
Abstract
Aims
The principal aim of this research was to quantify retention of a single, realistic Se biofortification application (10 g ha−1) in contrasting soils over two growing seasons utilizing an enriched stable Se isotope (77Se) to discriminate between applied Se and native soil Se.
Methods
Isotopically enriched 77Se (Na2SeO4) was applied (10 g ha−1) to four replicate plots (2 m × 2 m) of winter wheat, on three contrasting soils on the University of Nottingham farm (UK), at early stem extension in May 2012. Labelled 77Se was assayed in soil and crop fractions by ICP-MS.
Results
Topsoil retained a proportion of applied Se at harvest (c. 15 – 31%) with only minor retention in subsoil (2-4%), although losses were 37 – 43%. Further analysis of topsoil 77Se, the following spring, and at second harvest, suggested that labelled Se retained in soil was fixed and uptake by a following crop was negligible.
Conclusions
Prolonged biofortification leads to accumulation of Se in soil but the retained Se has very low bioavailability and mobility. The time required to double the soil Se content would be about 500 years. However, reincorporation of cereal straw could provide a residual source of Se for a following crop, depending on timing and management.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Agronomic biofortification with Se-enriched fertilisers is an effective means of increasing dietary Se intake by humans and animals (Fordyce 2005; Broadley et al. 2010). In biofortification studies selenium is typically applied either as sodium selenite (Na2SeO3) or sodium selenate (Na2SeO4), although application of other forms (e.g. K2SeO4, BaSeO4), including commercial compound Se fertilisers (e.g. Selcote Ultra®, Top Stock®) has also been studied (Gissel-Nielsen 1998; Gupta and Gupta 2002; Valle et al. 2002; Broadley et al. 2010). Application of Se may be carried out either by incorporating Se-enriched granular fertilizers or by spraying a liquid drench directly onto the crop canopy or soil (Broadley et al. 2010). Chilimba et al. (2012a) observed that Se uptake by maize was similar for liquid and solid applications of selenate to soil. Despite the wide range of application rates employed in cereal crop Se biofortification studies, from <10 to 500 g ha−1 (Yläranta 1983), uptake of Se by arable crops is widely reported as being a linear function of application rate (Ducsay and Ložek 2006; Stroud et al. 2010b; Broadley et al. 2010, Chilimba et al. 2012a; Galinha et al. 2012; Wang et al. 2013); this is also true of herbage sward studies (Whelan 1989; Rimmer et al. 1990; Shand et al. 1992; Valle et al. 2002). It is also widely accepted that crop uptake of selenate (SeVI) exceeds that of selenite (SeIV) (Shand et al. 1992; Chen et al. 2002; Zhao et al. 2005; Galinha et al. 2012, Ros et al. 2016) mainly because selenate is more soluble in soil within the pH range of arable soils (Fleming 1980; Jacobs 1989; Neal 1995) making it more bioavailable (Zayed et al. 1998; Li et al. 2008; Stroud et al. 2010b). Yläranta (1983) observed that application of just 10-20 g ha−1 Na2SeO4 (SeVI) was sufficient to produce barley grain Se concentrations of 100-200 μg kg−1, whereas over 100 g ha−1 Na2SeO3 (SeIV) was needed to achieve similar concentrations. Gupta et al. (1993) observed that application of 10 g ha−1 Na2SeO4 increased barley grain Se concentrations by approximately 200 μg kg−1 above controls, whereas the same application rate of Na2SeO3 apparently had no measureable effect on grain Se concentration.
Whilst the uptake of Se by crops in biofortification studies has been studied extensively, the retention of residual Se in the soil has received limited attention (Keskinen et al. 2011). This issue needs to be addressed if Se biofortification is to be implemented on a large scale and over long time periods. Extended application of Se to soils has implications for water quality and for long-term micronutrient management and supply of this scarce resource. Currently, the fate of Se from enriched fertilisers, and the underlying mechanisms which dictate this, are not fully understood (Alfthan et al. 2011). Quantification of applied Se retained in soil is very difficult; this is due to the very small amounts of Se typically applied (c. 10-20 g ha−1). For example, complete retention of Se at a biofortification rate of 10 g Se ha−1 would result in an increase in concentration of just 4 μg Se kg−1, assuming a topsoil mass of 2500 t ha−1 (topsoil depth = 20 cm; bulk density = 1.25 g cm−3). This is negligible (0.56%) in comparison with the UK average soil Se concentration of 710 μg kg−1 (Rawlins et al. 2012) and would be virtually impossible to measure within the span of a short term research project. However, this problem can be overcome by using an enriched stable isotope of Se, such as 77Se (natural abundance 7.63%). If 77Se (100% abundance) were applied at a rate of 10 g ha−1 (4 μg 77Se kg−1) this would equate to a 7.5% increase in soil 77Se, bringing quantification of retention within analytical reach. Chilimba et al. (2012a) used enriched 74Se (natural abundance 0.89%) to determine residual fertilizer effects in maize grown on acidic soils in Malawi. In principle, the very low natural abundance of 74Se would make it an ideal tracer for studying the fate of Se. However mass interferences to inductively coupled plasma mass spectrometry (ICP-MS) analysis from 74Ge and incomplete removal of iron polyatomics (possibly 57Fe-OH and 56Fe-OH2) made quantification of 74Se in soil samples problematic. In a recent field incubation study of selenium fractionation and speciation changes in three soil types, Di Tullo et al. (2016) used enriched 77Se to distinguish between added Se and native soil Se.
The primary aim of this paper was to quantify retention of a single, realistic Se biofortification application (10 g ha−1) in contrasting soils over two growing seasons utilizing an enriched stable Se isotope (77Se) to discriminate between applied Se and native soil Se. Secondary aims were to determine (i) the bioavailability of residual soil Se, (ii) the period in the growing season during which losses of added selenate from soil occurred, (iii) the rate of selenate immobilization in soil, and (iv) the likely contribution to available Se from biofortified wheat straw ploughed into soil. The stable enriched isotope 77Se was chosen to enable a full mass balance on the fate of Se applied to a wheat crop, as (i) it is the second least abundant isotope of Se (after 74Se) and (ii) at mass 77 it was thought that there would be a reduced chance of spectral interferences to ICP-MS analysis encountered at a mass:charge ratio (m/z) of 74.
Materials and methods
Site selection
Three fields on the University of Nottingham farms, with contrasting soil series, were selected for the biofortification experiment. These were, nominally (i) Dunnington Heath series (clay loam) Sutton Bonington Farm (grid ref.: 52°50′08.19″N-1°14′51.21″W), (ii) Newport series (sandy clay loam) Bunny farm (grid ref.: 52°51′28.63″N-1°07′25.36″W) and (iii) Worcester series (clay loam) Bunny Farm (grid ref.: 52°51′14.85″N-1°07′35.53″W). Each site was sown with winter wheat (Triticum aestivum) in September 2011 following a pre-existing farm management strategy. The varieties planted were JB Diego at the Dunnington Heath (DH) and Worcester (WOR) sites and Santiago at the Newport site (NEW). At each site four replicate treatment plots (2 m × 2 m) were established within a 2 × 8 m block between the field boundary/headland and the first cultivation tramline. Plot corners were recorded using line-of-site markers in the headland to enable relocation of the plots. Plough direction was recorded as ploughing causes a shift in the position of the topsoil, estimated by Lane and McGrath (1994) as 0.24 m parallel to, and 0.13 m perpendicular to, the original position of the topsoil in the direction of cultivation.
Preparation of selenate-77 stock solutions
Selenite-77 (77SeIV) and selenate-77 (77SeVI) were prepared from an elemental stock of enriched 77Se0 (50 mg; 99.2% atom %), purchased from Isoflex, USA, following the method described by Collins et al. (2006). Selenite-77 (SeIV) was prepared by dissolving 25 mg 77Se0 in 10 mL 70% HNO3 in a 100 mL conical flask and heating at 60 °C on a teflon-coated graphite block digester until dry. The resulting desiccated material was dissolved in 100 mL 2% HNO3 and stored in the dark at room temperature. Selenate-77 (SeVI) was prepared by dissolving 25 mg 77Se0 in 5 mL 30% H2O2 and 5 mL 2.0 M KOH in a 100 mL conical flask and heating at 90 °C on a teflon-coated graphite block digester until dry. The resulting desiccated material was dissolved in 10 mL 30% H2O2 and heated at 90 °C until dry twice more. Finally the remaining material was then dissolved in 100 mL of 2% HNO3 and stored in the dark at room temperature. Stock solution concentrations were calculated, from the original isotopic mass used, as 231 and 259 mg L−1 of 77SeIV and 77SeVI respectively. The purity of 77Se in each stock solution was confirmed by Se analysis by ICP-MS and the speciation of each 77Se stock solution was confirmed by coupled anion exchange liquid chromatography (LC-ICP-MS) analysis.
Selenium application to experimental plots
Selenium was applied to each plot as aqueous 77SeVI in mid-May 2012 at a rate of 10 g ha−1 (1.0 L m−2) using a knapsack sprayer (16 L Vermorel 2000 pro, Berthoud Jadin). Weather conditions experienced at the time of application at the DH site were dry, warm and still (14th May 2012), whereas conditions experienced at the NEW and WOR sites were intermittent rainfall, cool temperatures and moderate winds (17th May 2012). The timing of application (at all sites) coincided with early stem extension (growth stage 31/32, Zadoks et al. 1974; Feekes scale 6-7, Large 1954). Once applied, the crop was irrigated with 16 L de-ionized water at each site (1 mm depth) intended to limit retention of 77Se on the foliage.
Sample collection and processing
Soil samples were collected using a stainless steel auger from within the central square metre of each plot (2 m × 2 m). Topsoil was taken from a depth interval of 0-20 cm, and subsoils from a depth of 20-40 cm. Samples were collected from the four corners and centre of the central 1 m2 of each of the selenium application plots and combined. This was repeated for the four replicate plots and again repeated for the three study sites. The soil from each subplot was sieved to <2 mm, homogenised by the cone sampling method, finely ground, sub-sampled for analysis and the four sub-plots analysed separately. Prior to 77Se application, soils from each plot were sampled to act as individual plot controls. Following 77Se application, soils were sampled at (i) harvest 2012 (H1, mid-August), (ii) spring 2013 (S2, mid-May) and (iii) (residual) harvest 2013 (H2, mid-August), to quantify retention of 77Se in soil (i) at the end of a single growing season, (ii) after a winter of leaching and (iii) at the end of a second growing season, respectively. No subsoil samples were taken at H2.
Crops were sampled at H1 and H2 by removing the entire central square metre of each plot by hand at 10 cm above ground level with stainless steel scissors. Wheat ears and straw were separated following collection. Ears were dried at 40 °C in an oven (Scientific Laboratory Supplies Ltd) and then threshed to separate chaff and grain, which were milled separately in an ultra-centrifugal mill fitted with 0.5 mm titanium screen (Model ZM200, Retsch). Straw was sub-sampled (c. 20% of total), dried at 30 °C in a temperature-controlled room and chopped in a cutting mill (Model SM100, Retsch) fitted with a 1 mm stainless steel screen. As a check on possible local variation in the background 77Se:78Se isotopic ratio, crop samples were collected from within the same fields at each site, but outside the area of 77Se application (>30 m away).
Soil characterisation
Soil pH was determined using a pH meter (Model pH 209, HANNA Instruments) with combined glass electrode (Ag/AgCl) on a suspension of 5 g (± 0.5 g) air-dried soil (< 2 mm) in 12.5 mL of Milli-Q water (18.2 MΩ cm) after shaking end-over-end for 30 min at 27 rpm. Soil texture was determined by laser granulometry following initial treatment with 40% H2O2 and dispersion in 25 mL Calgon™ solution (3.5% sodium hexametaphosphate and 0.7% sodium carbonate). Following the study of Konert and Vandenberghe (1997) the clay fraction was taken as <8 μm to permit comparison with traditional methods. Total soil carbon concentration (SC) was determined by combustion at 900 °C in an Elemental Analyser (Model Flash EA1112, CE Instruments). Soil inorganic carbon (SIC) was determined in a Shimadzu TOC-VCPH instrument coupled with an SSM-5000A solids module (Shimadzu UK Ltd). Soil organic carbon (SOC) was determined by difference between SC and SIC. Soil iron (Fe), aluminium (Al) and manganese oxide (Mn) contents were determined by inductively coupled plasma mass spectrometry (ICP-MS) following dissolution in citrate bicarbonate dithionite (CBD). Olsen phosphate concentration was measured following extraction in 0.5 M NaHCO3. Total soil Se concentrations (control plot soils only) were determined by ICP-MS following HNO3-HClO4-HF digestion of 0.2 g in 2 mL 70% TAG HNO3, 1 mL 60% HClO4 and 2.5 mL 70% hydrofluoric acid (HF) in perfluoroalkoxy (PFA) digestion vessels heated in a 48-place teflon-coated graphite block digester (Model A3, Analysco Ltd).
Release of Se to soil from straw decomposition
The potential contribution from incorporated crop residues to plant-available Se was investigated by measuring short-term (0-30 d) release of labelled Se (77Sefert) from wheat straw from harvest H1, following incorporation into moist topsoil samples. Several amounts of milled 77Se-enriched straw from H1 of the DH site (0, 200, 500 and 1000 mg) and 5 g (± 0.05 g) moist (80% AWC), <4 mm sieved soil (Wick series, arable topsoil, sampled at the University of Nottingham Farm, Sutton Bonington, grid ref.: 52°49′49.50″N-1°14′18.83″W) were incubated at 21 °C, for different lengths of time (5, 10, 15, 20 and 30 days). The 77Sefert concentration in the straw was 219 μg kg−1. At each time point, KH2PO4-extractable soil 77Sefert was determined according to the method developed by Zhao and McGrath (1994) for sulphate extraction, which was later adapted by Martens and Suarez (1997) for Se extraction. The extractable soil Se was solubilised by suspending 3.0 g (± 0.3 g) of air-dried <2 mm sieved soil in 9 mL 0.016 M KH2PO4 in 50 mL polypropylene centrifuge tubes and shaking end-over-end for 1 h at 27 rpm. Soil solutions were centrifuged at 2200 g for 20 min and filtered to <0.22 μm using a Millex syringe driven filter unit and stored in ICP tubes, at room temperature, in the dark, pending elemental and speciation analysis, without dilution, by ICP-MS and LC-ICP-MS.
The half-life of the fixation of 77Se released by decomposition of the enriched straw, at each rate of straw addition (200, 500 and 1000 mg) was calculated from Eq. 1,
where Set is the concentration of extractable 77SeP at time t (μg kg−1), Se0 is the initial concentration of extractable 77Sefert present following addition of straw to the soil, k is the reaction rate constant (d−1) and t is time (d).
The potential increase in soil 77Se at each site from the incorporation of 77Se-enriched crop residues (wheat straw) during ploughing after H1 and the subsequent release of 77Se by microbially driven straw decomposition was calculated using Eq. 2.
Where, ∆77SeS is the concentration increase of soil 77Se (μg kg−1), 77SeST is the concentration of 77Sefert in the straw (μg kg−1), YST is the recovered yield of straw per plot (g m2), YpR is the proportion (%) of YST which is not harvested at H1 (crop residue, estimated at 22% for winter wheat by Zając et al. 2013) and WS is the weight of soil per plot (250 kg m2, assuming a bulk density of 2500 t ha−1).
Kinetics of selenate reaction with soil
The equivalent of 1500 g DW, <4 mm sieved, field moist topsoil from the DH and NEW sites, was spiked with a stock solution of 77SeVI at a rate of 16 μg kg−1 of soil and gently mixed with a food mixer, to ensure thorough mixing of the 77SeVI spike solution with the soil. Spiked soils were split equally into three replicate 1 L glass Duran bottles (500 g DW soil in each bottle), and incubated at 10 °C (Model MIR-254, Sanyo) in the dark. Sequential extractions were performed (at pre-determined incubation time points) on 5 g (± 0.1 g) spiked soil, with 0.01 M Ca(NO3)2 and 0.016 M KH2PO4 in a temperature-controlled room at 10 °C; the sum of which was defined as labile 77Se. Calcium nitrate extractable soil Se was solubilised by suspending 3.0 g (± 0.3 g) of air-dried <2 mm sieved soil in 20 mL 0.01 M Ca(NO3)2 in 50 mL polypropylene centrifuge tubes and shaking end-over-end for 1 h at 27 rpm. Suspensions were filtered to <0.22 μm using a Millex syringe driven filter unit and pipetted (1.5 mL) into HPLC vials, for immediate speciation analysis, without dilution, by LC-ICP-MS. ‘Adsorbed’ 77Se was then solubilised by re-suspending the soil in 9 mL 0.016 M KH2PO4 in 50 mL polypropylene centrifuge tubes and shaking end-over-end for 1 h at 27 rpm. Soil solutions were centrifuged at 2200 rpm for 20 min and syringe-filtered to <0.22 μm. The proportion of 77Sefert from the spike that was fixed in the soil was determined by subtracting the concentration of labile 77Sefert (sum of the 77Sefert from the spike in calcium nitrate and phosphate extractions) from the concentration of 77SeVI initially added to the system (16 μg kg−1 soil).
Selenium isotope analysis
Determination of residual 77Sefert in soils was initially attempted following HNO3-HClO4-HF digestion, however the dilution of digests required for ICP-MS analysis (2.5 L g−1 of soil), and the levels of interference encountered, were too great to enable discrimination of applied 77Se against background. Therefore, residual 77Sefert in soils was determined following extraction with 10% tetra methyl ammonium hydroxide (TMAH), according to the method developed by Watts and Mitchell (2009) for determination of total soil iodine concentration. TMAH-extractable soil Se was solubilised by suspending 1.0 g (± 0.1 g) of air-dried <2 mm sieved soil in 10 mL 10% TMAH in 50 mL polypropylene centrifuge tubes and heating to 70 °C for 3 h in an oven (Scientific Laboratory Supplies Ltd). Milli-Q water (5 mL) was added and the soil suspensions were centrifuged at 2200 g for 20 min. The resulting supernatant solution was diluted to 1% TMAH with Milli-Q water to produce an effective soil:solution (w/v) ratio of 1:100 prior to analysis by ICP-MS. Soil nickel (Ni) and germanium (Ge) content were also determined in these extractions to help assess a possible interference at m/z 77 from the polyatomic species 60Ni16O1H and the hydride 76Ge1H.
Total Se content of straw, chaff and grain was determined on oven-dried plant material (at 30 °C) after grinding in an ultra-centrifugal mill fitted with a 0.5 mm titanium screen. Approximately 0.2 g (± 0.02 g) of oven-dried, milled material was suspended in 6 mL 70% HNO3 in pressurised PFA vessels and digested via microwave heating (Model Multiwave 3000 fitted with a 48-place rotor; Anton Paar). Digested samples were diluted to 20 mL with Milli-Q water for storage and further diluted to 2% HNO3 prior to analysis by ICP-MS.
Elemental analysis
Elemental analysis was undertaken on an ICP-MS (X-SeriesII, Themo Fisher Scientific). Samples were introduced at 1 mL min−1 through a concentric glass venturi nebuliser and peltier-cooled (3 °C) spray chamber. Instrumental drift was corrected using internal standards (72Ge, 103Rh and 185Re). A hydrogen collision-reaction cell (H2-cell) was used to reduce interference from polyatomic species during Se analysis (e.g. 38Ar-40Ar interference with 78Se). For analysis of 77Se enriched samples, Ge was not used as an internal standard due to the interference at mass 77 caused by 76Ge hydride formation (76Ge natural abundance 7.76%). Methanol (c. 4%) was added to all samples to increase the ionisation efficiency of Se in the plasma. Selenium speciation analysis of KH2PO4 soil extractions (77SeIV and 77SeVI) was undertaken by LC-ICP-MS following in-line chromatographic separation (Dionex Model ICS-3000) in isocratic mode with a Hamilton PRP-X100 anion exchange column (250 × 4.6 mm; 5 μm particle size) fitted with a guard column (PEEK, PRP X-100). The mobile phase (flow rate 1.3 mL min−1) was 50 mmol L−1 NH4NO3, 1 × 10−5 mol L−1 Na2-EDTA, 2% b.v. methanol with pH adjusted to 9.4 with Tris buffer. Sample processing was undertaken using PlasmaLab software with peaks of individual species manually integrated. A standard was analysed after every six samples to enable correction for instrumental drift by linear interpolation.
Data processing and quantification of selenium-77 enrichment
Raw intensity data were exported as ‘counts-per-second’ (cps) values at m/z 77 and 78 corrected for blanks values and drift. The 78(m/z) signal was first corrected for hydride formation (77SeH). When measuring Se isotopes by ICP-MS in H2-cell mode, formation of Se hydride (SeH) occurs. This is irrelevant to Se analysis if the isotopic abundances of standards and samples are identical but a possible source of error when working with non-standard isotopic abundances. In the case of 77Se, the 77(m/z) signal is therefore subject to loss from the formation of 77SeH+, which in turn increases the 78(m/z) signal, and also gain from the formation of 76SeH+. The contribution from 76SeH+ to the 77(m/z) signal was quantified after the 78(m/z) signal was corrected for 77SeH+ formation because the 76(m/z) signal was inferred from the corrected 78(m/z) signal on the basis of expected background isotopic ratios. The contribution of 77SeH+ to the 78(m/z) signal was established by analysing pure 77Se standards (c. 100% isotope abundance) and calculating the ratio of 77SeH+ (reported as 78 m/z) to 77Se (reported as 77 m/z) using Eq. 3:
where, Hr is the hydride ratio and 78(m/z) and 77(m/z) are the signals (cps) measured in the pure 77Se standard. The 78(m/z) signal in samples was corrected for 77SeH+ production, arising from the (enriched) 77Se spike, using an approximate solution shown in Eq. 4:
where 78Secps is the corrected 78(m/z) signal and 77Seia and 78Seia are the naturally occurring isotopic abundances of 77Se and 78Se respectively.
The 76Se signal (76Secps) was then inferred from 78Secps on the basis of the natural isotopic ratio 76Seia:78Seia using Eq. 5. However, values of 76Secps are slightly overestimated due to the greater sensitivity of 78Se, therefore a mass discrimination factor (MDF, calculated in Eq. 6) must also be applied to correct for this:
where, 76Seia is the isotopic abundance of 76Se. The signal for native 77Secps was also calculated in this manner.
In Eq. 6, the value 0.9577 is the MDF per mass unit, determined from analysis of a normal multi-isotope calibration standard, which is multiplied by a mass difference of 2 (78-76).
It is reasonable to assume that the extent of hydride formation is similar for all Se isotopes (Nelms, S. Thermo Scientific, personal communication) therefore the 77(m/z) signal was also corrected for 76SeH+ formation and converted to represent the total 77Se concentration (77Setot expressed in μg L−1) using Eq. 7:
Where, 77Sesen is the measured sensitivity of 77Se expressed in cps ppb−1.
The concentration of 77Setot represents all 77Se in an extract, from both native and added 77Se. In order to establish the concentration of 77Se, which was from the enriched isotope fertilizer, the native 77Se concentration present (77SeN, μg L−1) was calculated first, using Eq. 8:
Where, 77SeN,cps is the native 77Se signal inferred from the 78Secps signal, on the basis of natural isotopic abundance.
The concentration of 77Se from addition of enriched 77Se isotope (77Sefert) was then calculated using Eq. 9:
The concentration of 77Sefert (μg L−1) was then converted to a gravimetric basis (μg kg−1) for individual samples of soil or plant material.
Rainfall data
Rainfall data were obtained from the University of Nottingham, School of Biosciences, weather station (grid ref.: 52°50′11.34″N-1°15′04.25″W) operated in partnership with the Met Office (metoffice.gov.uk).
Statistical analysis
Paired T-tests were performed in Minitab (version 16.2.2) and Duncan’s multiple range test was performed in ANOVA Options within Genstat (version 15.1.0.8035).
Results
Soil characteristics
Soil characteristics for the three field sites are shown in Table 1. The DH and WOR topsoil samples were both clay loams and the NEW topsoil was a sandy clay loam. Soil pH was similar in the DH and WOR topsoils (7.29 and 7.34, respectively) but lower in the NEW topsoil (6.95). Soil organic carbon (SOC) content was similar in the NEW and WOR topsoils (1.03 and 1.01%, respectively) but approximately double in the DH topsoil (2.07%). Manganese oxide concentration was lowest in the NEW topsoil and was approximately half that of the WOR topsoil (1.36 and 2.66 g kg−1, respectively), which was the greatest. Aluminium oxide concentration was comparable in the DH and WOR topsoils (0.460 and 0.475 g kg−1), but greatest in the NEW topsoil (0.527 g kg−1). Olsen phosphate concentration was highly variable between the three topsoils, with 16.5 mg kg−1 in the WOR, 37.5 mg kg−1 in NEW and 70.1 mg kg−1 in DH topsoil.
Selenium-77 recovery at first harvest (H1)
Based on harvest from the central 1.0 m2 from each plot, grain yields were 8.6, 8.5 and 9.7 t ha−1 for DH, NEW and WOR sites respectively; corresponding straw yields were 13.6, 11.1 and 12.0 t ha−1. Recovery of applied 77Se (77Sefert) in wheat grain at H1 (Fig. 1a) was comparable across treatment sites (DH 12.4%; NEW 11.9%; WOR 15.2%). Addition of 10 g 77SeVI ha−1 was sufficient to boost total grain Se concentration to 4-13 times that of controls (Table 2). Recovery of 77Sefert in wheat straw (Fig. 1a) was marginally greater than recovery in wheat grain at the NEW and WOR sites but substantially greater at the DH site (29.7% in straw; 12.4% in grain). Recovery of 77Sefert in wheat chaff (Fig. 1a) was very small at all treatment sites (<2.5%). Total recovery in the crop (sum of grain, straw and chaff) varied between all sites (DH 44.5%; NEW 25.9%; WOR 33.0%) with differences in recovery in the straw being the major discriminator. Retention of 77Sefert in topsoil was observed at all treatment sites (Fig. 1a) but varied between sites (DH 15.0%; NEW 30.9%; WOR 20.5%), whereas recovery in subsoils was small and comparable between sites (DH 3.57%; NEW 2.62%; WOR 3.49%). Apparent losses of 77Sefert from the system (Fig. 1a) were similar across all sites (DH 36.9%; NEW 40.6%; WOR 43.0%).

a Recovery and losses (% and g ha−1) of 77Se from fertiliser application (77Sefert) in grain, straw, chaff, topsoil and subsoil, at first harvest (H1) (soil concentrations are corrected for control values) and b concentration (g ha−1) and contribution (% listed above each bar) of native soil Se (SeN; all isotopes) to total Se in grain, straw and chaff, at H1. Error bars show the standard error of four replicate analyses
The concentration of native soil Se (SeN) in the crop was greatest in the straw at all treatment sites (Fig. 1b). Crop partitioning of SeN followed broadly the same pattern as 77Sefert (Fig. 1a) despite differences in the timing of supply. The contribution of native soil Se to total Se in the crop was comparable (within 5%) between crop fractions at each site, none of which exceeded 25%; the majority (>75%) of Se in each crop fraction originated from application of 77Sefert. The concentration and contribution of native soil 77Se (77SeN) to each crop fraction was greatest at the DH site and lowest at the WOR site (Fig. 1b).
Retention of selenium-77 in soil
All topsoil samples from 77Se-treated plots showed an increase in the raw 77(m/z):78(m/z) ratio above that of the field control topsoil and therefore measureable concentrations of residual 77Sefert (Fig. 2). This increase persisted across the three topsoil sampling events at harvest in the year of 77Se application (H1), the following spring (S2) and at the end of the following growing season (H2). All sampled subsoils, with the exception of the WOR subsoil sampled in 2013, showed only marginal concentrations of 77Sefert above that of the field control topsoil. The DH and NEW field control soils exhibited very similar raw 77(m/z):78(m/z) ratios (0.335 and 0.333, respectively), whereas the ratio in the WOR control soil was greater (0.343). However, the WOR control soil also exhibited an apparent measureable concentration of 77Sefert (Fig. 2). Coincidentally, during transition through the ICP-MS, the combined effect of hydride generation, which acts to increase the apparent 77(m/z):78(m/z) ratio, and mass discrimination which has the opposite effect, almost cancelled each other out.

Concentration (μg kg−1) of 77Se from fertiliser application (77Sefert) in control soils, subsoils and topsoils at H1, S2 and H2. Letters above bars denote statistically different groups (p < 0.05) following Duncan’s multiple range test. Error bars show the standard error of four replicate analyses
Residual selenium-77 in crops
Crop rotation occurred at the NEW and WOR sites after H1 (wheat): maize and oilseed rape were sown in September 2012 at the NEW and WOR sites, respectively, whereas winter wheat was re-sown at the DH site. Negligible concentrations of 77Sefert were detected in crop straw at H2 at all sites, compared to wheat straw at H1 (Table 3).
Fixation of selenium-77 in soil
The time-dependent fixation of selenate (77SeVI; 16 μg kg−1) incubated with soils from the DH and NEW sites is shown in Fig. 3. ‘Fixed’ Se (77SeVI Fixed) was calculated as the difference between the initial addition of selenate and the measured concentrations of inorganic SeIV and SeVI in a two-stage extraction with 0.01 M Ca(NO3)2 and 0.016 KH2PO4 solutions. There was clear evidence of reduction to selenite from the added selenate (16 μg kg−1) with 77SeIV concentrations ranging from 0.03 – 0.42 μg kg−1 and 0.03 – 0.54 μg kg−1 in phosphate extractions of the DH and NEW topsoils respectively. The fixed Se was assumed to be mainly bound to humus. The fixation rate was approximately 0.1 μg kg−1 d−1 in both soils so that 80% was fixed after 160 days.

Proportion (%) of added 77SeVI fert that is fixed in soils DH and NEW (77SeVI Fixed) as a function of incubation time; calculated from the proportion (%) of 77Sefert, added at 16 μg kg−1, that remains labile (ie extractable in 0.01 M Ca(NO3)2 and 0.016 M KH2PO4 solutions combined)
Release of selenium-77 to soil from straw decomposition
The concentrations of labelled selenate (77SeVI fert) and total 77Sefert, extractable by phosphate (0.016 M) from 77Se-enriched straw, incubated in moist soil, were positively correlated with straw application rate and all decreased steadily throughout the time of incubation (30 days) following an irreversible first order decay trend (Fig. 4a; b) due to fixation in the soil. The average proportion of 77Sefert released from the soil-incubated enriched straw that was present as selenate (77SeVI fert) increased with the level of straw application to 13.4, 45.7 and 48.7% of 77Sefert for straw additions of 200, 500 and 1000 mg per 5 g field moist soil respectively. The concentration of labelled selenite (77SeIV fert; not shown) released from 77Se-enriched straw was negligible (<1 μg kg soil−1) across all straw application levels, throughout the time of incubation (30 days).

Concentration of phosphate-extractable a) 77SeVI fert and b) total 77Sefert, released from 77Se-enriched wheat straw (from first harvest, H1, of the DH site) added at three application levels: 200 mg (○), 500 mg (▲) and 1000 mg (□) in 5 g moist soil, as a function of incubation time (30 days). The initial 77Sefert concentration in the straw was 219 μg kg−1. Solid lines represent the fit of an irreversible first order equation. Error bars show the standard error of three replicate analyses
Discussion
Recovery of 77Sefert at harvest 2012
The recovery of 77Sefert at H1 in wheat grain (Fig. 1a) was comparable to previous studies comparing Se treatments with controls. Broadley et al. (2010) applied SeVI in liquid form (as Na2SeO4) at six different application rates (in the range 5-100 g ha−1) to wheat at four sites in the UK and reported a Se recovery range of 10.1 to 17.3% in the grain. Lyons et al. (2004) and Curtin et al. (2008) reported average recovery of SeVI (added as Na2SeO4) in Australian wheat grain at 13.5% and New Zealand wheat grain at 17.0%, respectively. Chilimba et al. (2012a) reported recovery of SeVI (added as Na2SeO4) across successive application years in Malawian maize grain at 16-18%. However, Chilimba et al. (2012b) reported lower recovery levels of 74SeVI (added as Na2 74SeO4) in maize grain at 6.5% and 10.8% across two treatment sites in Malawi and Stephen et al. (1989) observed 2-6% recovery of SeVI (added as Na2SeO4) in wheat grain in New Zealand, which is in a similar range to results from the long-term Finnish biofortification programme (<10%), reported by Eurola (2005). Taking the average daily per-capita intake of cereal and cereal-based products for the UK population as 110 g (PHE and FAO 2014), the biofortified wheat grain produced in this study would provide, on average, 19.1 μg person−1 d−1 of Se (Table 2), which equates to 25.6% and 31.9% of the UK recommended nutrient intake (RNI, BNF 2001), for men and women, respectively.
Recoveries of 77Sefert in wheat straw and grain were similar at the NEW and WOR sites (Fig. 1a), which exhibited total crop recoveries of 25.1 and 33.0%, respectively. Broadley et al. (2010) reported marginally greater recovery of Se (applied as aqueous Na2SeO4 at six different application rates spanning 5 to 100 g ha−1) in winter wheat straw than in grain (mean 13.4 and 12.6%, respectively). The greater recovery of 77Sefert in wheat straw compared to grain at the DH site, can be attributed to two factors. Firstly, the straw:grain concentration ratio for 77Sefert was almost double that observed at the NEW and WOR sites (DH 1.53; NEW 0.818; WOR 0.847), suggesting that transfer of inorganic 77SeVI from the straw to protein- or amino acid-bound Se in the grain may have been restricted. Secondly, the straw:grain yield ratio was also greatest at the DH site (DH 1.60; NEW 1.31; WOR 1.23). The DH site also exhibited the lowest recovery of 77Sefert in topsoil (15.4%, less than half that observed at the other two sites, Fig. 1a). At the DH site, the greater recovery of 77Sefert in the straw and the lower recovery in the topsoil may suggest direct foliar adsorption 77Sefert at the time of application. Despite irrigating the crop canopy immediately after application of 77Se, the dry, warm weather conditions at the time of application at the DH site may have contributed to enhanced foliar adsorption of 77Sefert, which was not observed following the heavy rainfall at the time of application at the NEW and WOR sites.
The NEW site exhibited the greatest retention of 77Sefert in the topsoil (40%), which had the lowest pH and the greatest concentration of Fe and Al oxides (pH 6.95, 29.3 and 0.527 g kg−1, respectively, Table 1). Both factors are expected to increase initial retention of 77Sefert in topsoil through stronger binding to hydrous oxides at lower pH. Furthermore, results from the soil incubation experiment (Fig. 3), showed that the proportion of added 77SeVI (c. 40 g ha−1) that was fixed in incubated topsoils was approximately 50% for the DH and NEW sites, 95 days after addition. Therefore, considering the time elapsed between application of 77SeIV and first harvest (DH, 93 days; NEW and WOR, 96 days), it is likely that that 77Sefert retained in topsoil at these sites, would be present in organically bound form or possibly bound within oxides in a form not extractable by phosphate. Certainly Fig. 3 shows that these soils have the capacity to convert most soluble selenate additions to soil, in spring, to fixed forms within a 3–4 month period.
The relatively small recoveries of 77Sefert in subsoil (<4%) and comparable losses between sites (Fig. 1a) suggests rapid leaching of 77SeVI after application (c. 40% of the application), with the remainder being distributed between crop and mainly humus-bound forms in the topsoil. It should also be acknowledged that some of the applied 77SeVI may have been converted to forms that are not extractable with 10% TMAH – so retention in topsoil may be underestimated. Leaching losses raise the possibility of a potential toxic hazard. The WHO provisional Se standard for drinking water quality is 40 μg L−1, based on an intake (in 2 L water per day) of 20% of the upper safe intake level (400 μg day−1 of Se) (WHO 2011). It may be assumed that selenate being leached in the soil solution moves initially in a simple piston flow pattern, having mixed with soil pore water in the topsoil (0-20 cm). Thus, for a soil with a volumetric water content of 40% at field capacity (c. 800 m3 water in topsoil per hectare), a loss of 40% Se from an application of 10 g ha−1 (4 g ha−1) would produce a solution concentration of 5.0 μg L−1. Given further dilution through mixing beyond the field drains it is therefore unlikely that such losses could present a risk to drinking water supplies.
The similar partitioning pattern of native soil Se and 77Sefert within the crop (Fig. 1a and b), suggests that re-distribution and transformation within the wheat plant was broadly the same for both soil- and fertilizer-derived Se. This is despite differences in (i) the quantity of Se taken up by the crop from each source and (ii) relative availability during the growing season.
The relative bioavailability of 77Sefert and native soil Se (SeN) to wheat grain at H1 was calculated on a ratio basis BR from Eq. 10:
Where, SeN is the concentration of native soil derived Se present at each site, 77Sefert-G is the concentration of applied 77Se recovered in wheat grain at H1 and SeN-G is the concentration of native soil Se recovered in wheat grain at H1. All concentrations are expressed in μg kg−1.
Bioavailability ratios of 77Sefert:SeN in wheat grain (DH = 338; NEW = 537; WOR = 1221) confirmed that 77Sefert was 2 – 3 orders of magnitude more bioavailable than SeN at all sites (p < 0.001). This was expected due to the highly soluble nature of newly added SeVI under neutral to alkaline soil pH conditions (Fordyce 2005) (Table 1). The bioavailability ratios of 77Sefert:SeN at the DH and NEW sites are of the same order of magnitude to those calculated from other field-based cereal biofortification studies, (126-405, Stroud et al. 2010a; 51.7-324, Chilimba et al. 2012a; 125-208, Galinha et al. 2012). The bioavailability ratios of 77Sefert:SeN were also significantly different between sites (T-test; P < 0.001), most likely due to differences in soil properties and weather conditions at the time of application, etc.
Release of selenium-77 (77Sefert) from straw decomposition and fixation in soil
The straw incubation data showed that 77SeVI accounted for almost all inorganic 77Se released from 77Se-enriched straw incubated in moist soil – less than 4% was present as selenite. On average, 35.9% (S.E. = 5.4%) of total 77Se was released over 30 days incubation. The speciation of the remaining 77Se released from straw was not determined but is likely to exist as in organic forms (selenomethionine; selenocystine). Stadlober et al. (2001) conducted a pot trial, in which a range of crops (winter wheat, spring wheat, summer barley, summer rye and durum wheat) were fertilised with Na2SeO4 at a rate equivalent to 56.3 g ha−1. Speciation analysis (by HPLC-ICP-MS) of the resulting wheat crop showed substantial conversion of applied SeVI to selenomethionine (69-86%) and to a lesser degree selenocystine (7-13%). The steady decrease in phosphate-extractable 77SeVI P (Fig. 4) suggests gradual incorporation of labile 77SeIV into complexes with soil humus or reduction to other recalcitrant forms (e.g. Se0) which resist extraction by KH2PO4. Results from the soil incubation experiment showed no measurable selenate (77SeVI) from spike addition in KH2PO4 extracts in the DH and NEW soils at any stage. In the context of this study, this suggests negligible adsorption of 77SeVI by hydrous oxides and instead direct incorporation of labile 77SeVI into organic complexes or reduction to recalcitrant forms. The majority (mean 69.9%) of the decrease in total 77Sefert released from the straw through time (Fig. 4) can be ascribed to the observed decrease in 77SeVI P. The apparent half-lives of fixation of 77Se released by decomposition of the enriched straw, at each rate of straw addition were 25.7 d (200 mg), 17.3 d (500 mg) and 30.1 d (1000 mg), (average = 23.1 d; S.D. = 0.0089 d). However, under field conditions (compared to those in the laboratory), the greater particle size of incorporated straw (chopped rather than milled) and the lower average soil temperature for the UK (10 °C rather than 21 °C) would likely result in a much slower rate of straw decomposition.
The potential increases in soil 77Se from decomposition of 77Se-enriched straw at each site were 0.335, 0.142 and 0.179 μg 77Se kg soil−1 year−1 at the DH, NEW and WOR sites, respectively. Broadley et al. (2010) reported similar potential increases of soil Se from incorporation of Se-enriched winter wheat crop residues (0.33-0.67 μg Se kg soil−1 year−1), from initial application rates of 5-100 g Na2SeO4 ha−1. Dhillon et al. (2007) reported that Se-enriched crop residues containing ≤137 mg Se kg−1 could be incorporated into non-seleniferous topsoils, without the risk of Se toxicity in subsequently grown cereal crops.
Retention of soil selenium-77(77Sefert) in topsoil
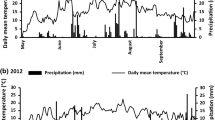
Monthly rainfall figures for the summer of 2012 in comparison to the long-term average are illustrated in Fig. 5. The summer of 2012 in the UK was the wettest for a century, which is apparent from the record rainfall figures recorded in June and July at Sutton Bonington (Fig. 5). Rainfall in the month of 77Se application (May 2012) was below the long-term average (Fig. 5), this suggests rainfall was low within the first two to three weeks post application before it increased sharply in June. Results from the incubation experiment (Fig. 4), showed that between 10 and 20% of 77SeVI from spike addition was likely to be fixed in the DH and NEW topsoils, within the first two to three weeks after spiking. As previously mentioned, the KH2PO4 extracts of the DH and NEW topsoils did not contain any 77SeVI from the spike application at any point during the incubation, suggesting the vast majority (>80%) of 77SeVI from application was present in the soil solution in a labile state, two to three weeks after spiking. In the context of this study, the measureable retention of 77Sefert observed in topsoil at all sites at H1 (15-31%, Fig. 2) was similar to that observed over the first two to three weeks in the parallel soil incubation experiment (Fig. 3), suggesting that fixation and crop uptake of 77Sefert occurred rapidly (within the first month after application) after which the sustained period of heavy rainfall is likely to have removed the remaining labile 77SeVI from the soil system via leaching.

Rainfall data (mm) collected at Sutton Bonington weather station (grid ref.: 52°50′11.34″N-1°15′04.25″W), for months April – August (inclusive) 2012, compared with the long term average (2002-2012). Error bars show the standard deviation of all monthly measurements from 2002 to 2012 (inclusive)
The apparent isotopic ratio (77Se/78Se) was slightly greater than the expected value (0.321) in TMAH extracts of the WOR control soil, producing an apparent fertilizer Se residue (Fig. 2). It was hypothesised that these differences were likely due to an interference at 77(m/z) during ICP-MS analysis, which artificially elevated the 77(m/z):78(m/z) ratio in all samples, including controls. The two most likely interferences at 77(m/z) are the polyatomic species 60NiOH+ (60Ni, 26.1% abundance) and the hydride 76GeH. The contribution of 60NiOH+ to the 77Se signal was investigated by Ni standard addition to TMAH extracts of the soils. However the interference from soil-derived Ni, estimated by extrapolation of the 77(m/z):78(m/z) ratio against Ni concentration, was very small and would only marginally increase the natural 77(m/z):78(m/z) ratios (DH 0.3224; NEW 0.3229; WOR 0.3235). Therefore, formation of 60NiOH+ species did not account for the elevated 77(m/z):78(m/z) ratios observed in the WOR soil. By contrast, analysis of the Ge/Se ratio in the TMAH extractions appeared to explain the elevated m/z 77 signal in the WOR soil. The Ge/Se ratios (from 72Ge and 78Se measurements) in the NEW, DH and WOR TMAH extracts were 0.37, 0.97 and 1.8 respectively and extrapolation of this ratio to Ge/Se = zero against the measured 77(m/z)/78(m/z) signals gave a ratio of 0.323. This is close to the expected value of 0.321 for 77Se/78Se in the absence of interference and suggests that the hydride species 76GeH was the cause of an apparently elevated 77Se isotopic abundance in the WOR soil. It is therefore recommended that future investigations quantify the extent of 76Ge hydride formation and implement a more direct correction on 77Se rather than relying on subtraction of apparent Se fertilizer residues in control soils. The alternative approach is simply to subtract ‘apparent’ 77Se residues measured in the control soils (as in Fig. 1a).
The concentration of 77Sefert in subsoils (at H1 and S2) was similar to those of control topsoils at all sites (Fig. 2), confirming minimal retention of 77Sefert in the subsoil. Statistical analysis by Duncan’s multiple range test (DMRT), indicated that the concentrations of 77Sefert in subsoils at the DH and NEW sites were not significantly different from those of controls. The concentration of 77Sefert in subsoil sampled at H1 at the WOR site was significantly different (p < 0.05) from subsoil sampled at S2, which was also significantly different from the control. The concentration of 77Sefert in all topsoil sampled was significantly different (p < 0.05) from controls, confirming measureable retention of 77Sefert, which was still present 15 months after application (H2). The difference in the concentration of 77Sefert in topsoil, between H1 and S2 was significant (p < 0.05) at the NEW and WOR sites. This decrease is likely to have occurred as a result of ploughing after H1, causing topsoil inversion. Fixation of 77Sefert is likely to have occurred primarily near the surface, producing a concentration gradient throughout the topsoil and into the subsoil. Ploughing would cause the most enriched layer (near the surface) to be buried at plough depth, which depending on plough depth and soil sampling depth may not be recovered, resulting in an incomplete audit of soil Se. Bhogal (1995) observed a similar effect with applied 15N at Ropsley. In the context of this study, it is worth noting that other losses of 77Sefert from topsoil may have occurred via leaching or reduction of 77Sefert to recalcitrant forms, which were not extracted with TMAH. The difference in concentration of 77Sefert in topsoil between S2 and H2 was significant (p < 0.05) at all sites (Fig. 2b). This increase cannot be ascribed to re-inversion of topsoil because sampling was undertaken prior to ploughing. However, decomposition of wheat straw residues from H1 enriched with 77Sefert, due to increased soil temperatures during the spring and summer, may have contributed to the apparent increase in the concentration of 77Sefert in topsoil at H2. Smith and Douglas (1971) observed that the content of nitrogen (N) in N-enriched wheat straw remained stable in soils below 4 °C and decreased significantly with increasing soil temperature above 4 °C due to increased rates of straw decomposition.
The potential increase in soil 77Sefert at each site from incorporation of 77Se-enriched wheat straw (calculated using Equation 2) would account for 147, 38.5 and 55.1% of the observed increase in 77Sefert concentration at the DH, NEW and WOR sites respectively between S2 and H2. However, this calculation makes the assumption that all 77Sefert present in crop residues is released by decomposition and subsequently retained by soil between S2 and H2, which may not be the case.
Residual crop selenium-77
Negligible residual 77Sefert was detected in crops harvested the following year after application, at all sites (Table 3). This confirmed that 77Sefert retained in topsoil from the initial application, and the minor incorporation of crop residue practiced on the experimental plots, was almost entirely present in a non-bioavailable form. Gupta et al. (1993) applied Na2SeO4 at three application rates (10, 20 and 40 g ha−1) to barley in Canada and observed no evidence of residual Se in crops the year following application (regardless of application rate). Stroud et al. (2010a) observed no significant difference in grain Se concentrations in winter wheat, grown the year after application with 10 or 20 g ha−1 Na2SeO4, compared to controls. Chilimba et al. (2012b) observed very small recoveries (2.0 and 0.78%) of isotopically labelled Se (applied as aqueous Na2 74SeO4 at a rate of 10 g ha−1) in the grain of maize crops grown the year after application, across two field sites in Malawi. However, at higher application rates (up to 100 g ha−1), the presence of applied Se in the grain of maize crops, grown the year after application, has been observed and also has a linear response to application rate (Chilimba et al. 2012b).
Conclusions
Application of 10 g 77SeVI ha−1 was sufficient to boost wheat grain concentrations by 4-13 times their control levels, equating to a recovery of 12-15% of the added fertilizer. Despite application during one of the wettest summers on record for the area, the timing of application was sufficiently far enough removed (2-3 weeks) from the period of intense rainfall for uptake by crops and fixation in soils to occur.
On average 22% (standard deviation = 8.07%) of 77Sefert was retained in the topsoil. However, virtually all the 77Se retained was non-bioavailable at the rate of application used in this study - there was no evidence of a significant residual effect on second crops. At the rate of residual Se accumulation observed it would take several centuries for soil Se concentration at the field trial sites to reach double the current level: DH = 621 y; NEW = 326 y; WOR = 456 y. Given such small annual additions to soil Se loading and the apparently rapid immobilization of selenate in the topsoil, it appears unlikely that long-term Se biofortification would present a significant toxic hazard to the soil environment. Perhaps the only remaining potential source of concern lies with the 77Se losses observed at all three sites; on average this amounted to 40.1% (std dev: 3.05%) of the original application. This was not recovered in the subsoil and so the suggestion is that it was lost from the soil through leaching
The negligible concentrations of 77Sefert detected in crops at second harvest (H2), suggests that biofortification of cereal crops in the UK with Se at a rate of 10 g SeVI ha−1, would have to be repeated annually to ensure effective grain enrichment. Although Se retention in soil does not provide a useful source for following crops, a possible source of residual Se may be the cereal straw, which on average absorbed 19.4% (std dev: 9.12%) of the applied Se. The primary species of Se released during the decomposition of Se-enriched straw was identified as SeVI, (selenate) which may provide a source of residual Se to subsequently grown crops. For this source of Se to provide an effective contribution to biofortification of a following crop the main issues to consider must be timing and extent of straw incorporation. The release rate will depend on the soil temperature and straw condition while uptake of released selenate will depend on the presence of a growing winter cereal crop, or possibly a catch crop to be ploughed in for a spring-sown cereal crop the following year. Stavridou et al. (2011, 2012) investigated several potential catch crops for Se and demonstrated both immobilization and mineralisation of Se following incorporation depending on the Se content of the catch crop. However, the usefulness of the catch crops in providing Se to spring-sown crops was generally limited by low Se uptake in Autumn. Further investigation should involve a field-based trial and should examine (i) the timing and extent of straw incorporation, (ii) alternative straw chopping and incorporation approaches, (iii) the alternative use of an Autumn-sown cereal or a fast growing catch crop for a subsequent Spring-sown cereal. Given the speciation of Se in straw (SeVI) and the proportion of Se originally applied (c. 20%) it seems likely that some residual effect from Se release from straw should be possible and it would be important to quantify this in the event of biofortification strategies being widely adopted in the UK.
References
Alfthan, G., Aspila, P., Ekholm, P., Eurola, M., Hartikainen, H., Hero, H., Hietaniemi, V., Root, T., Salminen, P., Venäläinen, E.R., Aro, A. (2011) Nationwide supplementation of sodium selenate to commercial fertilizers: history and 25-year results from the Finnish selenium monitoring programme. Thompson B, Amoroso L (eds) Combating micronutrient deficiencies: Food-based approaches 312-337
Bhogal (1995) Ph. D. thesis (Effects of long-term nitrogen applications on nitrogen cycling under continuous wheat). The University of Nottingham, pp. 205-208
British Nutrition Foundation (2001) Briefing Paper: Selenium and Health. https://www.nutrition.org.uk/attachments/145_Selenium and health.pdf
Broadley MR, Alcock J, Alford J, Cartwright P, Foot I, Fairweather-Tait SJ, Hart DJ, Hurst R, Knott P, Mcgrath SP, Meacham MC, Norman K, Mowat H, Scott P, Stroud JL, Tovey M, Tucker M, White PJ, Young SD, Zhao F (2010) Selenium biofortification of high-yielding winter wheat (Triticum aestivum L.) by liquid or granular Se fertilisation. Plant Soil 332:5–18
Chen L, Yang F, Xu J, Hu Y, Hu Q, Zhang Y, Pan G (2002) Determination of selenium concentration of rice in China and effect of fertilization of selenite and selenate on selenium content of rice. J Agric Food Chem 50:5128–5130
Chilimba ADC, Young SD, Black CR, Meacham MC, Lammel J, Broadley MR (2012a) Agronomic biofortification of maize with selenium (Se) in Malawi. Field Crop Res 125:118–128
Chilimba ADC, Young SD, Black CR, Meacham MC, Lammel J, Broadley MR (2012b) Assessing residual availability of selenium applied to maize crops in Malawi. Field Crop Res 134:11–18
Collins RN, Tran ND, Bakkaus E, Avoscan L, Gouget B (2006) Assessment of Isotope Exchange Methodology to Determine the Sorption Coefficient and Isotopically Exchangeable Concentration of Selenium in Soils and Sediments. Environ Sci Technol 40:7778–7783
Curtin D, Hanson R, Van der Weerden TJ (2008) Effect of selenium fertiliser formulation and rate of application on selenium concentrations in irrigated and dryland wheat (Triticum aestivum). N Z J Crop Hortic Sci 36:1–7
Dhillon SK, Hundal BK, Dhillon KS (2007) Bioavailability of selenium to forage crops in a sandy loam soil amended with Se-rich plant materials. Chemosphere 66:1734–1743
Di Tullo P, Pannier F, Thiry Y, Le Hécho I, Bueno M (2016) Field study of time-dependent selenium partitioning in soils using isotopically enriched stable selenium tracer. Sci Total Environ 562:280–288
Ducsay L, Ložek O (2006) Effect of selenium foliar application on its content in winter wheat grain. Plant Soil Environ 52:78–82
Eurola, M.H. (2005) Twenty years of selenium fertilization. MMT agrifoods research reports 69, Jokioinen, Finland
Fleming GA (1980) Essential Micronutrients 11: Iodine and Selenium. In: Davis BE (ed) Applied Soil Trace Elements. Wiley, New York, pp 199–234
Fordyce F (2005) Selenium deficiency and toxicity in the environment. In: Selninus O, Alloway B, Centeno J, Kinkelman R, Fuge R, Lindh U, Smedley P (eds) Essentials of Medical Geology. Elsevier, London, pp 373–415
Galinha G, Freitas MC, Pacheco AMG, Coutinho J, Maçãs B, Almeida AS (2012) Determination of selenium in bread-wheat samples grown under a Se-supplementation regime in actual field conditions. Radioanal Nuclear Chem 291:231–235
Gissel-Nielsen G (1998) Effects of selenium supplementation of field crops. In: Frankenberger WT, Engberg RA (eds) Environmental Chemistry of Selenium. Marcel-Dekker, New York, pp 99–112
Gupta UC, Winter KA, Sanderson JB (1993) Selenium content of barley as influenced by selenite-enriched and selenate-enriched fertilizers. Commun Soil Sci Plant Anal 24:1165–1170
Gupta UC, Gupta SC (2002) Quality of animal and human life as affected by selenium management of soils and crops. Commun Soil Sci Plant Anal 33:2537–2555
Jacobs, L.W., 1989. Selenium in Agriculture and the Environment [Soil Science Society of America Special Publication no. 23]. Madison: American Society of Agronomy and Soil Science Society of America
Keskinen R, Räty M, Yli-Halla M (2011) Selenium fractions in selenate-fertilized field soils of Finland. Nutr Cycl Agroecosyst 91:17–29
Konert M, Vandenberghe J (1997) Comparison of laser grain size analysis with pipette and sieve analysis: a solution for the underestimation of the clay fraction. Sedimentology 44:523–535
Lane PW, Mcgrath SP (1994) Modelling the effect of cultivation on the dispersion of pollutants in soil. Statistician 4:537–549
Large EC (1954) Growth in cereals – illustration of the Feekes scale. Plant Pathol 3:128–129
Li H, Mcgrath SP, Zhao F (2008) Selenium uptake, translocation, and speciation in wheat supplied with selenate or selenite. New Phytol 178:92–102
Lyons GH, Lewis J, Lorimer MF, Holloway RE, Brace DM, Stangoulis JCR, Graham RD (2004) High-selenium wheat: Agronomic biofortification studies to improve human nutrition. Food Agric Environ 2:171–178
Martens DA, Suarez DL (1997) Selenium speciation of soil/sediment determined with sequential extractions and hydride generation atomic absorption spectrophotometry. Environ Sci Technol 31:133–139
Neal RH (1995) Selenium. In: Alloway BJ (ed) Heavy Metals in Soils. Blackie Academic & Professional, London, pp 260–283
Public Health England and The Food Standards Agency (2014). National diet and nutrition survey. Results from years 1-4 (combined) of the rolling programme (2008/2009 and 2011/12) revised February 2017. Available at: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/594361/ndns_y1_to_4_uk_report_full_text_revised_february_2017.pdf
Rawlins, B.G., Mcgrath, S.P., Scheib, A.J., Breward, N., Cave, M., Lister, T.R., Ingham, M., Gowing, C., Carter, S., 2012. The advanced soil geochemical atlas of England and Wales. British Geological Survey, Keyworth, available from: www.bgs.ac.uk/gbase/advsoilatlasEW.html
Rimmer DL, Shiel RS, Syers JK (1990) Effects of soil application of selenium on pasture composition. J Sci Food Agric 51:407–410
Ros GH, Van Rotterdam AMD, Bussink DW, Bindraban PS (2016) Selenium fertilization strategies for bio-fortification of food: an agro-ecosystem approach. Plant Soil 404:99–112
Shand C, Coutts G, Duff E, Atkinson D (1992) Soil selenium treatments to ameliorate selenium deficiency in herbage. Sci Food Agric 59:27–35
Smith JH, Douglas CL (1971) Wheat straw decomposition in the field. Soil Soc Am Proc 35:269–272
Stadlober M, Sager M, Irgolic KJ (2001) Effects of selenate supplemented fertilisation on the selenium level of cereals – identification and quantification of selenium compounds by HPLC-ICP-MS. Food Chem 73:357–366
Stavridou E, Thorup-Kristensen K, Young SD (2011) Assessment of selenium mineralization and availability from catch crops. Soil Use Manag 27:305–311
Stavridou E, Young SD, Thorup-Kristensen K (2012) The effect of catch crop species on selenium availability for succeeding crops. Plant Soil 351:149–160
Stephen RC, Saville DJ, Watkinson JH (1989) The effects of sodium selenate applications on growth and selenium concentrations in wheat. N Z J Crop Hortic Sci 17:229–237
Stroud JL, Li HF, Lopez-Bellido FJ, Broadley MR, Foot I, Fairweather-Tait SJ, Hart DJ, Hurst R, Knott P, Mowat H, Norman K, Scott P, Tucker M, White PJ, Mcgrath SP, Zhao FJ (2010b) Impact of sulphur fertilisation on crop response to selenium fertilisation. Plant Soil 332:31–40
Stroud JL, Broadley MR, Foot I, Fairweather-Tait SJ, Hart DJ, Hurst R, Knott P, Mowat H, Norman K, Scott P, Tucker M, White PJ, Mcgrath SP, Zhao FJ (2010a) Soil factors affecting selenium concentration in wheat grain and the fate and speciation of Se fertilisers applied to soil. Plant Soil 332:19–30
Valle G, Mcdowell L, Prichard D, Chenoweth P, Wright D, Martin D, Kunkle B, Wilkinson N (2002) Selenium concentration of fescue and bahiagrasses after applying a selenium fertilizer. Commun Soil Sci Plant Anal 33:1461–1472
Wang JW, Wang ZH, Mao H, Zhao HB, Huang DL (2013) Increasing Se concentration in maize grain with soil- or foliar-applied selenite on the Loess Plateau in China. Field Crop Res 150:83–90
Watts MJ, Mitchell CJ (2009) A pilot study on iodine in soils of Greater Kabul and Nangarhar provinces of Afghanistan. Environ Geochem Health 31:502–509
Whelan BR (1989) Uptake of selenite fertiliser by subterranean clover pasture in Western Australia. Aust J Exp Agric 29:517–522
World Health Organisation., 2011. Selenium in drinking water. Background document for development of WHO Guidelines for Drinking-water Quality
Ylaranta T (1983) Sorption of selenite and selenate in the soil. Annales Agriculturae Fenniae 22:29–39
Zadoks JC, Chang TT, Konzak CF (1974) Decimal code for growth stages of cereals. Weed Res 14:415–421
Zając T, Oleksy A, Stokłosa A, Klimek-Kopyra A, Macuda J (2013) Vertical distribution of dry mass in cereals straw and its loss during harvesting. Int Agrophys 27:89–95
Zayed A, Lytle CM, Terry N (1998) Accumulation and volatilization of different chemical species of selenium by plants. Planta 206:284–292
Zhao CY, Ren JG, Xue CZ, Lin ED (2005) Study on the relationship between soil selenium and plant selenium uptake. Plant Soil 277:197–206
Zhao FJ, Mcgrath SP (1994) Extractable sulfate and organic sulfur in soils and their availability to plants. Plant Soil 164:243–250
Acknowledgements
This project was funded by the Lawes Agricultural Trust, Rothamsted Research (Charity number 208228) and the University of Nottingham, UK.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Ismail Cakmak.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mathers, A.W., Young, S.D., McGrath, S.P. et al. Determining the fate of selenium in wheat biofortification: an isotopically labelled field trial study. Plant Soil 420, 61–77 (2017). https://doi.org/10.1007/s11104-017-3374-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-017-3374-y