
Abstract
In physiological conditions, the adipose organ resides in well-defined areas, where it acts providing an energy supply and as an endocrine organ involved in the control of whole-body energy metabolism. Adipose tissue adipokines connect the body’s nutritional status to the regulation of energy balance. When it surrounds organs, it provides also for mechanical protection. Adipose tissue has a complex and heterogenous cellular composition that includes adipocytes, adipose tissue-derived stromal and stem cells (ASCs) which are mesenchymal stromal cells, and endothelial and immune cells, which signal to each other and to other tissues to maintain homeostasis. In obesity and in other nutrition related diseases, as well as in age-related diseases, biological and functional changes of adipose tissue give rise to several complications. Obesity triggers alterations of ASCs, impairing adipose tissue remodeling and adipose tissue function, which induces low-grade systemic inflammation, progressive insulin resistance and other metabolic disorders. Adipose tissue grows by hyperplasia recruiting new ASCs and by hypertrophy, up to its expandability limit. To overcome this limitation and to store the excess of nutrients, adipose tissue develops ectopically, involving organs such as muscle, bone marrow and the heart. The origin of ectopic adipose organ is not clearly elucidated, and a possible explanation lies in the stimulation of the adipogenic differentiation of mesenchymal precursor cells which normally differentiate toward a lineage specific for the organ in which they reside. The chronic exposition of these newly-formed adipose depots to the pathological environment, will confer to them all the phenotypic characteristics of a dysfunctional adipose tissue, perpetuating the organ alterations. Visceral fat, but also ectopic fat, either in the liver, muscle or heart, can increase the risk of developing insulin resistance, type 2 diabetes, and cardiovascular diseases. Being able to prevent and to target dysfunctional adipose tissue will avoid the progression towards the complications of obesity and other nutrition-related diseases. The aim of this review is to summarize some of the knowledge regarding the presence of adipose tissue in particular tissues (where it is not usually present), describing the composition of its adipogenic precursors, and the interactions responsible for the development of organ pathologies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 The pathological expansion of adipose tissue throughout the body
Obesity is recognized as a multifactorial chronic disease in which the qualitative and quantitative changes of the histological architecture of the adipose organ and the consequent functional alterations account for the severity of the disease and the development of its complications [1,2,3]. The primary cause of obesity is likely an altered relationship between the environment and the genetic heritage of the control systems which is at the basis of the regulation of energy metabolism and influences the bidirectional pathways of the energy transfer between the environment and the body [4]. Diverse evolutionary changes acting on the nuclear, mitochondrial and microbiota genome may impact genetic predisposition to obesity [5]. Obesity can also be considered an intrinsic path represented by the alteration of the dialogue between the center and the periphery or between the control systems present at the level of the Central nervous system (CNS) and the energy depots of the body, where the adipose organ is the most important source [6, 7].
Obesity and adipose organ dysfunction are the trigger of several complications such as cardiovascular disease, type 2 diabetes (T2D), and cancer, whose pathophysiological bases are very complex, in which the metabolic, hormonal, inflammatory and immune aspects coexist along with alteration of cellular composition and between cell communication [8, 9]. Adipose tissue (AT) is composed not only of adipocytes and their precursors, but also of an array of immune cells and an exclusive extracellular matrix, whose behavior under acute and chronic hypercaloric states is quite different [10].
The adipose organ could be defined as a diffuse organ and in fact not only does it exist in anatomical structures that are easily dissectable and separable from the context of other organs [11], but it can also be located both within organs in which physiologically it should not be [12], or surround organs and tissues in anatomical continuity or be clearly distinguished from them by anatomical borders (Fig. 1). As discussed by Zwick et al. [13], AT could assume different, often non-traditional functions depending on its proximity to other organs in relation to their physiological and pathological conditions. Moreover, in stem cell-rich skin, bone marrow, and mammary glands, adipocytes signal to and modulate organ regeneration and remodeling. In mammary glands and heart, adipocytes supply lipids to neighboring cells for nutritional and metabolic functions, respectively. AT near the surface of skin and intestine senses and responds to bacterial invasion, contributing to the body’s innate immune barrier. As the recognition of diverse adipose depot functions increases, novel therapeutic approaches centered on tissue-specific adipocytes are likely to emerge for a range of cancers and regenerative, infectious, and autoimmune disorders.

Pathological adipose tissue deposition. In humans during obesity and ageing, adipose tissue (AT) increases in mass, both as subcutaneous (SAT) and visceral (VAT). Moreover, it accumulates also outside from its physiological conserved regional location. The expansion nearby others organs cause their functional impairment and the appearance of comorbidities. (1) subcutaneous and visceral AT; (2) intermuscular AT; (3) bone marrow AT; (4) pericardial AT; (5) mammary AT; (6) mesenteric AT; (7) thymic AT. Created with BioRender.com
The purpose of this review is to consider the pathogenetic interactions between the presence of AT in particular sites, and in any case where it is usually not present, and the development of organ pathology such as sarcopenia and osteoporosis.
2 Adipose organ and the pathophysiological basis of adiposopathy
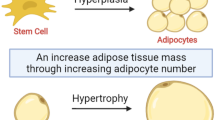
AT grows by hypertrophy, which consists in the enlargement of cell size by accumulation of triglycerides (TG) in lipid droplets, and by hyperplasia, recruiting and differentiating adipose progenitors into new mature cells [14]. While the first process refers to a metabolic adaptation of a single adipocyte to the environmental excess of nutrients, lipids in particular, and allows rapid and reversible change in AT mass, the second involves many cell types and their coordinated interactions and causes an increase in the number of adipocytes changing the functional properties of the resulting new AT. Both biological processes have an impact on the surrounding extracellular matrix, that influences them, in turn, through its composition and structure and thanks to the presence of other cell types.
In particular AT is crossed by many vessels supplying nutrients, oxygen and signal molecules to mature adipocytes [15], contains several nervous fibers cooperating to the functional shaping and tuning of the tissue [16], and includes some immune cells [17] and progenitor cells near to the capillary structures [18]. All these elements actively contribute to the healthy AT growth and functioning that are different and specific depending on the tissue color (white, brown and beige AT) and its anatomical localization (visceral, VAT, vs subcutaneous, SAT). In this way AT is able to efficiently fit the ever-changing environmental conditions (such as nutrient availability and temperature levels).
Adipose tissue stromal and stem cells (ASCs) have been isolated and characterized from both mice and humans proving their capacity to give rise to different lineages of mesenchymal origin in vitro and in vivo [19, 20]. In particular several researchers contribute to identify their surface markers and tissue localization by means of gold standard techniques such as cytometry, IHC and IF [21]. ASCs belong to the Mesenchymal stromal cells (MSCs) population and have been compared with Bone Marrow and Umbilical –derived stem cells especially for their immunomodulatory properties in the clinical trial setting [22, 23]. In humans, the combination of a few robust markers (CD34 + /CD31−/CD45−) allows for identifying and quantifying the ASC population ex-vivo to study its role in different pathological conditions [24]. Several studies, mainly conducted on animals, evidence a possible hierarchical relationship between ASCs, and suggest the existence of white, brown and beige precursors as well as depot-specific subpopulations of ASCs [25, 26]. Recent studies and new single-cell analysis underline the heterogeneity of ASCs describing new regulatory subpopulations controlling the AT expansion (ASC reg) [18]. The increasing data make the picture more complicated, but greatly enhance the range of possible therapeutic interventions.
The term “adiposopathy” indicates the disease of AT starting from damage to one or more of its components and causing a dysfunction of its metabolic and endocrine function [27], growth capacity and plasticity [28, 29]. Certainly, adiposopathy plays an important role, and according to some authors represents the primum movens in the genesis and development of obesity, a complex disease associated with several metabolic alterations involving different organs, tissues and cell types.
Two of the major signs of adiposopathy are represented by fibrosis and inflammation, which are well documented and deeply characterized in AT. Fibrosis is associated with a dramatic change in the composition and the physical characteristics of the AT extracellular matrix that affects several morphological and functional AT properties and is strongly linked with inflammation. Fibrosis increases in obesity and metabolic complications, both in animal models and humans, and probably involves the activation and proliferation of specific precursor cells (PDGRFα + /CD9hi) [30].
Many immune cells in AT have been described as participating in obesity-associated inflammation, thus obesity should be regarded as an inflammatory disease. Among them macrophages constitute a large proportion, are characterized by a pro-inflammatory M1-like phenotype and are recruited and activated in the AT by inflammatory cytokines and chemokines which are also secreted by dysfunctional adipocytes. Recently the phenotype plasticity of these macrophages has been thoroughly investigated, underlining the link between fuel bioenergetics and polarization, and evidencing their role in modulating lipid, catecholamine and iron availability [31]. Several T cell populations CD4 + (Th1, Th17, Tregs), CD8 + and iNKT are identified as resident immune cells in AT, especially in animal models, with specific pro-inflammatory or anti-inflammatory roles in obesity [17].
We recently showed that people with obesity, characterized by higher Body mass index (BMI), waist and LDL blood level than controls, displayed adipocytes larger in size and a higher number of ASCs both in VAT and in SAT compared to normal weight subjects, attesting that their white AT, independently of its anatomical localization, is able to expand by hypertrophy and hyperplasia to accommodate the excess nutrient intake. The number of capillaries per mm2 present in their AT is reduced compared to controls, probably due to the enlargement of the adipocytes rather than to a reduction in the angiogenic potential. Similar histomorphological evaluations performed on people with obesity and with a concomitant presence of early signs of metabolic complication (prediabetic condition) or stable diagnosed T2D, clearly showed that AT continues to expand by hypertrophy but does not sustain the new adipogenesis decreasing the number of ASCs [32]. Other studies performed both in animal models and human samples showed that hypertrophic adipocytes displayed alterations in metabolic and endocrine functions, expressing and secreting a different adipocytokine pattern with pro-inflammatory and pro-fibrotic effects [33]. Evidence has been collected so far indicating that while AT can preserve its adipogenic potential giving rise to new metabolic healthy small adipocytes, its expansion can be positive and functional [14]. In fact, a decrease in the AT stemness always corresponds to a hazardous unbalance in the growth, giving rise to lipid spillover from AT, alterations of AT endocrine signals and ultimately the development of metabolic complications.
3 Skeletal muscle and the origin of intermuscular adipose tissue
Skeletal muscle changes are not uniformly observed in individuals with obesity and the presence of heterogeneous phenotypes may contribute to their underestimation [34]. Indeed, a positive association between BMI and lean body mass has been reported in general population studies, and moderate increments in skeletal muscle mass may occur in obesity as a consequence of higher postural and ambulatory muscle work as well as potential direct anabolic effects of higher dietary protein intake. However, it is increasingly clear that profound skeletal muscle metabolism changes may occur in some people with obesity and may lead to altered body composition with higher fat mass and substantial impairment of muscle mass and quality [34].
Low muscle function and mass are currently addressed in obese individuals under the definition of sarcopenic obesity. The latter is based on the originally geriatric concept of sarcopenia, i.e. the age-associated combination of declining muscle mass and function (particularly muscle strength) [35]. The concept of sarcopenic obesity has been primarily applied in elderly people with obesity, but increasing evidence suggests that muscle impairment could occur at any age in individuals with obesity in the presence of metabolic complications, chronic comorbidities, acute or critical illness (including cancer), and following bariatric surgery or strict hypocaloric diet [34].
Despite the fact that common shared diagnostic criteria for sarcopenic obesity are still lacking [36], several alterations in skeletal muscle have been described [34]. In particular, lipid accumulation in skeletal muscle cells is considered a typical feature and it has been closely linked with tissue and systemic insulin resistance [37]. Mechanisms mediating metabolic lipotoxicity in skeletal muscle are complex, including direct pro-oxidative and inflammatory activities [38] as well as accumulation of metabolically toxic lipid moieties such as diacylglycerol and ceramides [39]. Lastly, ectopic lipid deposition may also compromise muscle protein turnover [40], promoting an ‘anabolic resistance’ state in which the response of skeletal muscle protein synthesis to nutrients is blunted and loss of muscle mass is promoted [34].
Parallel to lipid accumulation into myocells, the skeletal muscle of people with sarcopenic obesity is characterized by the “infiltration” of the muscular tissue by an increasing amount of adipocytes. The origin of the “new” adipocytes appearing in the muscle is still debated and several hypotheses have been made. Among the different mechanisms that could be responsible for the accumulation of fat in this site, the dysdifferentiation of muscle-derived stem cells or other mesenchymal progenitors has been postulated, turning them into cells with an adipocyte phenotype. In particular, muscle satellite cells (SCs) can acquire features of adipocytes, including the abilities to express adipocyte-specific genes and accumulate lipids. Failure to express the transcription factors that direct mesenchymal precursors into fully differentiated functionally specialized cells may be responsible for their phenotypic switch into the adipogenic lineage. Human SCs also possess this adipogenic potential, thus explaining the presence of mature adipocytes within skeletal muscle. Several pathways and factors (PPARs, WNT growth factors, myokines, GEF-GAP-Rho, p66(shc), mitochondrial ROS production, PKCβ) could be implicated in the adipogenic conversion of SCs [41,42,43,44,45,46]. Moreover, age, oxidative stress, injury, and regeneration can also activate adipogenic differentiation potential in myoblasts affecting fat accumulation in skeletal muscle [47]. This activation of the transdifferentiation of myoblasts into adipocytes can be mediated by several transcriptional factors (like Peroxisome proliferator‐activated receptors (PPARs), PR domain‐containing protein 16 (Prdm16), deletion of MyoD, Bone morphogenetic proteins (BMPs) and others), by the expression of specific MicroRNAs (miRNAs), and by nutritional factors [47]. The switch of muscular precursors from the myogenic to the adipogenic lineages of differentiation can be modulated also by the complex interplay between several interstitial cell types in the regenerating skeletal muscle. For instance, an interstitial CD142-positive cell population has recently been identified by single-cell RNA sequencing in mice and humans responsible for the inhibition of adipogenesis through GDF10 secretion [48]. This CD142-positive cell type is nearly absent in muscular dystrophy, where the interstitial cell composition is completely altered, muscle-cell regeneration is impaired and adipocytes abundant [48]. Moreover, adipogenesis was observed in Fibro-adipogenic cells (FAPs) [49, 50] which are muscle-resident non-myogenic progenitors of mesenchymal origin that proliferate in response to injury and are involved in muscle loss [51].
Alternatively to aberrant adipogenic differentiation of muscle stem cells, the adipocytes infiltrating the skeletal muscle in sarcopenic obesity could be derived by migration of adipocyte precursors from AT. Subcutaneous adipose tissue is able to release ASCs into circulation, under the regulation of the chemokine CXCL12 and its receptor CXCR4 [52]. Triggering the release of adipocyte precursors from subcutaneous AT with a high-fat diet and exposure to a CXCR4 antagonist directly promoted ectopic adipocyte formation in skeletal muscle in mice [53]. These data provide insights into the role of adipocyte progenitor trafficking in promoting ectopic fat deposition.
In conclusion, lipid accumulation and adipocyte infiltration in skeletal muscle is a part of the picture of sarcopenic obesity, typically occurring with aging or in the presence of metabolic diseases. These alterations in muscle structure have an important role in explaining insulin-resistance and loss of muscle strength and performance.
4 Adipose tissue in other organs
4.1 Bone marrow
AT localizes in Bone marrow (BM), where it is called Marrow adipose tissue (MAT) and exists in two forms: constitutive (cMAT) and regulated (rMAT), depending on the ability to respond to external stimuli. The first one is mainly present in the distal skeleton (yellow marrow) and its adipocytes develop soon after birth, possess high levels of CEBPA (CCAAT enhancer binding protein alpha) and CEBPB (CCAAT enhancer binding protein beta) and are filled by mono-unsaturated fatty acids. rMAT -adipocytes (which are relatively small) are enriched in the proximal, hematopoietic skeleton within the red marrow and form more gradually throughout life and expand or reduce in response to endogenous and exogenous stimuli, accumulating saturated fatty acids. The mass of MAT increases in response to pathological conditions, including aging, obesity, T2D and osteoporosis and during radiotherapy or glucocorticoid treatment [54].
Histological assessments and imaging methods describe that MAT in adult humans ranges from 30 to 70% of BM volume and collectively can also reach 8% of total body fat mass [54]. Although the unilocular adipocytes populating the BM were thought to be acting as passive space fillers for a long time, nowadays it is clear that they possess their own molecular signature, do not share any precursors with WAT and BAT and are functionally different from them [55].
Its high representation explains the multiple functions played in the regulation of systemic homeostasis of energy and endocrine mediated by adipokines such as leptin and adiponectin production [56, 57], by the release of free fatty acids [58] or by the regulation of basal glucose uptake [55].
The BM-MSCs are self-renewing and give rise to osteoblasts, chondrocytes and adipocytes [59]. Obviously, an extensive characterization of MAT adipocytes has been carried out, but a precise lineage hierarchy is still lacking. Lineage tracing analyses were not able to clarify whether MAT adipocytes arise from a single population or from a small number of different progenitors in the BM [60,61,62].
The tight constraint between cells in the BM microenvironment explains the influence of MAT on hematopoiesis and skeletal remodeling [63].
MAT is involved in the regulation of renewal, quiescence and differentiation of Hematopoietic stem cells (HSC) which provide for myeloid and lymphoid progenitors [64]. Its increase during ageing and a high-fat diet seems likely to be related to an increased myelopoiesis in association with a reduced lymphopoiesis [65, 66]. In vitro experiments demonstrated that the impairment linked to B-lineage progenitors is exerted by multiple actors: adipocyte-derived soluble factors, IL-1 and activation of inflammosome [67,68,69]. The switch toward myeloid populations induces tissue infiltration of monocytes and their differentiation into proinflammatory macrophages that prompt tissue inflammation and worsen metabolic disease [70]. In obesity, MAT is positively associated to ectopic fat deposition, serum lipid levels [71], and VAT [72,73,74]; it also increases with ageing in people with osteoporosis [75], and pelvic MAT has been inversely correlated with bone volume in younger and older adults [76, 77]. However, MAT displays a dynamic regulation involving the mineral metabolism [78]. Two possible theories have been supported by experimental findings: one is that a common BM progenitor (Osterix + /endosteal and/or LepR + /sinusoidal) is pushed toward adipogenesis at the expense of osteogenesis, sustaining a local feedback loop exacerbating abnormalities [61, 62]. Another possible explanation is that MAT’s endocrine properties change during pathological conditions modifying secreted factors (e.g., RANKL, CXCL1, CXCL2, IL-6, G-CSF) that directly regulate bone remodeling through fine tuning of the osteoblast-osteoclast activity [57], even in the presence of minor alterations in MAT mass.
5 Others
5.1 Heart
The mediastinal or intrathoracic VAT is classified as epicardial (EAT), paracardial (PAT), pericardial (which is epicardial plus paracardial), perivascular (PVAT) and intramyocardial AT. In normal physiological conditions, the EAT provides Free fatty acids (FFAs) to myocardium demands, it secretes vasoactive factors (i.e., leptin, adiponectin, nitric oxide, omentin) and it has thermogenic properties, expressing markers typical of the BAT (i.e., uncoupling protein 1, UCP-1). Several clinical studies have demonstrated a close association between the EAT and the occurrence of Coronary artery disease (CAD) [79] and Heart failure with preserved ejection fraction (HFpEF) [80]. The EAT volume, quantified by computed tomography, correlates with both the presence and the extent of calcified coronary artery plaques [81, 82], and is an independent predictor of early CAD [83]. EAT thickness is significantly correlated with obesity, insulin resistance, waist circumference, blood pressure, sleep apnea severity and dyslipidemia, suggesting that this AT depot could be considered an indicator of cardiovascular risk [84]. Indeed, caloric restriction and bariatric surgery has been demonstrated to reduce EAT [85], thus weight loss in obesity could be targeted to reduce EAT in order to obtain cardiovascular prevention. Coronary arteries are surrounded by PVAT, representing a mechanical support and being a vasocrine and paracrine source of cytokines and adipokines, it also participates in vascular remodeling. Recently, a new imaging approach has been developed for evaluating PVAT inflammation as a determinant of acute plaque rupture [86]. One of the possible involved mechanisms linking EAT and CAD is proximity, whereas the atherosclerotic lesions were preferentially localized in coronary arteries surrounded by AT. This closeness allows for a paracrine EAT-myocardium crosstalk, by direct diffusion of proinflammatory cytokines and other adipokines [87]. Conversely, cytokines from the EAT may be directly released into the vasa vasorum of the coronary arterial wall [88]. Consequently, it was hypothesized that EAT dysfunction, with a great number of inflammatory cells and a high expression of inflammatory cytokines, induces inflammation of adjacent arteries and thus contributes to the pathogenesis of atherosclerosis, plaque rupture, and thrombosis [89]. Several reports tried to address to the Cardiac adipocytes (CAs) origin-question, and recently it was demonstrated that PDGFRa+ and PDGFRb+ cardiac Mesenchymal cells (MCs) give rise to the intramyocardial CAs during development [90]. As in muscle, mesenchymal progenitors give rise to FAPs also in the heart [91] and cardiac stromal fibroblasts have been recognized as covering an important role both in health and disease. Acute ischemic injury or age seem to promote only fibrogenic differentiation, conversely, obesity pushes both fibrotic and adipogenic programs in FAPs [92].
5.2 Thymus
The thymus is one of the pillars of adaptive immunity. At the time of puberty, after an active period of production of new T-cells, the thymus loses the stroma and fills with fat (thymic AT, TAT). This involution decreases T-cell response to antigens. Conversely, the antigen-independent proliferation of T-cell is maintained and it may lead to dysfunctional cells, secreting proinflammatory factors [93]. Obesity accelerates the age-related reduction of T-cell receptors (TCRs). In fact, diet-induced obesity reduces thymocyte counts and significantly increases apoptosis of developing T-cell populations. Moreover, the TCR spectra typing analysis showed that obesity also compromised TCR repertoire diversity [94]. Indeed, it was demonstrated that leptin runs a thymoprotective role, not directly but rather as a secondary effect of suppression of AT expansion [95]. Lastly, the ASCs have been shown to increase expression of immunosuppressive markers in the thymus, preventing lymphocyte differentiation and promoting survival of T cells in a quiescent state [96].
6 Peritumoral adipose tissue
ASCs have a crucial role in cancer [97]. They can be chemoattracted to solid cancers and secrete several cytokines in a paracrine fashion, among them IL6 and Vascular endothelial growth factor (VEGF), which are able to promote the inflammatory microenvironment and further increase tumorigenesis, nutrient and oxygen metabolism [98], thus increasing cancer proliferation, survival and spreading.
Emerging evidence indicates a link between obesity and cancer progression [99]. Tumor microenvironment (TME) is defined as a unique metabolic niche, containing tumor, immune and stromal cells. Age-related immune deterioration is exacerbated by obesity in animal and human studies and impacts also on the metabolic landscape of TME. It was suggested that peritumoral AT (cancer associated adipocytes, CAAs) contributes to tumor initiation, growth and invasion in different kinds of tumors [100, 101].
Furthermore, peritumoral AT may promote, by paracrine signaling, the expression of depot-specific factors associated with therapeutic resistance [102]. A significant decrease in T cell proliferative capacity and a significant deficit in IFNγ and Tumor necrosis factor (TNF) production in diet-induced obese mice has been reported. This mechanism is partially mediated by leptin [103]. It was shown that a high-fat diet accelerates tumor growth and reduces the number and the function of intratumoral CD8 + T cells in mice due to altered fatty acid portioning and local depletion of essential metabolites [104].
7 Autocrine, paracrine and endocrine crosstalk alterations
The biological complexity of obesity is centered on information-based intricacy: the disruption of the information system controlling food intake and energy expenditure as well as the inter-organ crosstalk or dialogue could be seen as the major determinant in the obesity pathogenesis [2, 3, 6]. The fundamental nature of the communication process is characterized by the storage, transmission, and utilization of information, coding and decoding of the signals and their nature or origin [105, 106]. One of the principal contributions in the last three decades to the field of communication has been in the area of cybernetics, from which information theory is derived [107]. It can be considered today, together with artificial intelligence, as a system that acts to achieve a goal [108]. Cybernetics is the science of the feedback system, such as maintaining the temperature level or blood glucose concentration [109]. The system takes action to correct the differences between the current state and the goal through feedback signals. This process helps ensure stability when the perturbations threaten second order dynamic systems, which, observing the results of this perturbation within themselves, can change their goals. Second-order systems don’t just react; they can also learn, activating amongst them a conversation or a dialogue, an exchange on objectives and means.
It is possible to recognize in the various organs that make up the human body the single sections of an orchestra. The orchestra needs a code represented by the score to which each individual instrument refers, and also a conductor who moves all the sounds of the various instruments in a coordinated manner. The communication between the conductor and the sections (inter-organ crosstalk) and of the musicians amongst themselves (intra-organ crosstalk) is fundamental to harmonize every single note in a symphony. It is important to try to decode these communication systems to understand how homeostasis is preserved.
One of the most relevant reasons for a preferential inter-organ crosstalk is based on the developmental common origin of AT muscle and bone. Mesenchymal stem cells give rise to three main cell lineages by an early differentiation in osteoblast, adipoblasts and myoblasts which represent already committed stem cells of the three specific lineages which lead to the mature cell phenotype of osteocyte, adipocyte and myocyte [110].
AT, besides its ability to store blood nutrients as glucose and fatty acids, releases plenty of peptides, collectively called adipokines, and several metabolites as lactate and fatty acids and lipid signals / mediators (Fig. 2). The flux of lipid substrates to the adipose cell is a sensing mechanism of an increased availability of energy, transduced by specific receptors or transporters and evoking the release of leptin which transfers this information to the target hypothalamic structures [111,112,113]. The preferential channeling of fuel substrates toward AT rather than muscle could be determined by the decrease in oxidative capacity of skeletal muscle because of a sedentary lifestyle, aging or mitochondrial or genetic alterations [114]. This overflow of substrate continues until the expandability capacity of the adipose organ has been overcome due to the appearance of fat cell insulin resistance, when both long-term and short-term regulation and delivery of stored lipids in response to the peripheral energy need is no longer finely controlled and qualitative and/or quantitative changes in adipokine production are triggered.

Pathological adipose tissue endocrine signals. Adipose tissue synthesizes and delivery a complex array of molecules including cyto/adipokines, metabolites and lypokines, growth factors, extracellular vesicles (exosomes) and miRNAs which flow throughout the bloodstream and reach the target organs. The pathological increase of adipose tissue induces qualitative and quantitative changes in the production of its signals, provoking an overall alteration of the long-range communication, ending in the impairment of multiple organs and their functions. FABP4 fatty acid-binding protein 4, BCAAs branched-chain aminoacids, FAHFAs fatty acid esters of hydroxy fatty acids, diHOMEs dihydroxy octadecenoic acids, IL6 interleukin 6, TNFα tumor necrosis factor alpha, BMPs bone morphogenetic proteins, FGF21 fibroblast growth factor 21. Created with BioRender.com
In AT, autocrine and paracrine communication between the different adipocyte phenotypes, vasculo-stromal cells and immune cells could be of crucial importance in controlling AT biology, hypoxia, low‐grade chronic inflammation and fibrosis [115] (Fig. 3).

Pathological paracrine and autocrine signals mediated by adipocytes worsen functional and structural tissue remodeling. A Secreted adipocyte-factors (different color spheres) act on the diverse cellular component of adipose tissue. Pathological adipocytes induce recruitment and activation of macrophages causing a pro-inflammatory microenvironment. When they engage adipose progenitors, promote the differentiation of new mature cells inducing hyperplasia. During diabetes, the reduced ability to store glucose by dysfunctional adipocytes, increase capillary basement membrane thickness favoring hypoxia. Finally, adipocytes can directly signal to themselves promoting hypertrophy. B During obesity or ageing adipose tissue appears in muscle, involving satellite cells (SCs) conversion to adipocytes and recruiting fibroadipogenic precursors. These are able to reduce the differentiation of SCs in myocytes, activate a fibrotic program and to enroll inflammatory cells, all these events in turn induces muscle loss and reduced strength. C Pathological signals released by bone marrow adipocytes act on hematopoietic stem cells, impairing their self-renewal and differentiation abilities, promoting myelopoiesis. In the meantime, adipocytes stimulate their accumulation recruiting mesenchymal stromal cells toward new adipogenesis at the expense of osteogenesis. In addition, osteoblast and osteoclast activities are directly altered by bone marrow adipocytes which cause bone remodeling. Red arrow: activation, Blue arrow: inhibition, Created with BioRender.com
The accumulation of lipids and AT in skeletal muscles alters the secretion of several myokines acting both locally (IL-4, IL-6, VEGF, IGF-1 and irisin) and systemically (GDF8/myostatin, FGF21 and IL-6) which mediate the beneficial effects of exercise on other organs [116,117,118] and are involved in the regulation of energy homeostasis by AT itself, such as thermogenesis activation and lipid mobilization [119]. Moreover, a progressive decline in muscle myokines may contribute to the development of aging-related sarcopenia [120], as in the case of apelin which regulates muscle hypertrophy. In sarcopenic obesity the increased the presence of fat within skeletal muscle with structural and functional impairment of muscle may also affect bone density leading to the presence of osteoporosis. Myostatin, not only decreases muscle mass [121], but also negatively regulates bone mass. In adults, irisin was also (negatively) associated with vertebral fragility fractures [122] and sarcopenia in post-menopausal women, [123] suggesting that irisin plays a crucial role in bone metabolism to a greater extent when compared with its original effect on AT.
Osteocalcin (OCN) is a protein secreted in a carboxylated form (cOCN) by osteoblasts and is responsible for the matrix mineralization and bone formation, while the undercarboxylated form (ucOCN) controls numerous physiological processes in an endocrine fashion [124]. People who are overweight and people with obesity had a lower ucOCN and ucOC/OCN ratio negatively correlated to BMI. OCN is also expressed and released by subcutaneous and omental AT and during all stages of adipogenesis [125]. OCN is able to influence AT function by inducing adiponectin expression in adipocytes [125, 126]. The elevation in osteocalcin increases in humans during physical activity [127], increasing fatty acid transporter gene expression and β-oxidation, and promoting glucose uptake. During ageing its levels decrease, but experimental data displayed that the hormone prevents muscle loss regulating the protein turnover [128]. Bone and skeletal muscle could be considered as cooperating as a single functional unit. The presence of osteoporosis often parallels sarcopenia in many chronic diseases including obesity [34, 36], the decline in muscle function probably being the principal factor leading to the loss of bone mass. Moreover it cannot be excluded that the major trigger in inducing this dual impairment could be the development of adiposophaty and even more importantly the presence of ectopic AT within skeletal muscle and the excessive growth of AT inside the bone marrow, giving rise to a whole body or local autocrine/paracrine cytokine storm, supporting once again the hypothesis of inflammaging and metaflammation in the development of aging related diseases including osteoporosis and sarcopenia [129]. The mechanical loading due to the increased BMI results in increased bone mineral density, but its putative protective effect against fractures is not present in all skeletal sites, moreover it is well known that sarcopenic obese subjects display a reduction in bone mass. The relationship between obesity and bone health is complex and not completely elucidated. Obesity is thought to affect bone health through a variety of mechanisms such as mechanical loading, obesity phenotype, the distribution of AT, the secreted cytokines, gender, age, and bone sites. In fact, the risk for some non-vertebral fractures is higher, whereas the risk of vertebral and proximal femur fractures is lower in individuals with obesity as compared with subjects with normal weight [130, 131]. Obesity can initially increase bone mass, but later bone formation is unable to maintain the bone causing a reduction in its quality over the time [132].
8 Concluding remarks
Adiposopathy or adipose organ dysfunction, whose hallmarks are represented by qualitative and quantitative changes in fat depots whether it is subcutaneous or visceral, white or brown, leads to and resides also in the ectopic AT. The main features are characterized by changes in the localization and morphology (anatomical distribution, the brown to white fat differentiation, increased of adipocyte size, fibrosis), function (decreased insulin sensitivity, failure of the lipid storage rising peripheral lipotoxicity, mitochondrial impairment), secretion (altered adipokine production) and cellular composition (reduced neoangiogenesis, macrophage infiltration, stem cell abnormalities), which appear stepwise [115].
It is possible to think that the continuous overflow of energy substrates, once the storage capacity and the expandability property of AT have been overcome, could ultimately result in a lipid infiltration within organs such as the liver, muscle, and heart or evoke the adipogenic differentiation of cell precursors or adult stem cells normally able to differentiate toward lineage-specific cell types of the tissue or organ in which they reside, regulating homeostasis, regeneration, and aging of all tissues. The chronic exposition of these newly-formed adipose depots (mainly WAT) to a pathological environment, will confer to them all the phenotypic characteristics of a tissue affected by adiposopathy, perpetuating the organ damage. Adult stem cell senescence has emerged as an attractive theory for the decline in mammalian tissue and organ dysfunction of a variety of cardiovascular and metabolic diseases [133,134,135,136]. In this view, a better characterization of human AT progenitors and the precise definition of their recruitment and regulation could improve the possibility of targeting specific pathways acting in the AT expansion [28, 137, 138]. A further fascinating mechanism explaining the origin and growth of ectopic AT could be the colonization by ASCs of other organs, such as muscle or bone marrow. It is likely that this could happen because of the common embryological origin of the three tissues. The majority of adipocytes originate from the mesoderm and are organized into anatomically distinct depots. ASCs play important roles in tissue regeneration, remodeling, and homeostasis and are able to differentiate into different lineages, such as adipocytes, cardiomyocytes, osteoblasts, chondrocytes and myocytes [139, 140]. Recently, it was demonstrated in mice that the release of ASCs from subcutaneous AT directly affects ectopic adipocyte deposition in muscle, and ongoing studies focus on demonstrating that this mechanism is not restricted to skeletal muscle, suggesting that AT could bypass its expandability limitation allowing the colonization of non-AT organs, to store lipids in a competent cell [53].
Pathological conditions such as aging, as well as congenital lipodystrophy, and, to some extent, even obesity in humans, are accompanied by a reduction or loss or limitation in the expandability of fat with an accumulation of adipose cells and lipids in non-adipose depots, such as bone marrow, liver, and skeletal muscle. This accumulation worsens the functional performance of the target organs, especially in muscle and bone. For both, mass and strength decline with aging and evidence suggests that many of the factors observed to stimulate bone marrow adipogenesis (leptin deficiency, disuse atrophy, lack of estrogen and glucocorticoid treatment) also induce myosteatosis [141]. In the same way, many of the conditions that induce marrow adipogenesis and bone loss in men and women also stimulate the accumulation of adipocytes and Intramyocellular lipid (IMCL) in skeletal muscle [142]. Ectopic AT accumulation represents a predictor of organ disfunction, for example IMAT defines muscle metabolism and function but it also seems a modifiable risk factor, therefore, the clarification of the mechanisms at the basis of ectopic fat formation may provide new potential targets in the treatment of metabolic diseases. At present, exercise and physical activity appear to be effective countermeasures against the accumulation of AT, even if it is not currently possible to completely revert the effects of aging [143, 144]. Moreover, calorie restriction may also reduce fatty infiltration and potentiate the effect of exercise [145]. Pharmacological intervention combined with lifestyle changes to delay or reverse myosteatosis is still largely unexplored, for instance, treatment with an antimyostatin antibody capable of blocking myostatin signaling to treat people with muscle loss, but its effect on IMAT loss is still far from being considered an effective therapeutic strategy [146].
In conclusion, adipose tissue within as well as surrounding crucial organs has been identified as an important determinant of target organ dysfunction. Its stemness and composition, secretome, and location define its function in health and metabolic disease (Table 1). Preventing the appearance of dysfunctional AT is certainly the prerequisite to avoiding the progression towards the most important complications of obesity and other nutrition related diseases.
References
Choe SS, Huh JY, Hwang IJ, Kim JI, Kim JB. Adipose tissue remodeling: its role in energy metabolism and metabolic disorders. Front Endocrinol. 2016;7:30.
Vegiopoulos A, Rohm M, Herzig S. Adipose tissue: between the extremes. EMBO J. 2017;36:1999–2017.
Kahn CR, Wang G, Lee KY. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J Clin Invest. 2019;129:3990–4000.
Pillon NJ, Loos RJF, Marshall SM, Zierath JR. Metabolic consequences of obesity and type 2 diabetes: balancing genes and environment for personalized care. Cell. 2021;184:1530–44.
Qasim A, Turcotte M, de Souza RJ, Samaan MC, Champredon D, Dushoff J, et al. On the origin of obesity: identifying the biological, environmental and cultural drivers of genetic risk among human populations. Obes Rev. 2018;19:121–49.
Kim K-S, Seeley RJ, Sandoval DA. Signalling from the periphery to the brain that regulates energy homeostasis. Nat Rev Neurosci. 2018;19:185–96.
Schwartz MW, Porte D. Diabetes, obesity, and the brain. Science. 2005;307:375–9.
GBD 2015 Obesity Collaborators, Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, et al. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. 2017;377:13–27.
Longo M, Zatterale F, Naderi J, Parrillo L, Formisano P, Raciti GA, et al. Adipose tissue dysfunction as determinant of obesity-associated metabolic complications. Int J Mol Sci. 2019;20:E2358.
Schoettl T, Fischer IP, Ussar S. Heterogeneity of adipose tissue in development and metabolic function. J Exp Biol. 2018;221:jeb162958.
Cinti S. The adipose organ at a glance. Dis Model Mech. 2012;5:588–94.
Heilbronn L, Smith SR, Ravussin E. Failure of fat cell proliferation, mitochondrial function and fat oxidation results in ectopic fat storage, insulin resistance and type II diabetes mellitus. Int J Obes Relat Metab Disord. 2004;28(Suppl 4):S12-21.
Zwick RK, Guerrero-Juarez CF, Horsley V, Plikus MV. Anatomical, physiological, and functional diversity of adipose tissue. Cell Metab. 2018;27:68–83.
Ghaben AL, Scherer PE. Adipogenesis and metabolic health. Nat Rev Mol Cell Biol. 2019;20:242–58.
Paavonsalo S, Hariharan S, Lackman MH, Karaman S. Capillary rarefaction in obesity and metabolic diseases-organ-specificity and possible mechanisms. Cells. 2020;9.
Guilherme A, Henriques F, Bedard AH, Czech MP. Molecular pathways linking adipose innervation to insulin action in obesity and diabetes mellitus. Nat Rev Endocrinol. 2019;15:207–25.
Liu R, Nikolajczyk BS. Tissue immune cells fuel obesity-associated inflammation in adipose tissue and beyond. Front Immunol. 2019;10:1587.
Ferrero R, Rainer P, Deplancke B. Toward a consensus view of mammalian adipocyte stem and progenitor cell heterogeneity. Trends Cell Biol. 2020;30:937–50.
Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, et al. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. 2001;7:211–28.
Zuk PA, Zhu M, Ashjian P, De Ugarte DA, Huang JI, Mizuno H, et al. Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell. 2002;13:4279–95.
Zimmerlin L, Donnenberg VS, Rubin JP, Donnenberg AD. Mesenchymal markers on human adipose stem/progenitor cells. Cytometry A. 2013;83:134–40.
Ceccarelli S, Pontecorvi P, Anastasiadou E, Napoli C, Marchese C. Immunomodulatory effect of adipose-derived stem cells: the cutting edge of clinical application. Front Cell Dev Biol. 2020;8:236.
Strioga M, Viswanathan S, Darinskas A, Slaby O, Michalek J. Same or not the same? Comparison of adipose tissue-derived versus bone marrow-derived mesenchymal stem and stromal cells. Stem Cells Dev. 2012;21:2724–52.
Guimarães-Camboa N, Evans SM. Are perivascular adipocyte progenitors mural cells or adventitial fibroblasts? Cell Stem Cell. 2017;20:587–9.
Tchkonia T, Giorgadze N, Pirtskhalava T, Thomou T, DePonte M, Koo A, et al. Fat depot-specific characteristics are retained in strains derived from single human preadipocytes. Diabetes. 2006;55:2571–8.
Sanchez-Gurmaches J, Guertin DA. Adipocytes arise from multiple lineages that are heterogeneously and dynamically distributed. Nat Commun. 2014;5:4099.
Scheja L, Heeren J. The endocrine function of adipose tissues in health and cardiometabolic disease. Nat Rev Endocrinol. 2019;15:507–24.
Morigny P, Boucher J, Arner P, Langin D. Lipid and glucose metabolism in white adipocytes: pathways, dysfunction and therapeutics. Nat Rev Endocrinol. 2021;17:276–95.
Vishvanath L, Gupta RK. Contribution of adipogenesis to healthy adipose tissue expansion in obesity. J Clin Invest. 2019;129:4022–31.
Marcelin G, Silveira ALM, Martins LB, Ferreira AV, Clément K. Deciphering the cellular interplays underlying obesity-induced adipose tissue fibrosis. J Clin Invest. 2019;129:4032–40.
Caslin HL, Bhanot M, Bolus WR, Hasty AH. Adipose tissue macrophages: unique polarization and bioenergetics in obesity. Immunol Rev. 2020;295:101–13.
Belligoli A, Compagnin C, Sanna M, Favaretto F, Fabris R, Busetto L, et al. Characterization of subcutaneous and omental adipose tissue in patients with obesity and with different degrees of glucose impairment. Sci Rep. 2019;9:11333.
Liu F, He J, Wang H, Zhu D, Bi Y. Adipose morphology: a critical factor in regulation of human metabolic diseases and adipose tissue dysfunction. Obes Surg. 2020;30:5086–100.
Barazzoni R, Bischoff S, Boirie Y, Busetto L, Cederholm T, Dicker D, et al. Sarcopenic obesity: time to meet the challenge. Obes Facts. 2018;11:294–305.
Cruz-Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, et al. Sarcopenia: European consensus on definition and diagnosis: report of the European working group on Sarcopenia in older people. Age Ageing. 2010;39:412–23.
Donini LM, Busetto L, Bauer JM, Bischoff S, Boirie Y, Cederholm T, et al. Critical appraisal of definitions and diagnostic criteria for sarcopenic obesity based on a systematic review. Clin Nutr. 2020;39:2368–88.
Mingrone G, Rosa G, Di Rocco P, Manco M, Capristo E, Castagneto M, et al. Skeletal muscle triglycerides lowering is associated with net improvement of insulin sensitivity, TNF-alpha reduction and GLUT4 expression enhancement. Int J Obes Relat Metab Disord. 2002;26:1165–72.
Barazzoni R, Zanetti M, Gortan Cappellari G, Semolic A, Boschelle M, Codarin E, et al. Fatty acids acutely enhance insulin-induced oxidative stress and cause insulin resistance by increasing mitochondrial reactive oxygen species (ROS) generation and nuclear factor-κB inhibitor (IκB)-nuclear factor-κB (NFκB) activation in rat muscle, in the absence of mitochondrial dysfunction. Diabetologia. 2012;55:773–82.
Lipina C, Hundal HS. Lipid modulation of skeletal muscle mass and function. J Cachexia Sarcopenia Muscle. 2017;8:190–201.
Tardif N, Salles J, Guillet C, Tordjman J, Reggio S, Landrier JF, et al. Muscle ectopic fat deposition contributes to anabolic resistance in obese sarcopenic old rats through eIF2α activation. Aging Cell. 2014;13:1001–11.
Vettor R, Milan G, Franzin C, Sanna M, De Coppi P, Rizzuto R, et al. The origin of intermuscular adipose tissue and its pathophysiological implications. Am J Physiol Endocrinol Metab. 2009;297:E987–998.
De Coppi P, Milan G, Scarda A, Boldrin L, Centobene C, Piccoli M, et al. Rosiglitazone modifies the adipogenic potential of human muscle satellite cells. Diabetologia. 2006;49:1962–73.
Rossi CA, Pozzobon M, Ditadi A, Archacka K, Gastaldello A, Sanna M, et al. Clonal characterization of rat muscle satellite cells: proliferation, metabolism and differentiation define an intrinsic heterogeneity. PLoS One. 2010;5:e8523.
Scarda A, Franzin C, Milan G, Sanna M, Dal Prà C, Pagano C, et al. Increased adipogenic conversion of muscle satellite cells in obese Zucker rats. Int J Obes. 2010;34:1319–27.
Aguiari P, Leo S, Zavan B, Vindigni V, Rimessi A, Bianchi K, et al. High glucose induces adipogenic differentiation of muscle-derived stem cells. Proc Natl Acad Sci U S A. 2008;105:1226–31.
Thornell LE. Sarcopenic obesity: satellite cells in the aging muscle. Curr Opin Clin Nutr Metab Care. 2011;14:22–7.
Wang L, Shan T. Factors inducing transdifferentiation of myoblasts into adipocytes. J Cell Physiol. 2021;236:2276–89.
Camps J, Breuls N, Sifrim A, Giarratana N, Corvelyn M, Danti L, et al. Interstitial cell remodeling promotes aberrant adipogenesis in dystrophic muscles. Cell Rep. 2020;31:107597.
Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol. 2010;12:143–52.
Uezumi A, Fukada S, Yamamoto N, Ikemoto-Uezumi M, Nakatani M, Morita M, et al. Identification and characterization of PDGFRα+ mesenchymal progenitors in human skeletal muscle. Cell Death Dis. 2014;5:e1186.
Hogarth MW, Defour A, Lazarski C, Gallardo E, Diaz Manera J, Partridge TA, et al. Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nat Commun. 2019;10:2430.
Gil-Ortega M, Garidou L, Barreau C, Maumus M, Breasson L, Tavernier G, et al. Native adipose stromal cells egress from adipose tissue in vivo: evidence during lymph node activation. Stem Cells. 2013;31:1309–20.
Girousse A, Gil-Ortega M, Bourlier V, Bergeaud C, Sastourné-Arrey Q, Moro C, et al. The release of adipose stromal cells from subcutaneous adipose tissue regulates ectopic intramuscular adipocyte deposition. Cell Rep. 2019;27:323-333.e5.
Scheller EL, Cawthorn WP, Burr AA, Horowitz MC, MacDougald OA. Marrow adipose tissue: trimming the fat. Trends Endocrinol Metab. 2016;27:392–403.
Suchacki KJ, Tavares AAS, Mattiucci D, Scheller EL, Papanastasiou G, Gray C, et al. Bone marrow adipose tissue is a unique adipose subtype with distinct roles in glucose homeostasis. Nat Commun. 2020;11:3097.
Cawthorn WP, Scheller EL, Learman BS, Parlee SD, Simon BR, Mori H, et al. Bone marrow adipose tissue is an endocrine organ that contributes to increased circulating adiponectin during caloric restriction. Cell Metab. 2014;20:368–75.
Sulston RJ, Cawthorn WP. Bone marrow adipose tissue as an endocrine organ: close to the bone? Hormone molecular biology and clinical investigation. De Gruyter. 2016;28:21–38.
Scheller EL, Khandaker S, Learman BS, Cawthorn WP, Anderson LM, Pham HA, et al. Bone marrow adipocytes resist lipolysis and remodeling in response to β-adrenergic stimulation. Bone. 2019;118:32–41.
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–7.
Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Domínguez Á, et al. The bone marrow microenvironment at single-cell resolution. Nature. 2019;569:222–8.
Yue R, Zhou BO, Shimada IS, Zhao Z, Morrison SJ. Leptin receptor promotes adipogenesis and reduces osteogenesis by regulating mesenchymal stromal cells in adult bone marrow. Cell Stem Cell. 2016;18:782–96.
Mizoguchi T, Pinho S, Ahmed J, Kunisaki Y, Hanoun M, Mendelson A, et al. Osterix marks distinct waves of primitive and definitive stromal progenitors during bone marrow development. Dev Cell. 2014;29:340–9.
Ambrosi TH, Scialdone A, Graja A, Gohlke S, Jank AM, Bocian C, et al. Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell. 2017;20:771-784.e6.
Zhou BO, Yu H, Yue R, Zhao Z, Rios JJ, Naveiras O, et al. Bone marrow adipocytes promote the regeneration of stem cells and hematopoiesis by secreting SCF. Nat Cell Biol. 2017;19:891–903.
Bowers E, Singer K. Obesity-induced inflammation: the impact of the hematopoietic stem cell niche. JCI Insight. 2021;6:145295.
Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature. 2009;460:259–63.
Bilwani FA, Knight KL. Adipocyte-derived soluble factor(s) inhibits early stages of B lymphopoiesis. J Immunol. 2012;189:4379–86.
Kennedy DE, Knight KL. Inhibition of B lymphopoiesis by adipocytes and IL-1–producing myeloid-derived suppressor cells. J Immunol. 2015;195:2666–74.
Kennedy DE, Knight KL. Inflammatory changes in bone marrow microenvironment associated with declining B lymphopoiesis. JI. 2017;198:3471–9.
Griffin C, Eter L, Lanzetta N, Abrishami S, Varghese M, McKernan K, et al. TLR4, TRIF, and MyD88 are essential for myelopoiesis and CD11c+ adipose tissue macrophage production in obese mice. J Biol Chem. 2018;293:8775–86.
Bredella MA, Gill CM, Gerweck AV, Landa MG, Kumar V, Daley SM, et al. Ectopic and serum lipid levels are positively associated with bone marrow fat in obesity. Radiology. 2013;269:534–41.
Bredella MA, Torriani M, Ghomi RH, Thomas BJ, Brick DJ, Gerweck AV, et al. Vertebral bone marrow fat is positively associated with visceral fat and inversely associated with IGF-1 in obese women. Obesity. 2011;19:49–53.
Hasic D, Lorbeer R, Bertheau RC, Machann J, Rospleszcz S, Nattenmüller J, et al. Vertebral bone marrow fat is independently associated to VAT but not to SAT: KORA FF4—whole-body MR imaging in a population-based cohort. Nutrients. 2020;12:1527.
Fazeli PK, Bredella MA, Pachon-Peña G, Zhao W, Zhang X, Faje AT, Resulaj M, Polineni SP, Holmes TM, Lee H, O'Donnell EK, MacDougald OA, Horowitz MC, Rosen CJ, Klibanski A. The dynamics of human bone marrow adipose tissue in response to feeding and fasting. JCI Insight. 22 Jun 2021;6(12):e138636.
Justesen J, Stenderup K, Ebbesen EN, Mosekilde L, Steiniche T, Kassem M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology. 2001;2:165–71.
Shen W, Chen J, Gantz M, Punyanitya M, Heymsfield SB, Gallagher D, et al. MRI-measured pelvic bone marrow adipose tissue is inversely related to DXA-measured bone mineral in younger and older adults. Eur J Clin Nutr. 2012;66:983–8.
Bani Hassan E, Demontiero O, Vogrin S, Ng A, Duque G. Marrow adipose tissue in older men: association with visceral and subcutaneous fat, bone volume, metabolism, and inflammation. Calcif Tissue Int. 2018;103:164–74.
Fazeli PK, Faje AT, Bredella MA, Polineni S, Russell S, Resulaj M, et al. Changes in marrow adipose tissue with short-term changes in weight in premenopausal women with anorexia nervosa. Eur J Endocrinol. 2019;180:189–99.
Madonna R, Massaro M, Scoditti E, Pescetelli I, De Caterina R. The epicardial adipose tissue and the coronary arteries: dangerous liaisons. Cardiovasc Res. 2019;115:1013–25.
Ayton SL, Gulsin GS, McCann GP, Moss AJ. Epicardial adipose tissue in obesity-related cardiac dysfunction. Heart. 2021;heartjnl-2020-318242.
Rosito GA, Massaro JM, Hoffmann U, Ruberg FL, Mahabadi AA, Vasan RS, et al. Pericardial fat, visceral abdominal fat, cardiovascular disease risk factors, and vascular calcification in a community-based sample: the framingham heart study. Circulation. 2008;117:605–13.
Kim BJ, Kang JG, Lee SH, Lee JY, Sung KC, Kim BS, et al. Relationship of echocardiographic epicardial fat thickness and epicardial fat volume by computed tomography with coronary artery calcification: data from the CAESAR study. Arch Med Res. 2017;48:352–9.
Alexopoulos N, McLean DS, Janik M, Arepalli CD, Stillman AE, Raggi P. Epicardial adipose tissue and coronary artery plaque characteristics. Atherosclerosis. 2010;210:150–4.
Powell-Wiley TM, Poirier P, Burke LE, Després JP, Gordon-Larsen P, Lavie CJ, et al. Obesity and cardiovascular disease: a scientific statement from the American heart association. Circulation. 2021;143:e984-1010.
Rabkin SW, Campbell H. Comparison of reducing epicardial fat by exercise, diet or bariatric surgery weight loss strategies: a systematic review and meta-analysis. Obes Rev. 2015;16:406–15.
Goeller M, Achenbach S, Cadet S, Kwan AC, Commandeur F, Slomka PJ, et al. Pericoronary adipose tissue computed tomography attenuation and high-risk plaque characteristics in acute coronary syndrome compared with stable coronary artery disease. JAMA Cardiol. 2018;3:858–63.
Patel VB, Shah S, Verma S, Oudit GY. Epicardial adipose tissue as a metabolic transducer: role in heart failure and coronary artery disease. Heart Fail Rev. 2017;22:889–902.
Yudkin JS, Eringa E, Stehouwer CDA. Vasocrine signalling from perivascular fat: a mechanism linking insulin resistance to vascular disease. Lancet. 2005;365:1817–20.
Vilahur G, Ben-Aicha S, Badimon L. New insights into the role of adipose tissue in thrombosis. Cardiovasc Res. 2017;113:1046–54.
Jiang Z, Feng T, Lu Z, Wei Y, Meng J, Lin CP, et al. PDGFRb+ mesenchymal cells, but not NG2+ mural cells, contribute to cardiac fat. Cell Rep. 2021;34:108697.
Lombardi R, Chen SN, Ruggiero A, Gurha P, Czernuszewicz GZ, Willerson JT, et al. Cardiac fibro-adipocyte progenitors express desmosome proteins and preferentially differentiate to adipocytes upon deletion of the desmoplakin gene. Circ Res. 2016;119:41–54.
Soliman H, Rossi FMV. Cardiac fibroblast diversity in health and disease. Matrix Biol. 2020;91–92:75–91.
Moskalev A, Stambler I, Caruso C. Innate and adaptive immunity in aging and longevity: the foundation of resilience. Aging Dis. 2020;11:1363–73.
Yang H, Youm YH, Vandanmagsar B, Rood J, Kumar KG, Butler AA, et al. Obesity accelerates thymic aging. Blood. 2009;114:3803–12.
Sreenivasan J, Schlenner S, Franckaert D, Dooley J, Liston A. The thymoprotective function of leptin is indirectly mediated via suppression of obesity. Immunology. 2015;146:122–9.
Castro LL, Kitoko JZ, Xisto DG, Olsen PC, Guedes HLM, Morales MM, et al. Multiple doses of adipose tissue-derived mesenchymal stromal cells induce immunosuppression in experimental asthma. Stem Cells Transl Med. 2020;9:250–60.
Lazennec G, Lam PY. Recent discoveries concerning the tumor - mesenchymal stem cell interactions. Biochim Biophys Acta. 2016;1866:290–9.
Freese KE, Kokai L, Edwards RP, Philips BJ, Sheikh MA, Kelley J, et al. Adipose-derived stems cells and their role in human cancer development, growth, progression, and metastasis: a systematic review. Cancer Res. 2015;75:1161–8.
Strong AL, Burow ME, Gimble JM, Bunnell BA. Concise review: The obesity cancer paradigm: exploration of the interactions and crosstalk with adipose stem cells. Stem Cells. 2015;33:318–26.
Trevellin E, Scarpa M, Carraro A, Lunardi F, Kotsafti A, Porzionato A, et al. Esophageal adenocarcinoma and obesity: peritumoral adipose tissue plays a role in lymph node invasion. Oncotarget. 2015;6:11203–15.
Colleluori G, Perugini J, Barbatelli G, Cinti S. Mammary gland adipocytes in lactation cycle, obesity and breast cancer. Rev Endocr Metab Disord. 2021;22:241–55.
Carraro A, Trevellin E, Fassan M, Kotsafti A, Lunardi F, Porzionato A, et al. Esophageal adenocarcinoma microenvironment: peritumoral adipose tissue effects associated with chemoresistance. Cancer Sci. 2017;108:2393–404.
Wang Z, Aguilar EG, Luna JI, Dunai C, Khuat LT, Le CT, et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat Med. 2019;25:141–51.
Ringel AE, Drijvers JM, Baker GJ, Catozzi A, García-Cañaveras JC, Gassaway BM, et al. Obesity shapes metabolism in the tumor microenvironment to suppress anti-tumor immunity. Cell. 2020;183:1848-1866.e26.
Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. General principles of cell communication. Molecular biology of the cell 4th edition [Internet]. Garland Science. 2002. Available from https://www.ncbi.nlm.nih.gov/books/NBK26813/. Accessed 13 Aug 2021.
Brier S. Finding an information concept suited for a universal theory of information. Prog Biophys Mol Biol. 2015;119:622–33.
Wiener N. Cybernetics. Sci Am. 1948;179:14–8.
Collins GS, Moons KGM. Reporting of artificial intelligence prediction models. Lancet. 2019;393:1577–9.
Alcocer-Cuarón C, Rivera AL, Castaño VM. Hierarchical structure of biological systems: a bioengineering approach. Bioengineered. 2014;5:73–9.
Gesta S, Tseng Y-H, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131:242–56.
Pagano C, Dorigo A, Nisoli E, Tonello C, Calcagno A, Tami V, et al. Role of insulin and free fatty acids in the regulation of ob gene expression and plasma leptin in normal rats. Obes Res. 2004;12:2062–9.
Zamboni M, Zoico E, Fantin F, Panourgia MP, Di Francesco V, Tosoni P, et al. Relation between leptin and the metabolic syndrome in elderly women. J Gerontol A Biol Sci Med Sci. 2004;59:396–400.
Nisoli E, Carruba MO, Tonello C, Macor C, Federspil G, Vettor R. Induction of fatty acid translocase/CD36, peroxisome proliferator-activated receptor-gamma2, leptin, uncoupling proteins 2 and 3, and tumor necrosis factor-alpha gene expression in human subcutaneous fat by lipid infusion. Diabetes. 2000;49:319–24.
Fabris R, Nisoli E, Lombardi AM, Tonello C, Serra R, Granzotto M, et al. Preferential channeling of energy fuels toward fat rather than muscle during high free fatty acid availability in rats. Diabetes. 2001;50:601–8.
Scheele C, Wolfrum C. Brown adipose crosstalk in tissue plasticity and human metabolism. Endocr Rev. 2020;41.
Pedersen BK, Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol. 2012;8:457–65.
Colaianni G, Cuscito C, Mongelli T, Pignataro P, Buccoliero C, Liu P, et al. The myokine irisin increases cortical bone mass. Proc Natl Acad Sci U S A. 2015;112:12157–62.
Colaianni G, Mongelli T, Cuscito C, Pignataro P, Lippo L, Spiro G, et al. Irisin prevents and restores bone loss and muscle atrophy in hind-limb suspended mice. Sci Rep. 2017;7:2811.
Laurens C, Bergouignan A, Moro C. Exercise-released myokines in the control of energy metabolism. Front Physiol. 2020;11:91.
Vinel C, Lukjanenko L, Batut A, Deleruyelle S, Pradère J-P, Le Gonidec S, et al. The exerkine apelin reverses age-associated sarcopenia. Nat Med. 2018;24:1360–71.
McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90.
Palermo A, Strollo R, Maddaloni E, Tuccinardi D, D’Onofrio L, Briganti SI, et al. Irisin is associated with osteoporotic fractures independently of bone mineral density, body composition or daily physical activity. Clin Endocrinol. 2015;82:615–9.
Park HS, Kim HC, Zhang D, Yeom H, Lim SK. The novel myokine irisin: clinical implications and potential role as a biomarker for sarcopenia in postmenopausal women. Endocrine. 2019;64:341–8.
Ducy P, Desbois C, Boyce B, Pinero G, Story B, Dunstan C, et al. Increased bone formation in osteocalcin-deficient mice. Nature. 1996;382:448–52.
Foresta C, Strapazzon G, De Toni L, Gianesello L, Calcagno A, Pilon C, et al. Evidence for osteocalcin production by adipose tissue and its role in human metabolism. J Clin Endocrinol Metab. 2010;95:3502–6.
Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130:456–69.
Mera P, Laue K, Ferron M, Confavreux C, Wei J, Galán-Díez M, et al. Osteocalcin signaling in myofibers is necessary and sufficient for optimum adaptation to exercise. Cell Metab. 2016;23:1078–92.
Mera P, Laue K, Wei J, Berger JM, Karsenty G. Osteocalcin is necessary and sufficient to maintain muscle mass in older mice. Mol Metab. 2016;5:1042–7.
Ponti F, Santoro A, Mercatelli D, Gasperini C, Conte M, Martucci M, et al. Aging and imaging assessment of body composition: from fat to facts. Front Endocrinol. 2019;10:861.
Hou J, He C, He W, Yang M, Luo X, Li C. Obesity and bone health: a complex link. Front Cell Dev Biol. 2018;8:600181.
Fassio A, Idolazzi L, Rossini M, Gatti D, Adami G, Giollo A, et al. The obesity paradox and osteoporosis. Eat Weight Disord. 2018;23:293–302.
Lecka-Czernik B, Stechschulte LA, Czernik PJ, Dowling AR. High bone mass in adult mice with diet-induced obesity results from a combination of initial increase in bone mass followed by attenuation in bone formation; implications for high bone mass and decreased bone quality in obesity. Mol Cell Endocrinol. 2015;410:35–41.
Luo H, Mu WC, Karki R, Chiang H-H, Mohrin M, Shin JJ, et al. Mitochondrial stress-initiated aberrant activation of the NLRP3 inflammasome regulates the functional deterioration of hematopoietic stem cell aging. Cell Rep. 2019;26:945-954.e4.
Mohrin M, Shin J, Liu Y, Brown K, Luo H, Xi Y, et al. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. 2015;347:1374–7.
Zhang H, Ryu D, Wu Y, Gariani K, Wang X, Luan P, et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352:1436–43.
Yoshino M, Yoshino J, Kayser BD, Patti GJ, Franczyk MP, Mills KF, et al. Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women. Science. 2021;372:1224–9.
Sanna M, Borgo C, Compagnin C, Favaretto F, Vindigni V, Trento M, et al. White adipose tissue expansion in multiple symmetric lipomatosis is associated with upregulation of CK2, AKT and ERK1/2. Int J Mol Sci. 2020;21(21):7933.
Borgo C, Milan G, Favaretto F, Stasi F, Fabris R, Salizzato V, et al. CK2 modulates adipocyte insulin-signaling and is up-regulated in human obesity. Sci Rep. 2017;7:17569.
Locke M, Windsor J, Dunbar PR. Human adipose-derived stem cells: isolation, characterization and applications in surgery. ANZ J Surg. 2009;79:235–44.
Badimon L, Cubedo J. Adipose tissue depots and inflammation: effects on plasticity and resident mesenchymal stem cell function. Cardiovasc Res. 2017;113:1064–73.
Veldhuis-Vlug AG, Rosen CJ. Clinical implications of bone marrow adiposity. J Intern Med. 2018;283:121–39.
Hamrick MW, McGee-Lawrence ME, Frechette DM. Fatty infiltration of skeletal muscle: mechanisms and comparisons with bone marrow adiposity. Front Endocrinol. 2016;7:69.
Thompson D, Karpe F, Lafontan M, Frayn K. Physical activity and exercise in the regulation of human adipose tissue physiology. Physiol Rev. 2012;92:157–91.
Addison O, Marcus RL, Lastayo PC, Ryan AS. Intermuscular fat: a review of the consequences and causes. Int J Endocrinol. 2014;2014:309570.
Miller CT, Fraser SF, Levinger I, Straznicky NE, Dixon JB, Reynolds J, et al. The effects of exercise training in addition to energy restriction on functional capacities and body composition in obese adults during weight loss: a systematic review. PLoS One. 2013;8:e81692.
Lee SJ. Targeting the myostatin signaling pathway to treat muscle loss and metabolic dysfunction. J Clin Invest. 3 May 2021;131(9):e148372.
Goloviznina NA, Xie N, Dandapat A, Iaizzo PA, Kyba M. Prospective isolation of human fibroadipogenic progenitors with CD73. Heliyon. 2020;6:e04503.
Suárez-Calvet X, Fernández-Simón E, Piñol-Jurado P, Alonso-Pérez J, Carrasco-Rozas A, Lleixà C, et al. Isolation of human fibroadipogenic progenitors and satellite cells from frozen muscle biopsies. FASEB J. 2021;35:e21819.
Russo V, Yu C, Belliveau P, Hamilton A, Flynn LE. Comparison of human adipose-derived stem cells isolated from subcutaneous, omental, and intrathoracic adipose tissue depots for regenerative applications. Stem Cells Transl Med. 2014;3:206–17.
Funding
Open access funding provided by Università degli Studi di Padova within the CRUI-CARE Agreement. This work was funded by Progetti di Rilevante Interesse Nazionale (PRIN) from the Italian Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR) Grant number 2017L8Z2EM to RV.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Favaretto, F., Bettini, S., Busetto, L. et al. Adipogenic progenitors in different organs: Pathophysiological implications. Rev Endocr Metab Disord 23, 71–85 (2022). https://doi.org/10.1007/s11154-021-09686-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-021-09686-6