
Abstract
Polyethylene glycol (PEG) hydrogels are widely used in a variety of biomedical applications, including matrices for controlled release of biomolecules and scaffolds for regenerative medicine. The design, fabrication, and characterization of PEG hydrogels rely on the understanding of fundamental gelation kinetics as well as the purpose of the application. This review article will focus on different polymerization mechanisms of PEG-based hydrogels and the importance of these biocompatible hydrogels in regenerative medicine applications. Furthermore, the design criteria that are important in maintaining the availability and stability of the biomolecules as well as the mechanisms for loading of biomolecules within PEG hydrogels will also be discussed. Finally, we overview and provide a perspective on some of the emerging novel design and applications of PEG hydrogel systems, including the spatiotemporal-controlled delivery of biomolecules, hybrid hydrogels, and PEG hydrogels designed for controlled stem cell differentiation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Multiple physical, chemical, and biological cues are known to act cooperatively and/or synergistically to affect cellular function during tissue regeneration in vitro and in vivo (1). To design successful biomaterials for facilitating tissue regeneration, one must thoughtfully consider the interplay between the targeted cells/tissues and these environmental cues. Important factors often include soluble growth factors, cell–cell and cell–material interactions, and mechanical properties of the microenvironment (1). Among these, the delivery of bioactive molecules has been the subject of intensive research, and the targeted molecules are diverse, including low molecular weight drugs, nucleic acids, peptides, and proteins, for the accelerated regeneration of tissues. With respect to their role as delivery vehicles, hydrogels are a class of materials with numerous advantages to simultaneously encapsulate cells and biomolecules, and numerous gel systems allow one to intimately control the release characteristics through systematic changes in the gel’s physical and chemical structure (2). With respect to tailoring gel properties, those derived from synthetic polymers, such as poly(ethylene glycol) (PEG), poly(vinyl alcohol) (PVA), poly(N-isopropylacrylamide) (PNIPAAM), are particularly useful and have been used in numerous regenerative medicine applications (3). Of these, the non-ionic, hydrophilic PEG gel systems are increasingly used and represent the focus of this review.
Over the past few decades, poly(ethylene glycol) (PEG) hydrogels have been extensively used as matrices for controlling drug delivery, as well as cell delivery vehicles for promoting tissue regeneration (4–7). The versatility of the PEG macromer chemistry (8), together with its excellent biocompatibility, has spurred the development of numerous intelligently-designed hydrogel systems for regenerative medicine applications. Many of these studies have produced encouraging pre-clinical and clinical results. In the context of controlled delivery, properly designed PEG hydrogels play an important role in directing cellular functions that are important for survival, proliferation, secretory properties, and even differentiation. The objectives and design principles for this purpose are two-fold: to provide local and extended release of the loaded therapeutics to augment the therapeutic effect; and to decrease the adverse reactions and preserve the bioactivity of the therapeutics. To achieve these goals, one has to carefully consider several critical factors, including the physiological environments of the target tissues, gelation and molecule loading/release mechanisms, molecular characteristics of the therapeutics to be delivered, as well as potential interactions with the polymeric hydrogels.
PEG hydrogels provide a unique niche for cell encapsulation, as they are highly biocompatible to the cells under proper polymerization conditions (9). Through co-polymerization with other macromolecules, multiple functional moieties are readily introduced to suppress or promote cell survival and function. For example, the integrin binding peptide Arg-Gly-Asp (or RGD) is often introduced as a pendant functional group within otherwise bio-inert PEG hydrogels to promote the survival of adherent-dependent cells, such as osteoblasts (10). While PEG hydrogel environments are generally highly permissible and allow for facile diffusion of nutrients, this property often hinders the localized delivery and therapeutic efficacy of soluble factors targeted to the encapsulated cells, as the inert gel networks are equally permeable to the co-encapsulated therapeutics. The means by which bioactive molecules are presented to encapsulated cells within PEG hydrogel networks (both temporally and spatially) is, therefore, a major challenge in the design of hydrogel delivery systems and is currently a subject of intense research.
As we entered into the proteomic era, the increasing number of discovered therapeutically-relevant factors (11) has encouraged the development of material systems to fulfill the requests of delivering these newly identified therapeutic factors. Readers are referred to excellent reviews for the application of hydrogels on controlled drug delivery in tissue engineering and regenerative medicine (2,3,12). In this review, we will focus on an increasingly useful class of hydrogels developed in recent years for controlled release and regenerative medicine applications. Namely, hydrogels formed from the polymerization of macromolecular PEG precursors and the subsequent challenges in the incorporation and release of biologically important molecules for tissue engineering applications. Finally, we will also provide a perspective on current trends in delivery strategies to affect cell survival, proliferation, and differentiation using these PEG-based materials.
PEG HYDROGEL SYSTEMS
Mechanism of Hydrogel Formation: Step-Growth, Chain-Growth, or Mixed-Mode Polymerization
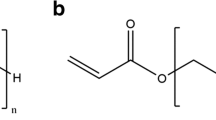
While various methods of gelation (i.e., physical, ionic, or covalent interactions) can be used to form PEG gels, chemically or covalently-crosslinking leads to relatively stable hydrogel structures with tunable physicochemical properties such as permeability, molecular diffusivity, equilibrium water content, elasticity, modulus, and degradation rate (2,3) (Fig. 1). Furthermore, the introduction of degradable linkers into the covalent crosslinks permits the fabrication of well-defined network structures with tailorable properties in time. Generally, the synthesis of covalently crosslinked PEG gels falls into one of the following categories, according to the crosslinking reaction mechanism (13): chain-growth, step-growth, or mixed-mode chain and step growth (Fig. 2 ). Fig. 2A illustrates the network structure resulting from the chain-growth polymerization of macromolecular PEG precursors. Typically, these networks are formed from functional PEG molecules, such as PEG-di(meth)acrylate (Fig. 3 ). Polymerization is initiated by reactive centers, such as radicals, generated from thermal energy, redox reactions, or the photo-cleavage of initiator molecules. These free radicals propagate through unsaturated vinyl bonds on the PEG macromolecular monomers and chain polymerization occurs. The propagation of free radicals through multiple carbon–carbon double bonds on the constituting PEG monomers results in covalently crosslinked, high molecular weight kinetic chains. The functionalities of the chain-growth polymerized hydrogels can be expanded through the copolymerization of other functional (meth)acrylated macromers. One disadvantage of chain-growth polymerization, compared to step-growth mechanism, is that it can lead to lower conversion of the functional groups. Therefore, when it is used in in situ polymerization, un-reacted monomers and/or functional groups remaining in the body may cause local inflammatory reaction or systematic immune response (14). Furthermore, hydrogels made from chain-growth polymerization usually contain certain network non-idealities (15) (Fig. 1A) that may adversely affect drug release performance and material properties.

A Simplified crosslinked hydrogel structure. Black dots represent crosslinking point; ξ represents mesh size of the gel. B Hydrogel property as a function of gel crosslinking density.

Schematic structures of PEG hydrogels formed via: A chain-growth, B step-growth, and C mixed-mode step and chain growth polymerization. (Components not scale to actual size).

Chemical structures of PEG macromer and its di(meth)acrylate derivatives that often solution polymerize to form hydrogel networks useful for cell encapsulation and other biomaterial applications.
Of all the chain polymerization schemes, photopolymerization is one of the preferable ways to fabricate hydrogels. Hydrogels formed via photopolymerizations usually take minutes to complete, and hence, avoid persistent exposure of biomolecules to thermal energy, a characteristic of thermally initiated polymerization and a condition generally not suitable for in situ encapsulation of fragile proteins and cells. The ability of photopolymerizations to produce stable hydrogel networks mildly and rapidly provides a convenient and efficient method for a variety of regenerative medicine applications.
In contrast to the chain growth mechanism, step-growth gelation occurs when at least two multifunctional monomers with mutually reactive chemical groups are reacted together in either stoichiometric balanced or imbalanced ratio, and the average monomer functionality is greater than 2 (16). Not only can this conjugation reaction be performed under ambient conditions without the use of free-radical initiators, it also produces fewer structural defects during network formation (17) (Fig. 2B), which permits more precise control over the gel crosslinking density and subsequent material properties. This aspect is quite important in the delivery of therapeutics, as it allows accurate mathematical predictions prior to the delivery, which is highly beneficial for determining appropriate drug dosing and release kinetics. Hubbell and colleagues developed a series of degradable hydrogels formed via step-growth Michael-type addition reaction between acrylated star PEG polymer and dithiol (18–21). The degradation products of these gels do not produce high molecular weight kinetic chains, which may not only exceed the glomerular filtration limit but may also induce host inflammatory or immune response. From the standpoint of protein delivery, Michael-type addition reactions decrease possible protein damage due to propagating free radicals as occurred in chain-growth polymerizations, but the presence of thiol groups may reduce the native disulfide bonds of the encapsulated proteins and cause protein denaturation, leading to decreased bioactivity and increased immunogenicity. Although Michael-type hydrogels have been successfully used in protein delivery (18–21) and synthetic extracellular matrix (ECM) for cell in-growth as shown by Hubbell group (19,22,23), these hydrogels do not permit spatial and temporal control over the network structure during network gelation.
Recently, PEG hydrogels formed via “Click” chemistry, another type of step-growth mechanism, have elicited tremendous amount of interests due to its rapid and specific reaction, as well as its versatility with respect to bioconjugation. In this typical reaction scheme, macromers bearing azide and alkyne functional groups are “clicked” together in the presence of catalysts (usually copper ions) to form stable covalent linkages (24,25). The well-defined network structures of click hydrogels open an avenue for creating hydrogels with excellent mechanical properties, while simultaneously permitting the conjugation of biomolecules after the hydrogels are crosslinked. In a recent report by Polizzotti and Anseth (26), spatial and temporal patterning of click hydrogels was made possible by a unique two-step fabrication process. PEG hydrogels were formed via “click” chemistry by reacting tetraazide-functionalized PEG with diacetylene-functionalized allyl ester polypeptide containing photo-reactive acrylate groups in the backbone of the polypeptide. After gelation, photopolymerization was utilized to subsequently pattern the click gels with a polypeptide functionality. This unique hydrogel fabrication method not only produces PEG hydrogels with exceptional mechanical properties but also permits the control over the physical and chemical properties of the PEG hydrogels independently.
One major drawback for conventional click hydrogels, however, is that these cycloaddition reactions are usually catalyzed by copper ion. Due to the cytotoxicity of copper, conventional click chemistry is not suitable for creating hydrogel networks with cell encapsulation in situ. Recently, copper-free click chemistry has been reported by the Bertozzi group (27,28). The specificity and fidelity of copper-free click chemistry offers great potential for a variety of regenerative medicine applications, including live cell imaging, cell surface engineering, and cell encapsulation.
In addition to chain-growth and step-growth mechanisms, PEG hydrogel networks can also be formed from mixed-mode polymerizations that exhibit characteristics between chain and step-growth polymerizations as shown in Fig. 2C (29–31). This rapid gelation of mixed-mode polymerization overcomes the long polymerization time needed in most of the Michael-type addition reaction. Furthermore, functional macromers, such as peptides, can be incorporated at lower concentrations (30,31). Anseth, Bowman and their coworkers have developed a new type of PEG hydrogels based on the mixed-mode polymerization of acrylated monomers and multifunctional thiols (29,32–35). In this approach, solutions with different molar ratios of thiol to acrylate groups were prepared, and the polymerization kinetics were examined either with or without the presence of initiators (29,32–35). Differing from thiol-acrylate Michael addition reaction, thiol-acrylate photopolymerization involves chain transfer of growing polymer chains to thiol monomers (34). The polymerization rates and acrylate conversion increased in the presence of photoinitiators, as more thiol groups were added. Unlike chain-growth photopolymerization, the use of initiator is not required in mixed-mode photopolymerizations; the initiator free mechanism has been studied by others (33,35). The network structure that results from this mechanism is directly impacted by the ratio of complimentary reactive groups. As the ratio of thiol to acrylate groups increases, the networks transition from being chain-like to more step-like. In the case of degradable mixed-mode hydrogels, the changes in material properties during degradation, such as swelling, can also be controlled by variations in thiol-acrylate ratios. This change further permits the control of the molecular weight distributions of the degradation products, which is favorable in many regenerative medicine applications because the degradation products can be naturally eliminated by renal filtration. Finally, spatial and temporal control is possible with the mixed-mode photopolymerization reactions.
Hydrogel Properties and Their Importance in Regenerative Medicine Applications
The essence of using PEG hydrogels to deliver therapeutically-relevant factors for regenerative medicine stems from its hydrophilicity (3). A loosely crosslinked PEG hydrogel can easily contain more than 95% of water in its total mass. The high water-content resembles natural soft tissue and, hence, creates a suitable environment for many encapsulated biomolecules and cells. For tissue regeneration, this is essential as it offers opportunity for facile nutrient-waste exchange that is critical for supporting long-term cell/tissue viability. High water-content, on the other hand, also creates challenges to the encapsulated cells, as it usually cannot prevent smaller cytotoxic molecules from passing through the hydrogel barrier. These cytotoxic molecules, including cytokines and reactive oxygen species, trigger the cellular apoptotic pathway that eventually leads to the failure of the encapsulating cells and are often present at the site of injury/implantation. From the perspective of controlled release, the high water-content of PEG hydrogels is critical in maintaining the bioactivity of hydrophilic biomolecules, but it also creates high permeability for the encapsulated small molecules (2). Consequently, rapid release occurs, which not only shortens the efficacy of the delivery device, but may cause a “dose-dumping” effect that is potentially harmful for the surrounding tissue.
Another beneficial feature of PEG hydrogels comes from its “stealth” or anti-fouling property that repels non-specific protein adsorption and cell adhesion (36,37). This property is intriguing as it allows for creating functional patterns either on PEG surfaces or within three-dimensional PEG hydrogels when modified with proper adhesion molecules. Similar to the high water-content property of PEG hydrogels, the ability of PEG molecules to prevent non-specific biomolecular interactions has its pros and cons. For tissue regeneration, anti-fouling of PEG hydrogels reduces the adhesion of inflammatory cells onto the hydrogel surface and decrease the chances of capsule formation. However, this property also hinders the adsorption of bioactive molecules, such as extracellular matrix (ECM) proteins that supports the growth and function of the encapsulated cells. This subsequently reduces the viability of encapsulated cells that require interactions with the surrounding matrix. Fortunately, functional moieties can be easily incorporated within PEG hydrogels through co-polymerization. A variety of ECM-derived cell adhesion peptide sequences have been incorporated within PEG hydrogels to promote the survival of encapsulated cells, including pancreatic β-cell (38), human mesenchymal stem cells (18,19,31,39,40), etc.
The applicability of PEG hydrogels in regenerative medicine can be further augmented by incorporating degradable segments within the PEG hydrogel networks (41). In particular, the incorporation of hydrolytically degradable poly(lactic acid) (PLA) groups greatly increase the potential applications of PEG hydrogels. When PEG hydrogels are to be used solely as cell delivery scaffolds, the ability to control their degradation rate is critical for enhancing tissue regeneration. For example, tri-block copolymers of PLA-PEG-PLA, containing polylatic acid (PLA) blocks and acrylate end groups, have been used to create photo-polymerizable and hydrolytically degradable hydrogels for controlled release and regenerative medicine applications (15,42–44). The degradation rate of the PLA-PEG-PLA hydrogels can be readily pre-engineered by changing either the polymer content of the hydrogels or the length of the degradable PLA segments.
Although hydrolytic degradation allows one to pre-engineer the hydrogel degradation rate, the ability to modify the gel degradation rate post-fabrication is limited. To overcome this, Hubbell and colleagues designed Michael-type PEG hydrogels with MMPs (matrix metalloproteinases) cleavable linkages to facilitate cell-responsive hydrogel degradation (45) as well as growth factor release (46). This “cell-dictated” hydrogel breakdown is highly beneficial for tissue regeneration as it eliminates the risks of erroneous/improper engineering of network degradation and therapeutic liberation profiles. Rice and Anseth, on the other hand, developed a PEG hydrogel system incorporating enzyme-labile caprolactone units for chondrocyte photo-encapsulation (47,48). This unique enzyme-responsive PEG hydrogel allows one to exogenously control material degradation through the delivery of a lipase, and manipulate the gel degradation in response to the regeneration of tissue.
CONTROLLED RELEASE OF BIOMOLECULES IN PEG HYDROGELS
Design Criteria for Controlled Release
Many fundamental studies have revealed the structure-property relationships of PEG-based hydrogels, especially related to equilibrium swelling, mechanics, and transport properties (3,7). Stemming from these studies, numerous PEG-based hydrogel systems have been developed for controlled delivery of many biomolecules, ranging from small molecular weight drugs to large biomacromolecules, such as nucleic acids, peptides, and proteins. Because of the diversity in the chemistry and size of the delivered molecules, the design criteria for controlled release in PEG-based hydrogels can differ widely from one application to another. Nevertheless, availability and stability of the therapeutics are two major concerns when designing PEG hydrogels for controlled release applications (Table I). To achieve desired therapeutic efficacy in vivo or in vitro, PEG hydrogels should be capable of delivering therapeutics at the right dosage (availability) with preserved molecular bioactivity (stability).
Availability of Biomolecules
The mechanisms by which drugs are released from PEG hydrogels depend upon multiple factors, including the method of drug loading, the size and molecular characteristics of the drug, and the dosage and release profile needed for specific therapeutic application. In general, the molecular release mechanisms in PEG hydrogels include diffusion-controlled, swelling-controlled, and chemically-controlled delivery. Readers are advised toward other reviews for detailed mechanistic descriptions of these release mechanisms (2,3). When releasing small molecular weight therapeutics such as synthetic drugs, small peptides and proteins, the high permeability of PEG hydrogels does not permit facile control over the release kinetics. One easy way to control molecular release kinetics is to tune the gel permeability by adjusting the network crosslinking density. However, this approach relies on size-exclusion and is not effective when controlling the release of small molecules. It is also not desirable for most tissue engineering applications, as a higher crosslinking density often represents decreased hydrophilicity and hence decreased cytocompatibility. Therefore, other controlling mechanisms have to be intelligently implemented if one wishes to maintain the preferential hydrophilic environments provided by PEG hydrogels. For example, a variety of “smart” hydrogels that change their volumetric swelling ratio in response to external stimuli have been developed for controlled release applications (3,49–51). Under physiological stimuli (such as pH, temperature, and ionic strength), these intelligently designed stimuli-responsive hydrogels can switch their physical characteristic from a collapsed to swollen state. In their collapsed state, the encapsulated drugs are protected from external harmful factors such as proteolytic degradation, and the encapsulated drugs are subsequently released in the swollen state under a proper stimulus.
Another strategy for delivering drugs without sacrificing preferential hydrophilicity of PEG hydrogels is the “pro-drug” technique. In this approach, therapeutics can be covalently tethered within PEG hydrogels via pendant functional groups. The incorporation of degradable linkers between the tether and the drug allows for pre-determined liberation and release rates. A variety of linker degradation mechanisms have been proposed including hydrolytic (52,53) and enzymatic degradation (54–56). Although the “pro-drug” technique is an effective means of controlling drug availability with pre-determined release rates while preserving hydrophilicity of PEG hydrogels, one drawback of this approach is that the covalent conjugation of drugs to the degradable linker may decrease the bioactivity of the drugs, especially when fragile proteins and peptides are the target therapeutics. Furthermore, the chemistry of the linker has to be carefully designed and be biocompatible such that no unwanted host immune responses will occur due to the remnants of the partially degraded linker (57,58).
Recently, “affinity” hydrogels have been suggested as an alternative for controlling drug availability in highly swollen PEG hydrogels (59–61). By discovering ligands that reversibly bind to target therapeutics and co-polymerizing them within PEG hydrogels, one can render inert PEG hydrogels to exhibiting specific affinity toward therapeutics. Other than facile controlling over drug release in highly permeable hydrogel environments, the use of affinity hydrogels also discards the needs of direct chemical modification on fragile therapeutics such as proteins. A number of affinity mechanisms have been shown to be effective at controlling drug availability in PEG hydrogels. For instance, cyclodextrin can be tethered into PEG hydrogels for controlled release of small molecular weight hydrophobic drugs (62,63). Heparin, a highly sulfated glycosaminoglycan (GAG), has also been extensively used to fabricate affinity hydrogels due to its reversible affinity to a broad variety of growth factors (59,60,64,65). However, affinity binding through heparin–protein interactions mainly relies on non-specific electrostatic interactions. The potential immunogenicity of heparin and the fact that it binds to numerous proteins further complicate the release outcome in vivo. Thus, peptide mimetics with specific affinity to target proteins have been explored recently as an alternative to heparin-based affinity binding (66,67).
Stability of Biomolecules
One challenging task in controlled delivery of biomacromolecules is how to maintain their bioactivity during hydrogel fabrication and throughout the entire course of release. Maintaining drug stability is of critical importance not only for therapeutic purposes but also for biocompatibility concerns, as it has been shown that denatured proteins/peptides can be immunogenic in vivo (68). It is well known that when encapsulating proteins within hydrophobic polymers (e.g., poly(lactide-co-glycolide) or PLGA), protective agents have to be incorporated within the polymer capsules to reduce protein denaturation due to acidic degradation products (lactic acid and glycolic acid) of PLGA (69–71). Similarly, when proteins or nucleic acids are encapsulated within PEG hydrogels during radical-based hydrogel crosslinking, the presence of highly reactive free radicals, as well as undesired side-reactions between reactive polymer chains and biomolecules, can damage the encapsulated biomolecules. Quick and Anseth have revealed the detrimental effect of free radicals on encapsulated DNA during in situ photoencapsulation and demonstrated that the addition of radical scavengers can effectively preserved the integrity of the encapsulated nucleic acids (72,73). Proper protein protection strategies (21,74) are also beneficial for preserving bioactivity of in situ loaded protein therapeutics. Research from Metters (74,75) as well as from Amsden et al. (76) revealed that the presence of propagating carbon free radicals attacks bioactive sites of proteins during in situ encapsulation. For example, Lin and Metters have shown that the concentration and characteristic of photoinitiators greatly affect the bioactivity and availability of hydrogel encapsulated protein and that the presence of acrylated monomers reinstates protein bioactivity due to the higher reactivity ratio between free radicals and acrylate bonds (74,75).
Loading and Release Mechanisms in PEG Hydrogels
The means by which the drugs are loaded directly impact the availability of the drugs during release. For example, drugs can be loaded into hydrogels through entrapment (Fig. 4A). Since hydrogels are highly swollen polymer networks, drugs can be dispersed uniformly within hydrogel matrices through either post-fabrication equilibrium partitioning (drugs are loaded through incubating gels in concentrated drug solutions) or in situ encapsulation (drugs are encapsulated during network crosslinking). While the former method ensures preserved drug stability (no potential chemical reactions between drugs and hydrogel network during crosslinking/polymerization processes), it does not permit accurate control over the amount of drug loading due to partition limitations. Furthermore, it is difficult to prepare hydrogels loaded with multiple drugs at controllable levels for sophisticating regenerative medicine applications where cooperative/synergistic actions of multiple factors are usually required. On the other hand, in situ encapsulation, a process in which drug loading and hydrogel network crosslinking are achieved simultaneously, can be used to prepare hydrogels loaded with multiple, high quantity drugs in a rapid and mild manner. More importantly, it can also be used to simultaneously encapsulate cells under mild physiological conditions for regenerative medicine applications. The disadvantage of in situ loading, however, is that the polymerization reactions can potentially induce undesired reactions on fragile biomolecules or between the encapsulated molecules and polymer network (see previous section). Peppas and coworkers developed a series of pH-sensitive poly(methacrylic acid)-grafted-ethylene glycol (poly(MAA-g-EG)) hydrogel microparticles for oral insulin delivery (7,77–84). The interaction between pendant methacrylic acid and the grafted ethylene glycol chains provides the pH-responsiveness of the hydrogels. At low pH (stomach) environment, these gels collapse due to the protonation of pendant MAA groups inducing hydrogen bonding with the oxygen on PEG chains. At higher pH environments, such as in the small intestines, this hydrogen bonding is disrupted because of the deprotonation of MAA groups. As a result, the gels swell and permit diffusion of insulin. The physical encapsulation of insulin limits proteolytic degradation of insulin before it reaches the targeted delivery site, preferentially the colon. In addition, the carboxylic acids tethered on the polymer chains chelate metal ions that are essential for enzymatic functions, thus further increasing the bioavailability of the encapsulated insulin. Poly(MAA-g-EG) hydrogels also exhibit mucoadhesiveness (83), which enhances the adsorption of hydrogel microparticles and promotes the utilization of encapsulated insulin. Two factors contribute to this mucoadhesiveness. First, carboxylic acids on poly(MAA) interact with the glycoprotein component of the mucus membrane. Second, the pendant MAA and PEG chains interpenetrate into the mucin layer. The use of poly(MAA-g-EG) microparticles represents a substantial success for orally delivering insulin. In a recent report focusing on oral administration of insulin, hypoglycemic effects were observed for both type 1 and type 2 diabetic rats (84).

Schematic of methods for loading of therapeutics into PEG hydrogels: A entrapment: Drugs are loaded into hydrogels via in situ entrapment or post-fabrication equilibrium partitioning. B Tethering: Drugs are modified with a crosslinkable and cleavable linker that can be liberated once the tethers are degraded hydrolytically or enzymatically. C Multiphase loading: Drugs are pre-loaded into microparticles that are subsequently entrapped in hydrogels. (Components not scale to actual size).
In addition to entrapment, drugs can also be incorporated within PEG hydrogels through tethering (Fig. 4B). Hubbell and coworkers have tethered growth factors within PEG-based hydrogels through enzyme-sensitive peptide linkers. In this approach, therapeutic proteins such as vascular endothelial growth factor (VEGF) can be covalently immobilized within hydrogel networks by enzyme-sensitive oligopeptides (54–56). The release of VEGF is mediated by proteases (e.g., matrix metalloproteinases or MMPs) secreted by migrating cells. The cell-demanded VEGF release matches the release profiles with cellular activity that is critical during tissue regeneration. These enzymatically degradable gels can be formed by photopolymerization or Michael-type addition reaction. The gel characteristics, such as gel formation and swelling, are shown to be unaffected by the incorporation of bulky oligopeptides. Integrin-binding domain (RGD) can also be incorporated into the synthetic biomimetic hydrogel matrices, as pendant chains to facilitate cell adhesion (54). These peptide-containing PEG hydrogel systems can be degraded by proteolytic enzymes secreted by migrating cells, such as collagenase, plasmin, and MMPs. The degradation of the peptide substrate is only sensitive to the corresponding protease. For example, the degradation of the peptide substrate Pro-Leu-Gly-Leu-Trp-Ala-D-Arg is sensitive to MMP-1 and MMP-9 (85). The main advantage of these approaches is that the cellularly controlled gel-degradation rate self-adjusts to the rate of cell infiltration, as gel degradation only occurs when specific enzymes are secreted by migrating cells. The cell-mediated gel degradation as well as growth factor release makes these gels extremely attractive for tissue regeneration applications, as these biomimetic materials only serve as provisional matrices and are degraded by invading cells.
Finally, therapeutics can also be loaded into hydrogels through the incorporation of micro or nanoparticles (Fig. 4C). The presence of multiphases (bulk hydrogel and dispersed microparticles) facilitates the incorporation and delivery of multiple therapeutics. For example, Mikos and colleagues have synthesized such composite hydrogel devices for dual growth factor delivery (86,87). Two growth factors, insulin-like growth factor 1 (IGF-1) and transforming growth factor-β (TGF-β), were incorporated separately within bulk hydrogels made of poly(propylene fumarate-co-ethylene glycol) (P(PF-co-EG)) or oligo(poly(ethylene glycol) fumarate (OPF) and gelatin microparticles embedded within bulk hydrogels (87). The delivery rates of the two growth factors were independently tuned by adjusting the loading positions or hydrogel crosslinking density. These hydrogels are biodegradable, injectable, in situ photocrosslinkable, and biocompatible. The degradation of these hydrogels occurs at the ester linkage within the poly(propylene fumarate) block and can be accelerated by decreasing pH and/or crosslinking density. Because of their excellent structural properties as well as the capability of independently controlling the delivery of multiple factors, these hydrogels hold great potential in drug delivery for regenerative medicine applications.
PEG HYDROGELS IN EMERGING REGENERATIVE MEDICINE
Spatiotemporal-Controlled Delivery for Controlling Cell Fate
Advances in drug delivery technologies for tissue regeneration have emerged from delivering single therapeutics for tissue regeneration (88,89) to temporal regulation of two factors to promote vascularization (90) to increasingly sophisticated strategies to present survival or differentiation cues for stem/progenitor cell differentiation (91–93). While much effort has been dedicated to identifying which biochemical cues are most critical and fabricating appropriate material delivery systems, opportunities and challenges exist for developing advanced drug delivery strategies to accelerate differentiation processes toward committed pathways. Currently, the delivery of survival or differentiation cues for controlling cell fate (either 2D or 3D culture) is usually through medium supplements (i.e., adding growth factors in cell culture medium). Although simple, this approach does not mimic the complex in vivo environments where the biochemical cues for tissue development and/or repair are usually presented in temporally and spatially-regulated manner. For tissue regeneration processes that require sequential presentation of multiple growth factors, PEG hydrogel matrices must be designed in a way that the delivery rates of multiple biochemical cues match the needs of neo-tissue development. Intelligent hydrogel matrices, such as the aforementioned multiphase composites and affinity hydrogels, should provide one such approach for temporally-controlled delivery of multiple factors.
Increasingly, the recruitment of multiple cell types such as inflammatory, immune cells and tissue-specific progenitor cells is acknowledged as important for the tissue regeneration process (94). A typical example is peripheral nerve regeneration where nerve cells migrate/proliferate toward increasing concentrations of neurotrophic factors. Although the exact mechanisms by which multiple cell types are attracted sequentially to the site of injury and the congruous interplay between these cell types are currently under intensive investigation, it is generally recognized that chemotaxis plays an important role in dictating cell migration (94). Therefore, it would be beneficial to create chemical gradients in biomaterials for facilitating migration and proliferation of host cells into the biomaterial environments. Toward this end, initial efforts on surface chemistry and engineering have lead to substantial success and numerous surface patterning and grafting techniques have been developed to generate chemical gradients for guiding cell migration (95–97). On the other hand, the creation of spatially-regulated biochemical cues presentation in 3D hydrogels faces more challenges. One way of circumventing 3D hydrogel patterning is through the use of single- or two-photon confocal laser scanning microscopy (CLSM). Due to the spatial precision of laser-guided patterning and the elimination/suppression of out-of-focus light, precise 3D patterning can be created within a 3D hydrogel structure. Taking advantage of CLSM, Luo and Shoichet have developed photolabile agarose hydrogel for spatially patterning RGD peptide for cell adhesion and migration (98). In this approach, a photolabile 2-nitrobenzyl group was used to protect thiol groups. Upon laser-guided UV exposure, the 2-nitrobenzyl groups were cleaved, leaving free thiol exposed. Maleimide-modified biomolecules were then spatially conjugated within the hydrogels through the thiol-maleimide reaction. Neuron cell migration and neurite outgrowth were observed only within patterned RGD channels. The use of CLSM to create spatially patterned chemical cues should prove useful in the design and fabrication of PEG hydrogels. In an alternative approach, West et al. have developed PEG hydrogels patterned with biochemical gradients that promote cell adhesion and migration (99–101). The patterns were created by forming partially crosslinked PEG hydrogels, followed by diffusing acrylated bioactive cues within the hydrogels. CLSM was subsequently used to photo-crosslink bioactive cues within PEG hydrogels. Using this simple approach, complex 3D geometries can be patterned within PEG hydrogels.
Recent advances in “click” chemistry also open avenues for spatially patterning biomolecules within PEG hydrogels. As mentioned in the previous section, a patterning scheme combining click chemistry and photopolymerization adopted by Polizzotti and Anseth offers novel approaches of creating spatially regulated biochemical cues presentation (26). This method should provide greater therapeutic applicability when copper-free click chemistry is incorporated.
Although laser-guided photopatterning techniques have proven effective in generating 3D hydrogels with spatially immobilized biochemical cues, one question remains to be answered. Can this technique be used to create hydrogel matrices loaded with spatially-distributed growth factors that are only needed temporally? Lessons from wound healing and organ/tissue development have revealed the importance of spatiotemporal distribution of biochemical cues. For example, recent studies have suggested that the persistent presence of the integrin-binding peptide motif Arg-Gly-Asp (RGD) hinders complete chondrogenic differentiation of human mesenchymal stem cells (hMSCs) within hydrogels (31,102,103). On the other hand, Salinas and Anseth designed PEG hydrogels immobilized with MMP-13 cleavable RGD motif for enhancing chondrogenesis of hMSCs (31). A ten-fold increased in glycosaminoglycans (GAGs) production (indication of chondrogenic differentiation) by encapsulated hMSCs was obtained when the linker to RGD motif was cleaved by MMP-13. Therefore, to design the next generation of bioactive hydrogels, new techniques capable of selectively removing biochemical cues should greatly facilitate stem cell differentiation and tissue regeneration.
Hybrid Hydrogels for Dynamic Sensing, Actuation, and Controlled Release
Stimuli-sensitive polymers have been successfully used to fabricate “smart” hydrogels for controlled release applications, including the pH-responsive gel systems for oral insulin delivery developed by Peppas et al. (78,80,84). More recently, hybrid hydrogels composed of synthetic polymers and biological components, such as proteins and peptides, are subjects of intense research (104,105). Differing from conventional stimuli-responsive synthetic polymers/hydrogels where the responsiveness usually comes from changes in physical parameters (e.g., temperature, pH, or hydrophobicity), the responsiveness of hybrid hydrogels is a result of biological recognitions or protein–ligand interactions. Typically, proteins with the ability to change their conformations upon ligand binding are first modified (either chemically or genetically) with crosslinkable moieties, followed by conjugating with synthetic polymer chains to form hybrid hydrogels. The uniqueness of this type of stimuli-responsive hydrogels is their ability to transform biological recognition events (protein–ligand binding) into mechanical displacements (hydrogel swelling).
Since the conception of hybrid hydrogels in the late 1990s by Kopecek et al. (104,105), several examples of such responsive systems have been proposed. For example, Daunert and co-workers designed acrylamide-based hydrogel containing genetically engineered protein–calmodulin (CaM) (106,107). The molecular recognition between CaM and calcium ions triggers the conformational change of CaM and its binding to phenothiazine (both CaM and phenothiazine were polymer-conjugated). This calcium-dependent binding subsequently results in reversible hydrogel swelling changes. Murphy et al. went on to develop PEG-based hybrid hydrogels that incorporate CaM molecules (108,109). The concept of biological-synthetic hybrid hydrogels opens avenues for the design and fabrication of stimuli-responsive microfluidic devices, biosensors, actuators, and miniaturized drug delivery systems.
In addition to the protein–ligand binding, another type of hybrid hydrogels developed for controlled release applications is based on protein-receptor binding. Kiick et al. developed receptor-responsive hydrogels assembled by heparin-binding growth factors and heparin-functionalized star PEG polymers for the delivery of vascular endothelial growth factor (VEGF) (110,111) and basic fibroblast growth factor (bFGF) (112). In this novel responsive hydrogel system, growth factors are not only therapeutics but also act as crosslinkers for hydrogel assembly. The receptor-responsive hybrid hydrogels are self-assembled via reversible binding between VEGF (or bFGF) and heparin conjugated on star PEG polymers. Upon reaching cell surfaces, the growth factors are removed due to binding to receptors. This causes the hydrogels to disassemble due to the loss of crosslinkers. Although this approach is intelligent and has been proven to be effective on promoting cell proliferation in vitro, the non-specific binding between heparin and a variety of growth factors makes it challenging to deliver growth factors in vivo as the heparin-bound growth factors may be replaced by other growth factors (or other biomolecules) exhibiting stronger affinity for heparin before the delivery vehicles reach the target site. The specificity and fidelity of this approach, however, can be expanded when affinity peptides with specific affinity for growth factors are discovered for binding and hybrid hydrogel assembly.
PEG Hydrogels for Controlled Stem Cell Differentiation
As briefly mentioned earlier in this section, PEG hydrogels have been shown to be useful in studying stem cell differentiation in 3-D environments. This is mainly due to the “blank slate” or “inert” PEG networks that make it possible to identify the effects of specific bioactive molecules on the encapsulated stem cell fate (113). For example, Anseth (31,40,53,91,113–118), Elisseeff (103,119–122) and their colleagues have extensively studied the effect of PEG hydrogel environments on controlling the osteogenic or chondrogenic differentiation of human mesenchymal stem cells (hMSCs) or embryonic stem cells (ESCs). To promote the survival of anchorage-dependent hMSCs or ESCs, cell-adhesion molecules, such as RGD peptide, were co-polymerized within PEG-based hydrogels (103,118). Furthermore, it has been shown that the incorporation of bioactive molecules within PEG-based hydrogels have a significant impact on the differentiation of hMSCs. For example, heparin (53,91) or phosphate (115,122) functionalized PEG hydrogels have been shown to promote osteogenic differentiation of hMSCs in the gel environments, possibly due to the retention of cell-secreted bioactive molecules within the functionalized hydrogels. With proper design and selection of functional groups, PEG hydrogels can also be used to induce chondrogenic (31,40,103,118) and adipogenic differentiation (123,124) from hMSCs.
Controlled stem cell differentiation can also be achieved by retaining cell-secreted biomolecules within PEG hydrogels. In in vivo environments, cells usually reside within dense extracellular matrix environments that can effectively retain cell-secreted bioactive molecules for autocrine signaling or other feedback mechanisms. The major benefits of retaining cell-secreted biomolecules, rather than covalently incorporating their synthetic or recombinant analogs within gel environments, are two fold. First, these molecules may only be needed temporarily during the differentiation processes. Secondly, the bioactivity of cell-secreted biomolecules may be more potent than synthetic or recombinant analogs. Although crosslinked hydrogel networks can effectively retain large, cell-secreted large ECM molecules due to the size-exclusion effect, small growth factors and other bioactive molecules secreted by the encapsulated cells may easily diffuse out of the crosslinked gels. As discussed in the previous sections, simply increasing gel crosslinking density is not an effective way of retaining small bioactive molecules within the highly permeable hydrogel environment. Therefore, identify affinity binding motif for specific biomolecules and covalently incorporate them within PEG hydrogels may greatly promote the survival, function, and differentiation of the encapsulated cells. The incorporation of peptide sequence KLER, a collagen II binding domain derived from decorin, has been shown to promote chondrogenic differentiation of hMSCs in PEG hydrogels (117). Although the binding between heparin and growth factors is not considered specific, the incorporation of heparin within PEG hydrogels can also be used as a binding mechanism for retaining cell-secreted growth factors (53).
CONCLUSION
With growing research interest in the use of PEG hydrogels for regenerative medicine applications, proper delivery strategies should be impregnated within PEG hydrogels to fulfill the diverse nature of tissue development/regeneration, as this will significantly affect the outcome of tissue regeneration. For delivering therapeutics to repair damage tissues, methods for tailoring availability and stability of bioactive molecules should be considered in parallel to augment the regeneration processes. For delivering survival and differentiation cues, general considerations should incorporate critical aspects of the dynamic in vivo environments and lessons from organ/tissue developments should provide valuable information regarding the criteria of hydrogel design. The development of in situ forming PEG hydrogel chemistry and fabrication techniques have offered a solid foundation on which current and future delivery strategies are built.
References
M. P. Lutolf, and J. A. Hubbell. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat. Biotechnol. 23:47–55 (2005). doi:10.1038/nbt1055.
C. C. Lin, and A. T. Metters. Hydrogels in controlled release formulations: Network design and mathematical modeling. Adv. Drug Deliv. Rev. 58:1379–1408 (2006). doi:10.1016/j.addr.2006.09.004.
N. A. Peppas, P. Bures, W. Leobandung, and H. Ichikawa. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 50:27–46 (2000). doi:10.1016/S0939-6411(00)00090-4.
N. A. Peppas, J. Z. Hilt, A. Khademhosseini, and R. Langer. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv. Mater. 18:1345–1360 (2006). doi:10.1002/adma.200501612.
J. L. Drury, and D. J. Mooney. Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials. 24:4337–4351 (2003). doi:10.1016/S0142-9612(03)00340-5.
M. B. Mellott, K. Searcy, and M. V. Pishko. Release of protein from highly cross-linked hydrogels of poly(ethylene glycol) diacrylate fabricated by UV polymerization. Biomaterials. 22:929–941 (2001). doi:10.1016/S0142-9612(00)00258-1.
N. A. Peppas, K. B. Keys, M. Torres-Lugo, and A. M. Lowman. Poly(ethylene glycol)-containing hydrogels in drug delivery. J. Control. Release. 62:81–87 (1999). doi:10.1016/S0168-3659(99)00027-9.
C. R. Nuttelman, M. A. Rice, A. E. Rydholm, C. N. Salinas, D. N. Shah, and K. S. Anseth. Macromolecular monomers for the synthesis of hydrogel niches and their application in cell encapsulation and tissue engineering. Prog. Polym. Sci. 33:167–179 (2008). doi:10.1016/j.progpolymsci.2007.09.006.
S. J. Bryant, C. R. Nuttelman, and K. S. Anseth. Cytocompatibility of UV and visible light photoinitiating systems on cultured NIH/3T3 fibroblasts in vitro. J. Biomater. Sci. Polym. Ed. 11:439–457 (2000). doi:10.1163/156856200743805.
J. A. Burdick, and K. S. Anseth. Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials. 23:4315–4323 (2002). doi:10.1016/S0142-9612(02)00176-X.
A. K. Pavlou, and J. M. Reichert. Recombinant protein therapeutics—success rates, market trends and values to 2010. Nat. Biotechnol. 22:1513–1519 (2004). doi:10.1038/nbt1204-1513.
K. A. Davis, and K. S. Anseth. Controlled release from crosslinked degradable networks. Crit. Rev. Ther. Drug Carr. Syst. 19:385–423 (2002). doi:10.1615/CritRevTherDrugCarrierSyst.v19.i45.30.
A. E. Rydholm, C. N. Bowman, and K. S. Anseth. Degradable thiol-acrylate photopolymers: polymerization and degradation behavior of an in situ forming biomaterial. Biomaterials. 26:4495–4506 (2005). doi:10.1016/j.biomaterials.2004.11.046.
E. Fournier, C. Passirani, C. N. Montero-Menei, and J. P. Benoit. Biocompatibility of implantable synthetic polymeric drug carriers: focus on brain biocompatibility. Biomaterials. 24:3311–3331 (2003). doi:10.1016/S0142-9612(03)00161-3.
A. T. Metters, K. S. Anseth, and C. N. Bowman. A statistical kinetic model for the bulk degradation of PLA-b-PEG-b-PLA hydrogel networks: Incorporating network non-idealities. J. Phys. Chem. B. 105:8069–8076 (2001). doi:10.1021/jp004083h.
G. Odian. Principles of polymerization. Wiley, New Jersey, 2004.
M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury, and C. J. Hawker. Synthesis of well-defined hydrogel networks using Click chemistry. Chem. Commun. 2774–2776 (2006). doi:10.1039/b603438a.
S. C. Rizzi, M. Ehrbar, S. Halstenberg, G. P. Raeber, H. G. Schmoekel, H. Hagenmuller, R. Muller, F. E. Weber, and J. A. Hubbell. Recombinant protein-co-PEG networks as cell-adhesive and proteolytically degradable hydrogel matrixes. Part II: Biofunctional characteristics. Biomacromolecules. 7:3019–3029 (2006). doi:10.1021/bm060504a.
S. C. Rizzi, and J. A. Hubbell. Recombinant protein-co-PEG networks as cell-adhesive and proteolytically degradable hydrogel matrixes. Part 1: Development and physicochernical characteristics. Biomacromolecules. 6:1226–1238 (2005). doi:10.1021/bm049614c.
D. L. Elbert, A. B. Pratt, M. P. Lutolf, S. Halstenberg, and J. A. Hubbell. Protein delivery from materials formed by self-selective conjugate addition reactions. J. Control. Release. 76:11–25 (2001). doi:10.1016/S0168-3659(01)00398-4.
P. van de Wetering, A. T. Metters, R. G. Schoenmakers, and J. A. Hubbell. Poly(ethylene glycol) hydrogels formed by conjugate addition with controllable swelling, degradation, and release of pharmaceutically active proteins. J. Control. Release. 102:619–627 (2005). doi:10.1016/j.jconrel.2004.10.029.
T. P. Kraehenbuehl, P. Zammaretti, A. J. Van der Vlies, R. G. Schoenmakers, M. P. Lutolf, M. E. Jaconi, and J. A. Hubbell. Three-dimensional extracellular matrix-directed cardioprogenitor differentiation: systematic modulation of a synthetic cell-responsive PEG-hydrogel. Biomaterials. 29:2757–2766 (2008). doi:10.1016/j.biomaterials.2008.03.016.
G. P. Raeber, M. P. Lutolf, and J. A. Hubbell. Molecularly engineered PEG hydrogels: a novel model system for proteolytically mediated cell migration. Biophys. J. 89:1374–1388 (2005). doi:10.1529/biophysj.104.050682.
H. C. Kolb, M. G. Finn, and K. B. Sharpless. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 40:2004–2020 (2001).
V. D. Bock, H. Hiemstra, and J. H. van Maarseveen. Cu–I-catalyzed alkyne-azide “click” cycloadditions from a mechanistic and synthetic perspective. Eur. J. Org. Chem. 51–68 (2005).
B. D. Polizzotti, B. D. Fairbanks, and K. S. Anseth. Three-dimensional biochemical patterning of click-based composite hydrogels via thiolene photopolymerization. Biomacromolecules. 9:1084–1087 (2008). doi:10.1021/bm7012636.
J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli, and C. R. Bertozzit. Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. U.S.A. 104:16793–16797 (2007). doi:10.1073/pnas.0707090104.
S. T. Laughlin, J. M. Baskin, S. L. Amacher, and C. R. Bertozzi. In vivo imaging of membrane-associated glycans in developing zebrafish. Science. 320:664–667 (2008). doi:10.1126/science.1155106.
A. E. Rydholm, S. K. Reddy, K. S. Anseth, and C. N. Bowman. Controlling network structure in degradable thiol-acrylate biomaterials to tune mass loss behavior. Biomacromolecules. 7:2827–2836 (2006). doi:10.1021/bm0603793.
C. N. Salinas, and K. S. Anseth. Mixed mode thiol-acrylate photopolymerizations for the synthesis of PEG-peptide hydrogels. Macromolecules. 41:6019–6026 (2008). doi:10.1021/ma800621h.
C. N. Salinas, and K. S. Anseth. The enhancement of chondrogenic differentiation of human mesenchymal stem cells by enzymatically regulated RGD functionalities. Biomaterials. 29:2370–2377 (2008). doi:10.1016/j.biomaterials.2008.01.035.
N. B. Cramer, and C. N. Bowman. Kinetics of thiol-ene and thiol-acrylate photopolymerizations with real-time Fourier transform infrared. J. Polym. Sci. A, Polym. Chem. 39:3311–3319 (2001). doi:10.1002/pola.1314.
N. B. Cramer, J. P. Scott, and C. N. Bowman. Photopolymerizations of thiol-ene polymers without photoinitiators. Macromolecules. 35:5361–5365 (2002). doi:10.1021/ma0200672.
N. B. Cramer, T. Davies, A. K. O'Brien, and C. N. Bowman. Mechanism and modeling of a thiol-ene photopolymerization. Macromolecules. 36:4631–4636 (2003). doi:10.1021/ma034072x.
N. B. Cramer, S. K. Reddy, M. Cole, C. Hoyle, and C. N. Bowman. Initiation and kinetics of thiol-ene photopolymerizations without photoinitiators. J. Polym. Sci. A, Polym. Chem. 42:5817–5826 (2004). doi:10.1002/pola.20419.
M. D. Scott, and K. L. Murad. Cellular camouflage: fooling the immune system with polymers. Curr. Pharm. Des. 4:423–438 (1998).
G. Csucs, R. Michel, J. W. Lussi, M. Textor, and G. Danuser. Microcontact printing of novel co-polymers in combination with proteins for cell-biological applications. Biomaterials. 24:1713–1720 (2003). doi:10.1016/S0142-9612(02)00568-9.
L. M. Weber, K. N. Hayda, K. Haskins, and K. S. Anseth. The effects of cell-matrix interactions on encapsulated beta-cell function within hydrogels functionalized with matrix-derived adhesive peptides. Biomaterials. 28:3004–3011 (2007). doi:10.1016/j.biomaterials.2007.03.005.
N. Gurav, M. P. Lutolf, G. P. Raeber, J. A. Hubbell, and L. Di Silvio. Differentiation of human bone marrow stem cells within RGD functionalised, proteolytically sensitive PEG gels. Tissue Eng. 13:1675–1675 (2007).
C. N. Salinas, B. B. Cole, A. M. Kasko, and K. S. Anseth. Chondrogenic differentiation potential of human mesenchymal stem cells photoencapsulated within poly(ethylene glycol)-arginine-glycine-aspartic acid-serine thiol-methacrylate mixed-mode networks. Tissue Eng. 13:1025–1034 (2007). doi:10.1089/ten.2006.0126.
G. D. Nicodemus, and S. J. Bryant. Cell encapsulation in biodegradable hydrogels for tissue engineering applications. Tissue Eng. Part B-Reviews. 14:149–165 (2008).
A. T. Metters, K. S. Anseth, and C. N. Bowman. Fundamental studies of a novel, biodegradable PEG-b-PLA hydrogel. Polymer. 41:3993–4004 (2000). doi:10.1016/S0032-3861(99)00629-1.
M. N. Mason, A. T. Metters, C. N. Bowman, and K. S. Anseth. Predicting controlled-release behavior of degradable PLA-b-PEG-b-PLA hydrogels. Macromolecules. 34:4630–4635 (2001). doi:10.1021/ma010025y.
K. S. Anseth, A. T. Metters, S. J. Bryant, P. J. Martens, J. H. Elisseeff, and C. N. Bowman. In situ forming degradable networks and their application in tissue engineering and drug delivery. J. Control. Release. 78:199–209 (2002). doi:10.1016/S0168-3659(01)00500-4.
M. P. Lutolf, J. L. Lauer-Fields, H. G. Schmoekel, A. T. Metters, F. E. Weber, G. B. Fields, and J. A. Hubbell. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: engineering cell-invasion characteristics. Proc. Natl. Acad. Sci. U. S. A. 100:5413–5418 (2003). doi:10.1073/pnas.0737381100.
M. Ehrbar, A. Metters, P. Zammaretti, J. A. Hubbell, and A. H. Zisch. Endothelial cell proliferation and progenitor maturation by fibrin-bound VEGF variants with differential susceptibilities to local cellular activity. J. Control. Release. 101:93–109 (2005). doi:10.1016/j.jconrel.2004.07.018.
M. A. Rice, J. Sanchez-Adams, and K. S. Anseth. Exogenously triggered, enzymatic degradation of photopolymerized hydrogels with polycaprolactone subunits: experimental observation and modeling of mass loss behavior. Biomacromolecules. 7:1968–1975 (2006). doi:10.1021/bm060086+.
M. A. Rice, and K. S. Anseth. Controlling cartilaginous matrix evolution in hydrogels with degradation triggered by exogenous addition of an enzyme. Tissue Eng. 13:683–691 (2007). doi:10.1089/ten.2006.0142.
N. A. Peppas, and B. Kim. Stimuli-sensitive protein delivery systems. Journal of Drug Delivery Science and Technology. 16:11–18 (2006).
M. E. H. El-Sayed, A. S. Hoffman, and P. S. Stayton. Smart polymeric carriers for enhanced intracellular delivery of therapeutic macromolecules. Expert Opin. Biol. Ther. 5:23–32 (2005). doi:10.1517/14712598.5.1.23.
A. S. Hoffman, and P. S. Stayton. Conjugates of stimuli-responsive polymers and proteins. Prog. Polym. Sci. 32:922–932 (2007). doi:10.1016/j.progpolymsci.2007.05.005.
J. W. DuBose, C. Cutshall, and A. T. Metters. Controlled release of tethered molecules via engineered hydrogel degradation: model development and validation. J. Biomed. Mater. Res. Part A. 74A:104–116 (2005). doi:10.1002/jbm.a.30307.
D. S. W. Benoit, S. D. Collins, and K. S. Anseth. Multifunctional hydrogels that promote osteogenic human mesenchymal stem cell differentiation through stimulation and sequestering of bone morphogenic protein 2. Adv. Funct. Mater. 17:2085–2093 (2007). doi:10.1002/adfm.200700012.
A. H. Zisch, M. P. Lutolf, M. Ehrbar, G. P. Raeber, S. C. Rizzi, N. Davies, H. Schmokel, D. Bezuidenhout, V. Djonov, P. Zilla, and J. A. Hubbell. Cell-demanded release of VEGF from synthetic, biointeractive cell-ingrowth matrices for vascularized tissue growth. FASEB J. 17:2260–2262 (2003).
M. Ehrbar, V. G. Djonov, C. Schnell, S. A. Tschanz, G. Martiny-Baron, U. Schenk, J. Wood, P. H. Burri, J. A. Hubbell, and A. H. Zisch. Cell-demanded liberation of VEGF(121) from fibrin implants induces local and controlled blood vessel growth. Circ. Res. 94:1124–1132 (2004). doi:10.1161/01.RES.0000126411.29641.08.
R. G. Schoenmakers, P. van de Wetering, D. L. Elbert, and J. A. Hubbell. The effect of the linker on the hydrolysis rate of drug-linked ester bonds. J. Control. Release. 95:291–300 (2004). doi:10.1016/j.jconrel.2003.12.009.
H. Soyez, E. Schacht, M. Jelinkova, and B. Rihova. Biological evaluation of mitomycin C bound to a biodegradable polymeric carrier. J. Control. Release. 47:71–80 (1997). doi:10.1016/S0168-3659(96)01617-3.
I. Watermann, J. Gerspach, M. Lehne, J. Seufert, B. Schneider, K. Pfizenmaier, and H. Wajant. Activation of CD95L fusion protein prodrugs by tumor-associated proteases. Cell Death and Differ. 14:765–774 (2007). doi:10.1038/sj.cdd.4402051.
S. E. Sakiyama-Elbert, and J. A. Hubbell. Controlled release of nerve growth factor from a heparin-containing fibrin-based cell ingrowth matrix. J. Control. Release. 69:149–158 (2000). doi:10.1016/S0168-3659(00)00296-0.
S. E. Sakiyama-Elbert, and J. A. Hubbell. Development of fibrin derivatives for controlled release of heparin-binding growth factors. J. Control. Release. 65:389–402 (2000). doi:10.1016/S0168-3659(99)00221-7.
C. C. Lin, and A. T. Metters. Metal-chelating affinity hydrogels for sustained protein release. J. Biomed. Mater. Res. Part A. 83A:954–964 (2007). doi:10.1002/jbm.a.31282.
J. Li, X. Li, X. P. Ni, X. Wang, H. Z. Li, and K. W. Leong. Self-assembled supramolecular hydrogels formed by biodegradable PEO-PHB-PEO triblock copolymers and alpha-cyclodextrin for controlled drug delivery. Biomaterials. 27:4132–4140 (2006). doi:10.1016/j.biomaterials.2006.03.025.
J. Li, X. P. Ni, and K. W. Leong. Injectable drug-delivery systems based on supramolecular hydrogels formed by poly(ethylene oxide) and alpha-cyclodextrin. J. Biomed. Mater. Res. Part A. 65A:196–202 (2003). doi:10.1002/jbm.a.10444.
S. S. Cai, Y. C. Liu, X. Z. Shu, and G. D. Prestwich. Injectable glycosaminoglycan hydrogels for controlled release of human basic fibroblast growth factor. Biomaterials. 26:6054–6067 (2005). doi:10.1016/j.biomaterials.2005.03.012.
D. B. Pike, S. S. Cai, K. R. Pomraning, M. A. Firpo, R. J. Fisher, X. Z. Shu, G. D. Prestwich, and R. A. Peattie. Heparin-regulated release of growth factors in vitro and angiogenic response in vivo to implanted hyaluronan hydrogels containing VEGF and bFGF. Biomaterials. 27:5242–5251 (2006). doi:10.1016/j.biomaterials.2006.05.018.
H. D. Maynard, and J. A. Hubbell. Discovery of a sulfated tetrapeptide that binds to vascular endothelial growth factor. Acta Biomaterialia. 1:451–459 (2005). doi:10.1016/j.actbio.2005.04.004.
S. M. Willerth, P. J. Johnson, D. J. Maxwell, S. R. Parsons, M. E. Doukas, and S. E. Sakiyama-Elbert. Rationally designed peptides for controlled release of nerve growth factor from fibrin matrices. J. Biomed. Mater. Res. Part A. 80A:13–23 (2007). doi:10.1002/jbm.a.30844.
J. B. Rothbard, and M. L. Gefter. Interactions between immunogenic peptides and Mhc proteins. Annu. Rev. Immunol. 9:527–565 (1991). doi:10.1146/annurev.iy.09.040191.002523.
J. C. Kang, and S. P. Schwendeman. Comparison of the effects of Mg(OH)(2) and sucrose on the stability of bovine serum albumin encapsulated in injectable poly(D,L-lactide-co-glycolide) implants. Biomaterials. 23:239–245 (2002). doi:10.1016/S0142-9612(01)00101-6.
G. Z. Zhu, S. R. Mallery, and S. P. Schwendeman. Stabilization of proteins encapsulated in injectable poly (lactide-co-glycolide). Nat. Biotechnol. 18:52–57 (2000). doi:10.1038/71916.
G. Z. Zhu, and S. P. Schwendeman. Stabilization of proteins encapsulated in cylindrical poly(lactide-co-glycolide) implants: mechanism of stabilization by basic additives. Pharm. Res. 17:351–357 (2000). doi:10.1023/A:1007513425337.
D. J. Quick, and K. S. Anseth. Gene delivery in tissue engineering: a photopolymer platform to coencapsulate cells and plasmid DNA. Pharm. Res. 20:1730–1737 (2003). doi:10.1023/B:PHAM.0000003368.66471.6a.
D. J. Quick, and K. S. Anseth. DNA delivery from photocrosslinked PEG hydrogels: encapsulation efficiency, release profiles, and DNA quality. J. Control. Release. 96:341–351 (2004). doi:10.1016/j.jconrel.2004.01.021.
C. C. Lin, and A. T. Metters. Enhanced protein delivery from photopolymerized hydrogels using a pseudospecific metal chelating ligand. Pharm. Res. 23:614–622 (2006). doi:10.1007/s11095-005-9395-x.
C. C. Lin, S. M. Sawicki, and A. T. Metters. Free-radical-mediated protein inactivation and recovery during protein photoencapsulation. Biomacromolecules. 9:75–83 (2008). doi:10.1021/bm700782c.
F. Gu, R. Neufeld, and B. Amsden. Maintenance of vascular endothelial growth factor and potentially other therapeutic proteins bioactivity during a photo-initiated free radical cross-linking reaction forming biodegradable elastomers. Eur. J. Pharm. Biopharm. 66:21–27 (2007). doi:10.1016/j.ejpb.2006.08.006.
M. Torres-Lugo, and N. A. Peppas. Preparation and characterization of P(MAA-g-EG) nanospheres for protein delivery applications. J Nanopart. Res. 4:73–81 (2002). doi:10.1023/A:1020137616302.
B. Kim, and N. A. Peppas. In vitro release behavior and stability of insulin in complexation hydrogels as oral drug delivery carriers. Int. J. Pharm. 266:29–37 (2003). doi:10.1016/S0378-5173(03)00378-8.
B. Kim, K. La Flamme, and N. A. Peppas. Dynamic swelling Behavior of pH-sensitive anionic hydrogels used for protein delivery. J. Appl. Polym. Sci. 89:1606–1613 (2003). doi:10.1002/app.12337.
K. Nakamura, R. J. Murray, J. I. Joseph, N. A. Peppas, M. Morishita, and A. M. Lowman. Oral insulin delivery using P(MAA-g-EG) hydrogels: effects of network morphology on insulin delivery characteristics. J. Control. Release. 95:589–599 (2004). doi:10.1016/j.jconrel.2003.12.022.
L. Serra, J. Domenech, and N. A. Peppas. Drug transport mechanisms and release kinetics from molecularly designed poly(acrylic acid-g-ethylene glycol) hydrogels. Biomaterials. 27:5440–5451 (2006). doi:10.1016/j.biomaterials.2006.06.011.
T. Yamagata, M. Morishita, N. J. Kavimandan, K. Nakamura, Y. Fukuoka, K. Takayama, and N. A. Peppas. Characterization of insulin protection properties of complexation hydrogels in gastric and intestinal enzyme fluids. J. Control. Release. 112:343–349 (2006). doi:10.1016/j.jconrel.2006.03.005.
L. Serra, J. Domenech, and N. A. Peppas. Design of poly(ethylene glycol)-tethered copolymers as novel mucoadhesive drug delivery systems. Eur. J. Pharm. Biopharm. 63:11–18 (2006). doi:10.1016/j.ejpb.2005.10.011.
M. Morishita, T. Goto, K. Nakamura, A. M. Lowman, K. Takayama, and N. A. Peppas. Novel oral insulin delivery systems based on complexation polymer hydrogels: Single and multiple administration studies in type 1 and 2 diabetic rats. J. Control. Release. 110:587–594 (2006). doi:10.1016/j.jconrel.2005.10.029.
G. M. McGeehan, D. M. Bickett, M. Green, D. Kassel, J. S. Wiseman, and J. Berman. Characterization of the peptide substrate specificities of interstitial collagenase and 92-kDa gelatinase—implications for substrate optimization. J. Biol. Chem. 269:32814–32820 (1994).
T. A. Holland, J. K. V. Tessmar, Y. Tabata, and A. G. Mikos. Transforming growth factor-beta 1 release from oligo(poly(ethylene glycol) fumarate) hydrogels in conditions that model the cartilage wound healing environment. J. Control. Release. 94:101–114 (2004). doi:10.1016/j.jconrel.2003.09.007.
T. A. Holland, Y. Tabata, and A. G. Mikos. Dual growth factor delivery from degradable oligo(poly(ethylene glycol) fumarate) hydrogel scaffolds for cartilage tissue engineering. J. Control. Release. 101:111–125 (2005). doi:10.1016/j.jconrel.2004.07.004.
K. Y. Lee, M. C. Peters, K. W. Anderson, and D. J. Mooney. Controlled growth factor release from synthetic extracellular matrices. Nature. 408:998–1000 (2000). doi:10.1038/35044106.
J. Y. Lee, S. H. Nam, S. Y. Im, Y. J. Park, Y. M. Lee, Y. J. Seol, C. P. Chung, and S. J. Lee. Enhanced bone formation by controlled growth factor delivery from chitosan-based biomaterials. J. Control. Release. 78:187–197 (2002). doi:10.1016/S0168-3659(01)00498-9.
T. P. Richardson, M. C. Peters, A. B. Ennett, and D. J. Mooney. Polymeric system for dual growth factor delivery. Nat. Biotechnol. 19:1029–1034 (2001). doi:10.1038/nbt1101-1029.
D. S. W. Benoit, A. R. Durney, and K. S. Anseth. The effect of heparin-functionalized PEG hydrogels on three-dimensional human mesenchymal stem cell osteogenic differentiation. Biomaterials. 28:66–77 (2007). doi:10.1016/j.biomaterials.2006.08.033.
E. K. Moioli, L. Hong, J. Guardado, P. A. Clark, and J. J. Mao. Sustained release of TGF beta 3 from PLGA microspheres and its effect on early osteogenic differentiation of human mesenchymal stem cells. Tissue Eng. 12:537–546 (2006). doi:10.1089/ten.2006.12.537.
J. Sohier, D. Hamann, M. Koenders, N. Cucchiarini, H. Madry, C. van Blitterswijk, K. de Groot, and J. M. Bezemer. Tailored release of TGF-beta(1) from porous scaffolds for cartilage tissue engineering. Int. J. Pharm. 332:80–89 (2007). doi:10.1016/j.ijpharm.2006.09.037.
A. J. Ridley, M. A. Schwartz, K. Burridge, R. A. Firtel, M. H. Ginsberg, G. Borisy, J. T. Parsons, and A. R. Horwitz. Cell migration: integrating signals from front to back. Science. 302:1704–1709 (2003). doi:10.1126/science.1092053.
T. J. Stefonek, and K. S. Masters. Immobilized gradients of epidermal growth factor promote accelerated and directed keratinocyte migration. Wound Repair Regen. 15:847–855 (2007). doi:10.1111/j.1524-475X.2007.00288.x.
M. Arnold, V. C. Hirschfeld-Warneken, T. Lohmuller, P. Heil, J. Blummel, E. A. Cavalcanti-Adam, M. Lopez-Garcia, P. Walther, H. Kessler, B. Geiger, and J. P. Spatz. Induction of cell polarization and migration by a gradient of nanoscale variations in adhesive ligand spacing. Nano Lett. 8:2063–2069 (2008). doi:10.1021/nl801483w.
B. M. Lamb, D. G. Barrett, N. P. Westeott, and M. N. Yousaf. Microfluidic lithography of SAMs on gold to create dynamic surfaces for directed cell migration and contiguous cell cocultures. Langmuir. 24:8885–8889 (2008). doi:10.1021/la801680d.
Y. Luo, and M. S. Shoichet. A photolabile hydrogel for guided three-dimensional cell growth and migration. Nature Materials. 3:249–253 (2004). doi:10.1038/nmat1092.
M. S. Hahn, J. S. Miller, and J. L. West. Three-dimensional biochemical and biomechanical patterning of hydrogels for guiding cell behavior. Adv. Mater. 18:2679–2684 (2006).
M. S. Hahn, L. J. Taite, J. J. Moon, M. C. Rowland, K. A. Ruffino, and J. L. West. Photolithographic patterning of polyethylene glycol hydrogels. Biomaterials. 27:2519–2524 (2006). doi:10.1016/j.biomaterials.2005.11.045.
S. H. Lee, J. J. Moon, and J. L. West. Three-dimensional micropatterning of bioactive hydrogels via two-photon laser scanning photolithography for guided 3D cell migration. Biomaterials. 29:2962–2968 (2008). doi:10.1016/j.biomaterials.2008.04.004.
J. T. Connelly, A. J. Garcia, and M. E. Levenston. Inhibition of in vitro chondrogenesis in RGD-modified three-dimensional alginate gels. Biomaterials. 28:1071–1083 (2007). doi:10.1016/j.biomaterials.2006.10.006.
N. S. Hwang, S. Varghese, Z. Zhang, and J. Elisseeff. Chondrogenic differentiation of human embryonic stem cell-derived cells in arginine-glycine-aspartate modified hydrogels. Tissue Eng. 12:2695–2706 (2006). doi:10.1089/ten.2006.12.2695.
C. Wang, R. J. Stewart, and J. Kopecek. Hybrid hydrogels assembled from synthetic polymers and coiled-coil protein domains. Nature. 397:417–420 (1999). doi:10.1038/16264.
C. Wang, J. Kopecek, and R. J. Stewart. Hybrid hydrogels cross-linked by genetically engineered coiled-coil block proteins. Biomacromolecules. 2:912–920 (2001). doi:10.1021/bm0155322.
J. D. Ehrick, S. K. Deo, T. W. Browning, L. G. Bachas, M. J. Madou, and S. Daunert. Genetically engineered protein in hydrogels tailors stimuli-responsive characteristics. Nature Materials. 4:298–302 (2005). doi:10.1038/nmat1352.
J. D. Ehrick, S. Stokes, S. Bachas-Daunert, E. A. Moschou, S. K. Deo, L. G. Bachas, and S. Daunert. Chemically tunable lensing of stimuli-responsive hydrogel microdomes. Adv. Mater. 19:4024–4027 (2007).
W. L. Murphy, W. S. Dillmore, J. Modica, and M. Mrksich. Dynamic hydrogels: translating a protein conformational change into macroscopic motion. Angew. Chem. Int. Ed. 46:3066–3069 (2007). doi:10.1002/anie.200604808.
Z. J. Sui, W. J. King, and W. L. Murphy. Protein-based hydrogels with tunable dynamic responses. Adv. Funct. Mater. 18:1824–1831 (2008). doi:10.1002/adfm.200701288.
N. Yamaguchi, L. Zhang, B. S. Chae, C. S. Palla, E. M. Furst, and K. L. Kiick. Growth factor mediated assembly of cell receptor-responsive hydrogels. J. Am. Chem. Soc. 129:3040–3041 (2007).
S. H. Kim, and K. L. Kiick. Heparin-mimetic sulfated peptides with modulated affinities for heparin-binding peptides and growth factors. Peptides. 28:2125–2136 (2007). doi:10.1016/j.peptides.2007.08.010.
T. Nie, A. Baldwin, N. Yamaguchi, and K. L. Kiick. Production of heparin-functionalized hydrogels for the development of responsive and controlled growth factor delivery systems. J. Control. Release. 122:287–296 (2007). doi:10.1016/j.jconrel.2007.04.019.
D. S. W. Benoit, M. P. Schwartz, A. R. Durney, and K. S. Anseth. Small functional groups for controlled differentiation of hydrogel-encapsulated human mesenchymal stem cells. Nature Materials. 7:816–823 (2008). doi:10.1038/nmat2269.
D. S. W. Benoit, C. R. Nuttelman, S. D. Collins, and K. S. Anseth. Synthesis and characterization of a fluvastatin-releasing hydrogel delivery system to modulate hMSC differentiation and function for bone regeneration. Biomaterials. 27:6102–6110 (2006). doi:10.1016/j.biomaterials.2006.06.031.
C. R. Nuttelman, D. S. W. Benoit, M. C. Tripodi, and K. S. Anseth. The effect of ethylene glycol methacrylate phosphate in PEG hydrogels on mineralization and viability of encapsulated hMSCs. Biomaterials. 27:1377–1386 (2006). doi:10.1016/j.biomaterials.2005.08.014.
C. R. Nuttelman, M. C. Tripodi, and K. S. Anseth. Dexamethasone-functionalized gels induce osteogenic differentiation of encapsulated hMSCs. J. Biomed. Mater. Res. Part A. 76A:183–195 (2006). doi:10.1002/jbm.a.30537.
C. N. Salinas, and K. S. Anseth. Decorin moieties tethered into PEG networks induce chondrogenesis of human mesenchymal stem cells. J Biomed. Mater. Res. Part A in press: (2008). doi:10.1002/jbm.a.32112.
C. N. Salinas, and K. S. Anseth. The influence of the RGD peptide motif and its contextual presentation in PEG gels on human mesenchymal stem cell viability. Journal of Tissue Engineering and Regenerative Medicine. 2:296–304 (2008). doi:10.1002/term.95.
S. Varghese, N. S. Hwang, A. C. Canver, P. Theprungsirikul, D. W. Lin, and J. Elisseeff. Chondroitin sulfate based niches for chondrogenic differentiation of mesenchymal stem cells. Matrix Biology. 27:12–21 (2008). doi:10.1016/j.matbio.2007.07.002.
B. Sharma, C. G. Williams, M. Khan, P. Manson, and J. H. Elisseeff. In vivo chondrogenesis of mesenchymal stem cells in a photopolymerized hydrogel. Plast. Reconstr. Surg. 119:112–120 (2007). doi:10.1097/01.prs.0000236896.22479.52.
N. S. Hwang, M. S. Kim, S. Sampattavanich, J. H. Baek, Z. J. Zhang, and J. Elisseeff. Effects of three-dimensional culture and growth factors on the chondrogenic differentiation of murine embryonic stem cells. Stem Cells. 24:284–291 (2006). doi:10.1634/stemcells.2005-0024.
D. A. Wang, C. G. Williams, F. Yang, N. Cher, H. Lee, and J. H. Elisseeff. Bioresponsive phosphoester hydrogels for bone tissue engineering. Tissue Eng. 11:201–213 (2005). doi:10.1089/ten.2005.11.201.
K. Hemmrich, and D. von Heimburg. Biomaterials for adipose tissue engineering. Expert Rev. Med. Dev. 3:635–645 (2006). doi:10.1586/17434440.3.5.635.
A. V. Vashi, E. Keramidaris, K. M. Abberton, W. A. Morrison, J. L. Wilson, A. J. O'Connor, J. J. Cooper-White, and E. W. Thompson. Adipose differentiation of bone marrow-derived mesenchymal stem cells using Pluronic F-127 hydrogel in vitro. Biomaterials. 29:573–579 (2008).
ACKNOWLEDGEMENTS
The authors wish to acknowledge the Howard Hughes Medical Institute for support of their time, and KSA wishes to extend her heartfelt thanks to Professor Nicholas Peppas for introducing her to photopolymerizations and hydrogel materials.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lin, CC., Anseth, K.S. PEG Hydrogels for the Controlled Release of Biomolecules in Regenerative Medicine. Pharm Res 26, 631–643 (2009). https://doi.org/10.1007/s11095-008-9801-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-008-9801-2