
Abstract
This study was carried out to determine the antibody responses and protective capacity of an inactivated recombinant vaccine expressing the fimbrial protein of Pasteurella multocida B:2 following intranasal vaccination against hemorrhagic septicemia in goats. Goats were vaccinated intranasal with 106 CFU/mL of the recombinant vaccine (vaccinated group) and 106 CFU/mL of pET32/LIC vector without fimbrial protein (control group). All three groups were kept separated before all goats in the three groups were challenged with 109 CFU/mL of live pathogenic P. multocida B:2. During the course of study, both serum and lung lavage fluid were collected to evaluate the antibody levels via enzyme-linked immunosorbent assay. It was found that goats immunized with the inactivated recombinant vaccine developed a strong and significantly (p < 0.05) higher specific IgA and IgG responses in both serum and lung lavage fluid samples compared to the control and unvaccinated groups. Following intratracheal challenge, the rate of isolation was 17% for the vaccinated group, 67% for the control group and 100% for the unvaccinated group. However, none of the goat from the vaccinated group had P. multocida B:2 in the liver, tonsil and heart. Therefore, the study revealed that an inactivated recombinant vaccine significantly provides significant protection against high dose challenge and enhances the stimulation of the local and systemic immunities.
Similar content being viewed by others
Introduction
Hemorrhagic septicemia (HS) caused by Pasteurella multocida serotype B:2 is an acute and highly fatal systemic disease of cattle and buffalo in tropical countries (Wijerwardana 1992; De Alwis 1995; Kamarudin 2005). It is characterized by a rapid course, high fever, loud, and stertorous breathing due to edematous swelling, petechial hemorrhages in the throat and brisket regions, profuse salivation, and severe depression leading to death within 24 h (Dawkins et al. 1990; Horadagoda et al. 2001).
Vaccination is the principal method of control for HS. Effective vaccine such as alum precipitated and oil adjuvant vaccines have been developed, (Chandrasekaran et al. 1994; Myint and Jones 2007) but difficulties in vaccine administration have led to low vaccination coverage and disease outbreaks (Saharee et al. 1993). One of the potential vaccine candidates is the fimbriae, which is a type surface protein. The fimbriae are key colonization factors and important protective antigens for the bacterium (Mattick 2002). Several reports have been described about the antigenic structure of the fimbrial protein and the fimbrial vaccines against a range of bacterial pathogens (Lepper et al. 1995; Ruffolo et al. 1997; Doughty et al. 2000; Ernie 2005). It was found that the fimbrial protein is antigenic and important to P. multocida, and has the capability to stimulate antibody production that can protect against challenge (Ruffolo et al. 1997; Ina-Salwany 2009).
Vaccine delivery plays an important role in vaccination and choosing the vaccine delivery depends on several factors such as man power, costing, safety, timing, and ease of administration. Intranasal vaccination is one of the most suitable alternatives for HS because this method not only activates the mucosal immunity, but also activates the systemic antibody response. This mode of vaccination has the advantage of being safer, less costly, and has fewer secondary effects than the systemic vaccination (Ogra et al. 2001). This study was carried out to determine the efficacy intranasal administration of a recombinant Escherichia coli cell vaccine that expresses a fimbrial protein of P. multocida B:2.
Materials and methods
Bacterial strains, plasmids, and culture conditions
The isolate of P. multocida B:2 PMX (Zamri-Saad et al. 2006) from a previous outbreak of hemorrhagic septicemia in cattle was grown at 37°C either on brain heart infusion (BHI) agar (Oxoid, Thermo Fisher Scientific Inc, UK) containing 10% goats blood or in BHI broth (Oxoid, Thermo Fisher Scientific Inc, UK) with agitation (200×g). A non-expression host, the E. coli strains NovaBlue GigaSingles™ (Merck, Darmstadt, Germany), and expression host, the E. coli strain BL21(DE3) (Merck, Darmstadt, Germany), were used for cloning and expression, respectively. Both E. coli strains were grown at 37°C with agitation (250×g) either in Luria–Bertani (LB) broth (Oxoid, Thermo Fisher Scientific Inc, UK) or on LB agar (Oxoid, Thermo Fisher Scientific Inc, UK). Whenever required, a total concentration of 50 μg/mL of ampicillin (Sigma, USA) was added. The overnight cultures in LB broth were kept in 25% (v/v) glycerol and stored at −80°C in cryo-vials until used. The expression vector, pET-32 Ek/LIC, was obtained from Merck, Darmstadt, Germany.
PCR amplification of the fimbrial gene of P. multocida B:2
To isolate the gene of interest, a set of primers flanking the fimbrial gene of P. multocida B:2 strain PMX was designed based on the published sequence of P. multocida B:2, AY788893 (Ernie 2005). The designated forward and reverse primer sequences were LIC-fimbrialF (5'-GACGACGACAAGATGAAAAAAGCCATTTTC-3') and LIC-fimbrialR (5'-GAGGAGAAGCCCGGTTAGCGCAAAATCCTGC-3'), respectively. The primers were designed to generate products with vector cohesive overhangs (bold), for efficient cloning onto an Ek/LIC vector, pET-32 Ek/LIC (Merck, Darmstadt, Germany). PCR was carried out in 50 μL volumes containing 5 μL of 10× PCR buffer, 3 μL of 25 mM MgSO4, 5 μL of 2 mM dNTPs (0.2 mM of each dNTPs), 1.5 μL of each 10 μM primer, 2.0 μL DNA template (8.0 ng/μL), and 1 U KOD XL DNA Polymerase (2.5 U/μL in 50 mM KCl; 50 mM Tris–HCl; 1 mM DTT; 0.1 mM EDTA; 50% glycerol; 0.1% Nonidet P-40; 0.1% Tween-20, pH 8.0; Merck, Darmstadt, Germany). The amplifications were carried out using the Mastercycler personal (Eppendorf, Germany) starting with denaturation at 95°C for 2 min, followed by 30 cycles consisting at 95°C for 20 s, 63°C for 10 s, and final elongation at 70°C for 10 s. After amplification, 5 μL samples mixed with 2 μL of loading dye (Merck, Darmstadt, Germany) were subjected to electrophoresis in a 1% agarose gel at 70 V for 1.5 h and stained with ethidium bromide (Merck, Darmstadt, Germany) to detect the presence of the amplified products (450 bp). The DNA band was observed under UV transilluminator and photographed by Alpha Imager, UK.
Plasmid construction
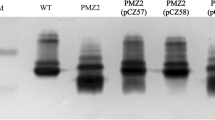
The blunt PCR products were purified and treated with T4 DNA polymerase in the presence of the dGTP and DTT to generate the specific vector cohesive overhangs. The insert was ligated in the linear vector according to the vector manufacturer protocol (Merck, Darmstadt, Germany). The competent E. coli NovaBlue GigaSingles™ (Merck, Darmstadt, Germany) is non-expression host and used for the propagation of the recombinant plasmids. The competent NovaBlue GigaSingles™ cells were used to transform the recombinant vectors and plated overnight at 37°C on LB agar containing 50 μg/mL of carbenicillin (Invitrogen, USA). The presence of plasmid carrying the inserts were screened for inserts using vector specific primers of upstream primer, S-tag and T7 terminator primer; 5′-GAACGCCAGCACATGGAC-3′ and 5'-GCTAGTTATTGCTCAGCGG-3′. Purified plasmids were isolated according to the protocol of the GeneJet Plasmid Miniprep Extraction Kit (Fermentas, USA) for further DNA sequencing and the positive plasmids containing the insert were then transformed into expression host, E. coli BL21(DE3). The “BioEdit” software was used to analyze the arrangement of the fimbrial gene in vector sequence and compared with others serotype of P. multocida (Fig. 1; Ina-Salwany 2009). The positive clone individually known as pET32/LIC-fimbrial was stored in LB broth with 50 μg/mL carbenicillin at −80°C.

ClustalW Multiple Alignment program (BioEdit ver. 7.0.5.3) was used to align the predicted amino acid sequences of the fimbrial genes of different isolates of P. multocida. The fimbrial gene designations are shown as follows (GenBank): PMX (FJ526996), Ernie B:2 (AY88893), ptfA B:2 (AY644678), PM70 (AE006044), ptfA D:1 (DQ788723), pftA A:1 (AF154834), and ptfA F:3 (DQ788724)
Production of recombinant proteins by isopropyl-beta-d-thiogalactopyranoside autoinduction
The Overnight Express Autoinduction System (Merck, Darmstadt, Germany) was used for protein expression study. Autoinduction was designed for high level protein expression with pET and other isopropyl-beta-d-thiogalactopyranoside (IPTG)-inducible bacterial expression systems without the need to monitor the cell growth (Merck, Darmstadt, Germany). The IPTG autoinduction was accomplished by inoculating with 50 μL of glycerol stock containing pET32/LIC-fimbrial into 50 mL of LB broth with 50 μg/mL carbenicillin in 250 mL flask and incubating overnight (∼16 h) with shaking (280×g) at 37°C as recommended by the manufacturer (Merck, Darmstadt, Germany). Pelleted cells were subjected to Bugbuster Protein Extraction Reagent plus Benzonase Nuclease (Merck, Darmstadt, Germany) to determine the expressed protein. This reagent is formulated for the gentle disruption of the cell wall of E. coli resulting in the liberation of soluble protein. After centrifugation, the soluble extracts were assayed for total protein concentration and analyzed by SDS-PAGE. Western immunoblotting was performed following SDS-PAGE to confirm the expressed proteins using anti-His tag and lung’s lavage fluids, respectively (Fig. 2a, b; Ina-Salwany 2009).

a Western blot analysis of the soluble fusion protein of the recombinant fimbrial after expression in E. coli BL21(DE3) using His.Tag MAb (Novagen, USA) as primary antibody. Lane 1 The host E. coli BL21(DE3). Lanes 2 and 4 The soluble cell protein of E. coli BL21(DE3) expressing pET32/LIC-fimbrial. Lane 3 The soluble cell proteins of E. coli BL21(DE3) expressing pET32/LIC without insert. The arrows indicate the presence of expressed protein, ∼33 kDa. b Immunoblot profiles of the recombinant 18 kDa fimbrial proteins of P. multocida B:2, reacted against goat’s lung lavage fluid showing positive reaction. Lane 1 Fimbrial protein of PMTB strain. Lane 2 Fimbrial protein of PMX strain. Lane M Standard molecular weight markers
Preparation of inactivated recombinant vaccine
After induction with IPTG autoinduction as described above, cultures of the recombinant E. coli BL21(DE3) expressing pET32/LIC-fimbrial were harvested and killed in 0.5% formalin in phosphate buffered saline (PBS, pH 7.4; Sigma, USA) overnight at 4°C. This was followed by washing three times in sterile PBS by centrifugation (5,000×g) at 4°C for 10 min to ensure that the formalin was completely removed. Finally, the inactivated recombinant cells were re-suspended in sterile PBS as stock vaccine seed. For preparation of vaccine, the stock vaccine seed was added into adequate amount of sterile PBS to give a final concentration of 1 × 106 colony forming unit (CFU)/mL using McFarland turbidity standard (Smibert and Kreig 1994). The sterility of the inactivated recombinant vaccine was tested by inoculating 0.1 mL of the vaccine onto the blood agar followed by incubation at 37°C for 24 h. The vaccine was considered sterile when no growth appeared on the blood agar.
Preparation of live P. multocida B:2 for challenge
P. multocida B:2 strain PMX was subcultured onto blood agar at 37°C for 24 h before three colonies was selected, inoculated into 100 mL of BHI broth for 18 h at 37°C with gentle shaking. Following incubation, the bacterial concentrations were determined using the standard plate count technique. Approximately 1 mL of the broth was serially diluted tenfolds before 0.1 mL of each dilution was poured and spread onto blood agar and incubated at 37°C for 24 h. Following incubation, the number of colonies, particularly those plates containing between 25 and 250 colonies, was counted before the concentration was expressed as CFU. The challenge dose was used immediately direct from BHI broth, and it contained 3.5 × 109 CFU/mL of live P. multocida B:2.
Experimental design
Animals
Sixty healthy goats of approximately 8 months old with no history of vaccination against HS were selected for the trials. All animals were kept in the same paddock. The goats were left to graze during daytime and kept in confined areas at night. While in confinement, they were fed with 300 g/animal/day supplemented feed while drinking water was available ad libitum. Immediately upon arrival at the experimental station, nasal swabs were collected twice a week for two consecutive weeks from all goats for bacterial isolation, particularly P. multocida. The trial started only when all goats were found free of P. multocida in their nasal cavity.
Vaccination procedure
The goats were divided into three groups. The goats in group 1 (n = 21) were vaccinated intranasal with 2 mL of inoculums containing 1 × 106 CFU/mL of recombinant E. coli expressing the fimbrial protein of P. multocida B:2. The goats in group 2 (n = 21) were exposed intranasal with 2 mL of inoculums containing 1 × 106 CFU/mL of E. coli expressing pET32/LIC vector without the fimbrial protein as control group, and group 3 (n = 18) were designated as unvaccinated group and were kept separated from two other groups (Fig. 3). Two weeks after the first vaccination, a booster dose was repeated on goats of groups 1 and 2, respectively. Throughout the 5-week study period, serum samples were collected prior to and at weekly intervals postvaccination (p.v.) from all goats for serological analysis. Three goats out of 15 from each group were randomly slaughtered each week and post-mortem were carried out immediately to examine the entire of respiratory tract before all lungs were lavaged by introducing 50 mL sterile PBS into the lungs through the trachea, then massaged gently before the fluid was recollected. The lung lavage fluid was centrifuged at 1,000×g for 15 min to remove the debris. Two weeks after the second exposure, all remaining goats, group 1, vaccinated goats (n = 6), group 2, control goats (n = 6), and group 3, unvaccinated goats (n = 3), were challenged intratracheal with 1 mL of inoculums containing 3.5 × 109 CFU/mL of live P. multocida B:2. Following the intratracheal challenge, all goats were monitored daily for any clinical signs and death before the goats were slaughtered on day 3 post-challenge. The experimental design was approved and compliance with the humane methods recommended by the Research Ethics Committee, Faculty of Veterinary Medicine, Universiti Putra Malaysia.

Antibody responses (serum: IgG) against P. multocida B:2 in goats following intranasal vaccination. Bars represent SEM of antibody for each animal tested. rF group which vaccinated with the inactivated recombinant vaccine expressing fimbrial protein of P. multocida B:2, rP control group which exposed to recombinant E. coli without fimbrial protein, cX unvaccinated group
Serology
The enzyme-linked immunosorbent assay was performed as previously described (Zamri-Saad et al. 2006) by analyzing the systemic immunoglobulin G (IgG) while the lung lavage fluid samples for the local secreting immunoglobulin A (sIgA). Briefly, systemic IgG levels were determined using 50 μL of the rabbit anti-goat IgG peroxidase conjugate (Sigma), diluted at 1:6,000. While, for determination of secreting IgA levels, 50 μL of the swine anti-goat/sIgA serum (Nordic, The Netherland) was diluted at 1:6,000 in PBS, was applied. Then, 50 μL of immunoconjugate, goat anti-swine/IgG(H + L)/PO (Nordic, The Netherland) with dilutions of 1:6,000 was used. After a final three-wash step with PBST, bound conjugate was detected using 100 μL of TMB One Solution substrate (Promega, USA), before stop with 0.2 mol/L sulfuric acid. Optical density values were measured at 450 nm wavelength in an Anthos Zenyth 340 st reader (Austria).
Bacterial isolation, biochemical test, and multiplex PCR
Samples of lung, liver, tonsil, and heart blood were collected from each goat immediately after slaughtered. On the same day, all the samples were cultured aseptically onto the agar (Oxoid, UK) and incubated at 37°C for 24 h for bacterial examination. Gram stain and biochemical tests were used to identify P. multocida (Carter and Chengappa 1980; Jablonski et al. 1996). Suspected colonies of P. multocida B:2 were confirmed by multiplex PCR as described earlier (Biswas et al. 2004; Zamri-Saad et al. 2006; Shafarin et al. 2007). Multiplex PCR was conducted using two sets of primer (Research Biolabs, Malaysia) designated from the sequence of the clones KMTI for the detection of P. multocida strain (KMTIT7-5′-ATCCGCTATTTACCCAGTGG-3′ and KMT1SP6-5′-GCTGTAAACGAACTCGCCAC-3′), while 6b primers were used for typing P. multocida type B:2 (Townsend et al. 1998) (KTT2-5′-AGGCTCGTTTGGATTATGAAG-3′) and (KTSP61-5′-ATCCGCTAACACACTCTC-3′). Briefly, 25 μL reaction mixture containing 10× PCR buffer (100 mM Tris–HCl, pH 8.8 at 25°C; 500 mM KCl, 0.8% Nonidet P40; Fermentas, USA), 2.0 mmol/L MgCI2, 200 μmol/L of each dNTP, 20 pmol of each primer, and 1 U Taq DNA polymerase (20 mM Tris–HCl (pH 8.0); 1 mM DTT, 0.1 mM EDTA, 100 mM KCl, 0.5% Nonidet P40, 0.5% Tween 20, and 50% glycerol; Fermentas, USA) was prepared, and one colony was picked from the plate as a DNA template and re-suspended in the PCR mixture (Zamri-Saad et al. 2006). The reaction mixture was subjected to amplification in a Thermocycler (Eppendorf, Germany) according to the following program: initial denaturation at 95°C for 4 min, denaturation at 95°C for 45 s, annealing at 55°C for 45 s, extension at 72°C for 45 s, which was repeated for 30 cycles, and a final extension at 72°C for 6 min. Amplified products were separated by agarose gel electrophoresis (1.0% agarose in 1× TBE stained with ethidium bromide) at 70 V for 1.5 h. The DNA band was observed under UV transillumination and photographed by Alpha Imager, UK.
Statistical analysis
Antibody titers and other data were analyzed by one way ANOVA using LSD all-pairwise comparisons test. All values were expressed as means ± SE. All data were analyzed using Statistix ver. 8 (Analytical software, USA) and tested at 5% level of significance.
Results
Clinical observations
Animals in all groups coughed moderately immediately following the intratracheal injection. However, none of the challenged goats died throughout the 3-day postinfection period but six goats (40%) from groups 2 and 3 showed clinical signs of respiratory distress such as mild to moderate coughing and sneezing.
Serological responses
Serum antibody responses
Prior to the vaccination, all animals in groups 1, 2, and 3 showed low serum IgG and IgA levels against P. multocida B:2. Following the first intranasal vaccination, the IgG responses by animals in the vaccinated group (group 1) increased significantly (p < 0.05) as compared to the control (group 2) and unvaccinated (group 3; Fig. 3). However, following the booster dose on week 2 p.v., the IgG levels in group 1 increased significantly (p < 0.05) when compared to group 3, but not significant (p > 0.05) when compared to group 2. The serum IgG levels of both groups 1 and 2 increased significantly (p < 0.05) at week 4 p.v. as compared to group 3, at the time of challenge with the live wild-type P. multocida B:2.
Antibody responses in lung lavage fluid
The lung IgA levels against P. multocida B:2 showed similar increasing pattern as lung IgG levels following the first intranasal vaccination. Following a booster dose, the lung IgA levels of group 1 increased and significantly (p < 0.05) higher when compared to groups 2 and 3, particularly at week 3. Following the challenge exposure to wild-type P. multocida B:2 on week 4, the lung IgA level of group 1 increased and significantly (p < 0.05) higher than group 2, but insignificant (p > 0.05) than the group 3 (Fig. 4). The result showed that the lung IgA levels in all groups (group 1, 2, and 3) increased significantly (p < 0.05) higher following challenge than the level following booster dose at week 2 p.v.

Antibody responses (lung lavage fluid: sIgA) against P. multocida B:2 in goats following intranasal vaccination. Bars represent SEM of antibody for each animal tested. rF group which vaccinated with the inactivated recombinant vaccine expressing fimbrial protein of P. multocida B:2, rP control group which exposed to recombinant E. coli without fimbrial protein, cX unvaccinated group
Bacterial isolation
Isolation of P. multocida B:2 was successfully made from the lung, liver, tonsil, and heart blood of goats that were humanely killed on the third day post-challenge (Table 1). Multiplex PCR produced the amplified products of ∼460 and ∼620 bp, which were considered specific for HS-causing P. multocida B:2. Isolation rate were more frequent from groups 2 and 3 than group 1. The isolation made from the lung posses only 17% (1/6) of group 1, 67% (4/6) of group 2, and 100% (3/3) of group 3. However, none of the group 1 had P. multocida B:2 in the liver, tonsil and heart. P. multocida B:2 was isolated from three (50%) out of six livers of group 2 and from two (67%) out of three tonsils or hearts of group 3.
Discussion
Recent advances in subunit vaccine development have been based on bacterial recombinants (Ertl and Xiang 1996). One of these approaches consists of expressing the foreign antigen on the surface of bacteria. High antibody titers have been obtained against a hepatitis B virus epitope inserted with the outer membrane LamB protein located on the surface of E. coli (Leclerc et al. 1989). Guilloteau et al. (1999) studied the humoral and cellular immune responses specific to Brucella melitensis induced by recombinant E. coli expressing the B. melitensis Omp 31 gene in mice. While, Sabri (2006) studied the in vivo heterologous immunogenicity of the inactivated recombinant vaccine containing 31 kDa OMPs of Mannheimia haemolytica A7 following subcutaneous injections in goats.
The fimbrial protein of P. multocida has been associated with the virulence of the organism through its function in the attachment to the host cells which enhances the establishment and infectivity of the organism (Ruffolo et al. 1997; Doughty et al. 2000). Thus, stimulating local antibody production against fimbrial protein is believed to be beneficial in preventing the infection through prevention of the fimbrial function in enhancing attachment (Ernie 2005; Ina-Salwany 2009). In this experimental trial, inactivated recombinant vaccine expressing antigenic fimbrial protein of P. multocida B:2 was studied to determine the potentiality and immunogenicity of the fimbrial protein in goat for a period of 5 weeks. In the other hand, recombinant E. coli alone was used as a control to study the efficacy of recombinant fimbrial protein in inducing immunity.
Vaccination increased the antibody levels as early as week 1 p.v. The antibody levels pattern were generally similar to that observed in IgA and IgG responses against P. multocida B:2 either in serum or lung lavage fluid. The antibody levels started to decline at week 2 p.v. as shown in the pattern of antibody levels of serum IgG; however, IgA in the lung lavage persistently increased until week 3. This pattern was similar to the OMP vaccines of P. multocida B:2 and M. haemolytica (Pati et al. 1996; Sabri et al. 2000). The reduction in antibody level indicated that a booster dose was required. However, the significantly high IgA levels against P. multocida B:2 was observed in the lung lavage fluid, as early week 1 p.v. and was still increasing at week 2 p.v. The same observation was made by Effendy et al. (1998) following intranasal exposures to formalin-killed P. hemolytica A2 and Donachie et al. (1986) following aerosol exposure in lambs. Husband (1987) also detected high levels of IgA in the respiratory tract mucosa 7 days after intratracheal exposures to antigen.
A booster dose of intranasal vaccination at week 2 p.v. gradually stimulated the antibody production, and it increased significantly for the next 2 weeks of p.v., i.e., at weeks 3 and 4 p.v. Even though, the lung lavage IgA responses were lower at week 3 p.v., they were still significantly higher as compared to the responses of the control and unvaccinated group. The results showed that the local immune response started by moderate initiation response by both IgG and IgA in the lower respiratory tract as well as systemic immune responses. The response became stronger following the booster vaccination. Responses of local immunity following intranasal immunization were reliable to initiate mucosal immune responses, and it is important to mucosa-associated lymphoid tissue (Kiyono et al. 1992). The increase of the IgG and IgA in serum and lung lavage after re-exposure of recombinant fimbrial cell vaccine suggested that IgG and IgA were functioning well in preventing establishment of pathogen.
The antibody levels of serum IgG responses at week 3 p.v. after booster dose observed in vaccinated animals was significantly lower compared to week 1 p.v. This observation showed that the vaccine could not boost appropriate memory cells to generate a significant level of secondary antibody following the administration of the booster dose. This is due to the fact that the vaccine was degraded too fast by the host defense mechanism without being given appropriate time for cloning the vaccine materials that were used by the memory cells (Isaguliants et al. 2004). Moreover, the increase of antibody levels was observed either in lung lavage fluid or serum in responding after challenge. It can be concluded that this procedure acted as another booster to the immune system resulting in even higher antibody levels. It is common to observe a rapid and high antibody response following challenge by live organism (Sabri et al. 2000). Pati et al. (1996) reported that the challenge of animals in the vaccination trial was most suitably done between days 28 and 35 p.v.
Basically, exposure of goats to P. multocida B:2 using different routes of infections revealed that more goats succumbed to HS following subcutaneous and intratracheal infections (Zamri-Saad and Shafarin 2007). However, intratracheal route took a longer time resulting in slightly lower death rate (Zamri-Saad and Shafarin 2007). Another study showed similar observations were made in cattle (De Alwis et al. 1990). In the present study, based on multiplex PCR assay, it has been revealed that P. multocida B:2 was able to localize in goat after 3 days post-challenge following intratracheal administration. Most isolation of P. multocida B:2 were made from the lungs. However, this study has failed to isolate P. multocida B:2 from the tonsil except 10% isolation from the unvaccinated group. The results differ from the previous report in which tonsil and lymph nodes associated with the respiratory tract are known to be good colonization sites for P. multocida B:2 in cattle and buffaloes (Wijerwardana et al. 1986; De Alwis et al. 1990; Saharee et al. 1993). Following intratracheal challenge, none of the vaccinated goats showed evidence of P. multocida B:2 in their tonsil, heart, and liver.
The intranasal route was chosen to immunize goats in view of previous reports (Effendy et al. 1998; Zamri-Saad et al. 2006). The most effective method to increase mucosal immunity in the upper respiratory tract is through intranasal administration of antigen (Todd 1973; Wilkie 1982; Hjerpe 1990). It appears that an immune response to a protein antigen administered via the mucosal route can be expressed as (a) mucosal immune response with sIgA antibody, (b) systemic immune responses priming, with development of serum IgG and IgM, and specific cellular immunity, and (c) development of mucosal tolerance with systemic immunologic hyporesponsiveness with or without any change in IgA-specific mucosal immune reactivity (Mowat and Weiner 1999).
On the whole, this study has revealed that the antibody levels of IgG and IgA in vaccinated goats were significantly higher (p < 0.05) as compared to control and unvaccinated goats either in serum or lung lavage fluid. Thus, this study has illustrated that the inactivated recombinant vaccine expressing the fimbrial protein of P. multocida B:2 provided high degree of protection against high dose challenge and was able to enhance the stimulation the local and systemic immunity which appeared to be an important role in the mucosal immune responses. However, the amount of different doses of expressed protein needs to determine in order to increase solid protection against the disease. In conclusion, the results have raised two vital points: (a) fimbrial protein was the suitable antigen that contributed to protective immunity to P. multocida B:2 infection and (b) a strong local and systemic antibody response was obtained by intranasal vaccination with inactivated recombinant vaccine expressing the fimbrial protein of P. multocida B:2.
References
Biswas, A., Shivachandra, S.B., Saxena, M.K., Kumar, A.A., Singh, V.P. and Srivastava, S.K., 2004. Molecular variability among strains of Pasteurella multocida isolated among outbreaks of haemorrhagic septicaemia in India, Veterinary Research Communication, 28, 287–298.
Carter, G.R. and Chengappa, M.M., 1980. Hyaluronidase production by type B Pasteurella multocida from cases of haemorrhagic septicaemia, Journal of Clinical Microbiology, 11, 94–96.
Chandrasekaran, S., Kennett, L., Yeap, P.C., Muniandy, N., Rani, B. and Mukkur, T.K.S., 1994. Characterization of immune response and duration of protection in buffaloes immunized with haemorrhagic septicaemia vaccine, Veterinary Microbiology, 41, 213–219.
Dawkins, H.J.S., Johnson, R.B., Spencer, T.L. and Patten, B.E., 1990. Rapid identification of Pasteurella multocida organism responsible for haemorrhagic septicaemia using an enzyme-linked immunosorbent assay, Research in Veterinary Science, 49, 261–267.
De Alwis, M.C.L., 1995. Haemorrhagic septicaemia in cattle and buffaloes. In: Donachie, W., Lainson, F.A. and Hodgson, J.C. (eds.), Haemophilus, Actinobacillus and Pasteurella, pp. 9–24 (Plenum Press, New York).
De Alwis, M.C.L., Wijewardana, T.G., Gomis, A.I.U. and Vipulasiri, A.A., 1990. Persistence of the carrier status in haemorrhagic septicaemia (Pasteurella multocida serotype 6:B infection) in Buffaloes, Tropical Animal Health and Production, 22, 185–194.
Donachie, W., Burrells, C., Sutherland, A.D., Gilmour, J.S. and Gilmour, N.J.L., 1986. Immunity of specific pathogen-free lambs to challenge with an aerosol of Pasteurella haemolytica biotype A serotype 2. Pulmonary antibody and cell responses to primary and secondary infections, Veterinary Immunology and Immunopathology, 11, 265–279.
Doughty, S.W., Ruffolo, L.G. and Adler, B., 2000. The type 4 fimbrial subunit gene of Pasteurella multocida, Veterinary Microbiology, 72, 79–90.
Effendy, A.W.M., Zamri-Saad, M., Puspa, R. and Rosiah, S., 1998. Efficacy of intranasal administration of formalin-killed Pasteurella haemolytica A2 against intratracheal challenge in goats, Veterinary Record, 142, 428–431.
Elleman, T.C. and Stewart, D.J., 1988. Efficacy against foot rot of a Bacteroides nodosus 265 (serogroup H) pilus vaccine expressed in Pseudomonas aeruginosa, Infection and Immunity, 56, 595–600.
Ernie, Z.A., 2005. Cloning and expression of fimbrial subunit gene of Pasteurella multocida type 6:B, isolated from cattle with Haemorrhagic septicaemia. Master’s Thesis. Universiti Putra Malaysia.
Ertl, H.C. and Xiang, Z., 1996. Novel vaccine approaches, Immunology, 156, 3579–3582.
Guilloteau, L.A., Laroucau, K., Vizcaino, N., Jacques, I. and Dubray, G., 1999. Immunogenicity of recombinant Escherichia coli expressing the omp31 gene of Brucella melitensis in BALB/c mice, Vaccine, 17, 353–361.
Hirabayashi, Y., Kurata, H., Funato, H., Nagamine, T., Aizawa, C., Tamura, S., Shimada, K. and Kurata, T., 1990. Comparison of intranasal inoculation of influenza HA vaccine combined with cholera toxin B subunit with oral or parenteral vaccination, Vaccine, 8, 243–248.
Hjerpe, C.A., 1990. Bovine vaccines and herd vaccination program, Veterinary Clinics of North American: Food Animal Practice, 6, 167–260.
Horadagoda, N.U., Hodgson, J.C., Moon, G.M., Wijewardana, T.G. and Eckersall, P.D., 2001. Role of endotoxin in the pathogenesis of haemorrhagic septicaemia in the buffalo, Microbial Pathogenesis, 30, 171–178.
Hultgren, S.J., Jones, C.H. and Normark, S., 1996. Bacterial adhesins and their assembly. In Escherichia coli and Salmonella: Cellular and Molecular Biology, 2nd edn, vol. II, Edited by Neidhardt, F.C. and others. Washington, DC, pp. 2730–2756 (American Society for Microbiology).
Husband, A.J., 1987. Perspectives in mucosal immunity: A ruminant model. Veterinary Immunology and Immunopathology, 17, 357–365.
Ina-Salwany, M.Y., 2009. Identification, cloning, sequencing, expression and protective capacity of the gene encoding a fimbrial protein of Pasteurella multocida B:2. Doctor of Philosophy Thesis, Faculty of Science and Technology, University Malaysia Terengganu.
Isaguliants, M.G., Petrakova, N.V., Kashuba, E.V., Suzdaltzeva, Y.G., Belikov, S.V., Mokhonov, V.V., Prilipov, A.G., Matskova, L., Smirnova, I.S., Jolivet-Reynaud, C. and Nordenfelt, E., 2004. Immunization with hepatitis C virus core gene triggers potent T-cell response, but affects CD4+ T-cells, Vaccine, 22, 1656–1665.
Jablonski, P.E., Jaworski, M. and Hovde, C.J., 1996. A minimal medium for growth of Pasteurella multocida, FEMS Microbiology Letters, 140, 165–169.
Kamarudin, M.I., 2005. Haemorrhagic septicaemia: eradication is a possibility. Proceedings of the Regional Symposium on Haemorrhagic Septicaemia, September, Putrajaya, 12–15.
Kiyono, H., Bienenstock, J., McGhee, J.R. and Ernst, P.B., 1992. The mucosal immune system: features of inductive and effector sites to consider in mucosal immunization and vaccine development, Regional Immunology, 4, 54–62.
Leclerc, C., Charbit, A., Molla, A. and Hofnung, M., 1989. Antibody response to a foreign epitope expressed at the surface of recombinant bacteria: importance of the route of immunization, Vaccine, 7, 242–248.
Lepper, A.W.D, Atwell, J.L., Lehrbach, P.R., Schwartzkoff, C.L., Egerton, J.R. and Tennent, J.M., 1995. The protective efficacy of cloned Moraxella bovis pili in monovalent and multivalent vaccine formulations against experimentally induced infectious bovine keratoconjunctivitis (IBK), Veterinary Microbiology, 45, 129–138.
Mattick, J.S., 2002. Type IV pili and twitching motility, Annual Review Microbiology, 56, 289–314.
Mowat, A.M. and Weiner, H.L., 1999. Oral tolerance: physiological basis and clinical applications. In: Ogra, P.L., Mestecky, J., Lamm, M.E., Strober, W., Bienenstock, J. and McGhee, J.R. (ed.), Mucosal immunology, 2nd ed., pp. 587–618 (Academic Press, New York).
Myint, A. and Carter, G.R., 1990. Field use of live haemorrhagic septicaemia vaccine, Veterinary Record, 126, 648.
Myint, A. and Jones, T.O., 2007. Efficacy of haemorrhagic septicaemia alum precipitated vaccine, Veterinary Record, 160, 172.
Myint, A., Jones, T.O. and Nyunt, H.H., 2005. Safety, efficacy and cross-protectivity of a live intranasal aerosol haemorrhagic septicaemia vaccine, Veterinary Record, 156, 41–45.
Novagen, 2000. Ek/LIC Vector Kits Manual, 9th ed., USA. (http://www.novagen.com).
Ogra, P.L., Faden, H. and Welliver, R. C., 2001. Vaccination strategies for mucosal Immune responses, Clinical Microbiology Reviews, 14, 430–445.
Pati, U.S., Srivastava, S.K., Roy, S.C. and More, T., 1996. Immunogenicity of outer membrane protein of Pasteurella multocida in buffalo calves, Veterinary Microbiology, 52, 301–311.
Ruffolo, C.G., Tennent, J. M., Michalski, W.P. and Adler, B., 1997. Identification, purification, and characterization of the type 4 fimbriae of Pasteurella multocida, Infection and Immunity, 65, 339–343.
Sabri, M.Y., 2006. Identification, cloning, sequencing, expression and protective capacity of the gene encoding a 31-kilodalton outer membrane protein of Mannheimia haemolytica. Doctor of Philosophy Thesis, Faculty of Veterinary Medicine, University Putra Malaysia.
Sabri, M.Y., Zamri-Saad, M., Mutalib, A.R., Israf, D.A. and Muniandy, N., 2000. Efficacy of an outer membrane protein of Pasteurella haemolytica A2, A7, or A9-enriched vaccine against intratracheal challenge exposure in sheep, Veterinary Microbiology, 73, 13–23.
Saharee, A.A., Salim, N.B., Rasedee, A. and Jainudeen, M.R., 1993. Haemorrhagic septicaemia carries among cattle and buffalo in Malaysia. Pasteurellosis in Production Animals. (ACIAR Proceedings no. 43), 89–91.
Shafarin, M.S., Zamri-Saad, M., Jamil, S.M., Siti Khairani, B. and Saharee, A.A., 2007. Experimental transmission of Pasteurella multocida 6:B in goats, Journal of Veterinary Medicine Series A, 54, 136–139.
Smibert, R.M. and Kreig, N.R., 1994. Phenotypic characterization section 25.4.9. In: Methods for General and Molecular Bacteriology. Gerhardt, P., et al. (ed.), pp. 607–654 (American Society for Microbiology, Washington, D.C.).
Stewart, D.J. and Elleman, T.C., 1987. A Bacteroides nodosus pili vaccine produced by recombinant DNA for prevention and treatment of foot-rot in sheep, Australian Veterinary Journal, 64, 79–81.
Todd, J.D., 1973. Immune response to parenteral and intranasal vaccinations, Journal of the American Veterinary Medical Association, 163, 807.
Townsend, K.M., Frost, A.J., Lee, C.W., Papadimitriou, J.M. and Dawkins, H.J.S., 1998. Development of PCR assays for species and type specific identification of Pasteurella multocida isolates, Journal of Clinical Microbiology, 36, 1096–1100.
Wijerwardana, T.G., 1992. Haemorrhagic septicaemia, Reviews in Medical Microbiology, 3, 59–63.
Wijerwardana, T.G., De Alwis, M.C.L. and Vipulasiri, A.A., 1986. An investigation into the possible role of the goat as a host in haemorrhagic septicaemia, Sri Lanka Veterinary Journal, 34, 24–32.
Wilkie, B.N., 1982. Respiratory tract immune response to microbial pathogens, Journal of the American Veterinary Medical Association, 181, 1074–1079.
Zamri-Saad, M., 2005. Attempts to develop vaccines against Haemorrhagic septicaemia: A review. Proceedings of the Regional Symposium on Haemorrhagic Septicaemia, September, Putrajaya, 1–8.
Zamri-Saad, M. and Shafarin, M.S., 2007. Response of goats to the different routes of infection by Pasteurella multocida B:2, Journal of Animal and Veterinary Advances, 6, 340–343.
Zamri-Saad, M., Ernie, Z.A. and Sabri, M.Y., 2006. Protective effect following intranasal exposure of goats to live Pasteurella multocida B:2, Tropical Animal Health and Production, 38, 541–546.
Acknowledgments
This work was supported by Ministry of Science, Technology and Innovation, Malaysia (MOSTI). We thank to all staff of Histopathology Laboratory, and Ruminant Research Centre, Faculty of Veterinary Medicine, Universiti Putra Malaysia for their excellent assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mohd Yasin, IS., Mohd Yusoff, S., Mohd, ZS. et al. Efficacy of an inactivated recombinant vaccine encoding a fimbrial protein of Pasteurella multocida B:2 against hemorrhagic septicemia in goats. Trop Anim Health Prod 43, 179–187 (2011). https://doi.org/10.1007/s11250-010-9672-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11250-010-9672-5