al. An 18F-FDG-PET maximum standardized uptake value > 10 represents a novel valid marker for discerning Richter's syndrome. Leuk Lymphoma. 2016;57:1474–7.
Mato AR, Wierda WG, Davids MS, et al. Analysis of PET-CT to identify Richter’s transformation in 167 patients with disease progression following kinase inhibitor therapy. Blood. 2017;130(suppl 1). Abstract 834.
Tsimberidou A-M, O’Brien S, Khouri I, et al. Clinical outcomes and prognostic factors in patients with Richter’s syndrome treated with chemotherapy or chemoimmunotherapy with or without stem-cell transplantation. J Clin Oncol. 2006;24:2343–51.
Jain P, Thompson PA, Keating M, Estrov Z, Ferrajoli A, Jain N, et al. Longterm outcomes for patients with chronic lymphocytic leukemia who discontinue ibrutinib. Cancer. 2017;123(12):2268–73.
Byrd JC, Blum KA, Burger JA, Coutre SE, Sharman JP, Furman RR, et al. Activity and tolerability of the Bruton’s tyrosine kinase (Btk) inhibitor PCI-32765 in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL): interim results of a phase Ib/II study [abstract]. J Clin Oncol. 2011;29(suppl 15):6508.
Davids MS, Huang Y, Rogers KA, Stern R, Brown JR, Thompson PA, et al. Richter’s syndrome (RS) in patients with chronic lymphocytic leukemia (CLL) on novel agent therapy [abstract]. J Clin Oncol. 2017;35(suppl 15):7505.
Anderson MA, Tam C, Lew TE, Juneja S, Juneja M, Westerman D, et al. Clinicopathological features and outcomes of progression of CLL on the BCL2 inhibitor venetoclax. Blood. 2017;129:3362–70.
Mato AR, Nabhan C, Barr PM, Ujjani CS, Hill BT, Lamanna N, et al. Outcomes of CLL patients treated with sequential kinase inhibitor therapy: a real world experience. Blood. 2016;128:2199–205.
Langerbeins P, Busch R, Anheier N, Dürig J, Bergmann M, Goebeler ME, et al. Poor efficacy and tolerability of R-CHOP in relapsed/refractory chronic lymphocytic leukemia and Richter transformation. Am J Hematol. 2014;89:E239–43.
Wierda WG, Kipps TJ, Mayer J, Stilgenbauer S, Williams CD, Hellmann A, et al. Ofatumumab as single-agent CD20 immunotherapy in fludarabine-refractory chronic lymphocytic leukemia. J Clin Oncol. 2010;28:1749–55.
Eyre TA, Clifford R, Bloor A, Boyle L, Roberts C, Cabes M, et al. NCRI phase II study of CHOP in combination with ofatumumab in induction and maintenance in newly diagnosed Richter syndrome. Br J Haematol. 2016;175:43–54.
Rogers KA, Salem G, Stephens DM, Andritsos LA, Awan FT, Byrd JC, et al. A single-institution retrospective cohort study of patients treated with R-EPOCH for Richter's transformation of chronic lymphocytic leukemia. Blood. 2015;126:2951.
Dabaja BS, O’Brien SM, Kantarjian HM, Cortes JE, Thomas DA, Albitar M, et al. Fractionated cyclophosphamide, vincristine, liposomal daunorubicin (daunoXome), and dexamethasone (hyper-CVXD) regimen in Richter’s syndrome. Leuk Lymphoma. 2001;42:329–37.
Tsimberidou AM, Kantarjian HM, Cortes J, Thomas DA, Faderl S, Garcia-Manero G, et al. Fractionated cyclophosphamide, vincristine, liposomal daunorubicin, and dexamethasone plus rituximab and granulocytemacrophage- colony stimulating factor (GM-CSF) alternating with methotrexate and cytarabine plus rituximab and GMCSF in patients with Richter syndrome or fludarabine refractory chronic lymphocytic leukemia. Cancer. 2003;97:1711–20.
Tsimberidou AM, Wierda WG, Plunkett W, Kurzrock R, O’Brien S, Wen S, et al. Phase I-II study of oxaliplatin, fludarabine, cytarabine, and rituximab combination therapy in patients with Richter’s syndrome or fludarabine-refractory chronic lymphocytic leukemia. J Clin Oncol. 2008;26:196–203.
Tsimberidou AM, Wierda WG, Wen S, Plunkett W, O'Brien S, Kipps TJ, et al. Phase I-II clinical trial of oxaliplatin, fludarabine, cytarabine, and rituximab therapy in aggressive relapsed/refractory chronic lymphocytic leukemia or Richter syndrome. Clin Lymphoma Myeloma Leuk. 2013;13:568–74.
Witzig TE, Flinn IW, Gordon LI, Emmanouilides C, Czuczman MS, Saleh MN, et al. Treatment with ibritumomab tiuxetan radioimmunotherapy in patients with rituximab-refractory follicular non-Hodgkin’s lymphoma. J Clin Oncol. 2002;20:3262–9.
Tsimberidou AM, Murray JL, O’Brien S, Wierda WG, Keating MJ. Yttrium-90 ibritumomab tiuxetan radioimmunotherapy in Richter syndrome. Cancer. 2004;100:2195–200.
Cwynarski K, van Biezen A, de Wreede L, Stilgenbauer S, Bunjes D, Metzner B, et al. Autologous and allogeneic stem-cell transplantation for transformed chronic lymphocytic leukemia (Richter's syndrome): a retrospective analysis from the chronic lymphocytic leukemia subcommittee of the chronic leukemia working party and lymphoma working party of the European Group for Blood and Marrow Transplantation. J Clin Oncol. 2012;30:2211–7.
Aulakh S, Reljic T, Yassine F, et al. Allogeneic hematopoietic cell transplantation is an effective treatment for patients with Richter syndrome: A systematic review and meta-analysis. Hematol Oncol Stem Cell Ther. 2020;20(S1658–3876):30097–2.
Tsimberidou AM, O'Brien S, Kantarjian HM, Koller C, Hagemeister FB, Fayad L, et al. Hodgkin transformation of chronic lymphocytic leukemia: the M. D Anderson Cancer Center experience. Cancer. 2006;107:1294–302.
Bockorny B, Codreanu I, Dasanu CA. Hodgkin lymphoma as Richter transformation in chronic lymphocytic leukaemia: a retrospective analysis of world literature. Br J Haematol. 2012;156:50–66.
Tadmor T, Shvidel L, Goldschmidt N, Ruchlemer R, Fineman R, Bairey O, et al. Hodgkin's variant of Richter transformation in chronic lymphocytic leukemia; a retrospective study from the Israeli CLL study group. Anticancer Res. 2014;34:785–90.
Martin WG, Ristow KM, Habermann TM, Colgan JP, Witzig TE, Ansell SM. Bleomycin pulmonary toxicity has a negative impact on the outcome of patients with Hodgkin’s lymphoma. J Clin Oncol. 2005;23:7614–20.
Johnson P, Federico M, Kirkwood A, Fossa A, Berkahn L, Carella A, et al. Adapted treatment guided by interim PET-CT scan in advanced Hodgkin’s lymphoma. N Engl J Med. 2016 June 23;374:2419–29 A large prospective randomized trial to define whether the omission of bleomycin after negative interim PET-CT scan determines noninferior progression-free survival rate at 3 years, in comparison with patients continuing standard ABVD.
Puente XS, Pinyol M, Quesada V, Conde L, Ordóñez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–5.
Kuruvilla J, Byrd JC, Flynn JM, Garzon R, Porcu P, Wagner-Johnston N, et al. The oral selective inhibitor of nuclear export (SINE) selinexor (KPT-330) demonstrates broad and durable clinical activity in relapsed/refractory non Hodgkin’s lymphoma (NHL). Blood. (ASH Annual Meeting Abstracts) 2014;124:396.
Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42.
Byrd JC, Brown JR, O’Brien S, Barrientos KC, Kay NE, Reddy NM, et al. RESONATE Investigators Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371:213–23.
Tsang M, Shanafelt TD, Call TG, Ding W, Chanan-Khan A, Leis JF, et al. The efficacy of ibrutinib in the treatment of Richter syndrome. Blood. 2015;125:1676–8.
Giri S, Hahn A, Yaghmour G, Martin MG. Ibrutinib has some activity in Richter's syndrome. Blood Cancer J. 2015;5(1):e277.
Master S, Leary C, Takalkar A, Coltelingam J, Mansour R, Mills GM, et al. Successful treatment of Richter transformation with Ibrutinib in a patient with chronic lymphocytic leukemia following allogeneic hematopoietic stem cell transplant. Case Rep Oncol. 2017;10(2):534–41.
Hillmen P, Schuh A, Eyre TA, Pagel JM, Brown JR, Ghia P, et al. Acalabrutinib monotherapy in patients with Richter transformation from the phase 1/2 ACE-CL-001 clinical study. Blood. 2016;128:2260.
Blakemore S, Kohlhaas V, Al-Maarri M, et al. Active AKT signaling triggers CLL towards Richter's transformation via over-activation of NOTCH1. EHA 25 virtual congress 2020; abstract S154. Recent evidences assessing the role of the functional status of a potentially oncogenic signaling pathway in aggressive transformation of indolent lymphomas.
Visentin A, Imbergamo S, Scomazzon E, et al. BCR kinase inhibitors, idelalisib and ibrutinib, are active and effective in Richter syndrome. Br J Haematol. 2019;185(1):193–7 A retrospective study reporting the results of potential new targeted treatment for RS.
Davids MS, Roberts AW, Seymour JF, Pagel JM, Kahl BS, Wierda WG, et al. Phase I first-in-human study of venetoclax in patients with relapsed or refractory non-Hodgkin lymphoma. J Clin Oncol. 2017:JCO2016704320.
Davids MA, Rogers KA, Tyekucheva S, et al. A multicenter phase II study of venetoclax plus dose-adjusted R-EPOCH (VR-EPOCH) for Richter’s syndrome. J Clin Oncol 38: 2020 (suppl; abstr 8004). Recent encouraging data about the new combination of venetoclax with R-EPOCH for patients with DLBCL variant RS.
Ding W, LaPlant BR, Call TG, Parikh SA, Leis JF, He R, et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood. 2017;129(26):3419–27.
Sagiv-Barfi I, Kohrt HE, Czerwinski DK, Ng PP, Chang BY, Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci U S A. 2015;112:E966–72.
Younes A, Brody J, Carpio C, et al. Safety and activity of ibrutinib in combination with nivolumab in patients with relapsed non-Hodgkin lymphoma or chronic lymphocytic leukaemia: a phase 1/2a study. Lancet Haematol. 2019;6(2):e67–78 A phase 1/2a study was designed to assess the safety and efficacy of ibrutinib in combination with nivolumab in patients with relapsed or refractory hematological malignancies including high-risk CLL/SLL, follicular lymphoma, diffuse large B-cell lymphoma, and Richter’s transformation. Overall response was seen in the Richter’s transformation cohort (13 [65%] of 20 patients), with two CRs and 11 PRs. The median duration of response was 6.9 months (1.4–NE) for the Richter’s transformation cohort.
Leiming X, Qian C, Qiao L, Tan L, Yi W, Yangyi B. The clinical study on CD19-directed chimeric antigen receptor-modified T cells in patient with Richter Syndrome. https://doi.org/10.1158/1538-7445.AM2017-CT041.
Evans A, Burack R, Rothberg PG, Porter D, Liesveld JL. Evolution to Plasmablastic lymphoma (PBL) after CAR-T cell therapy in a case of SLL/CLL with Richter’s transformation. Blood. 2014;124:5660.
Forconi F, Moss P. Perturbation of the normal immune system in patients with CLL. Blood. 2015 Jul 30;126:573–81.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–99.
Gauthier J, Hirayama AV, Purushe J, et al. Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood. 2020;135:1650–60 Recent data from a pilot study to evaluate the safety and feasibility of administering ibrutinib concurrently with CD19 CAR T-cell immunotherapy.
Funding
Open Access funding provided by Università della Svizzera italiana.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Neither of the authors has any potential conflicts of interest to disclose.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Lymphomas
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Condoluci, A., Rossi, D. Richter Syndrome. Curr Oncol Rep 23, 26 (2021). https://doi.org/10.1007/s11912-020-01001-x
Accepted:
Published:
DOI: https://doi.org/10.1007/s11912-020-01001-x