
Abstract
The HIT-SIOP-PNET4 randomised trial for standard risk medulloblastoma (MB) (2001–2006) included 338 patients and compared hyperfractionated and conventional radiotherapy. We here report the long-term outcome after a median follow up of 7.8 years, including detailed information on relapse and the treatment of relapse. Data were extracted from the HIT Group Relapsed MB database and by way of a specific case report form. The event-free and overall (OS) survival at 10 years were 76 ± 2 % and 78 ± 2 % respectively with no significant difference between the treatment arms. Seventy-two relapses and three second malignant neoplasms were reported. Thirteen relapses (18 %) were isolated local relapses in the posterior fossa (PF) and 59 (82 %) were craniospinal, metastatic relapses (isolated or multiple) with or without concurrent PF disease. Isolated PF relapse vs all other relapses occurred at mean/median of 38/35 and 28/26 months respectively (p = 0.24). Late relapse, i.e. >5 years from diagnosis, occurred in six patients (8 %). Relapse treatment consisted of combinations of surgery (25 %), focal radiotherapy (RT 22 %), high dose chemotherapy with stem cell rescue (HDSCR 21 %) and conventional chemotherapy (90 %). OS at 5 years after relapse was 6.0 ± 4 %. In multivariate analysis; isolated relapse in PF, and surgery were significantly associated with prolonged survival whereas RT and HDSCR were not. Survival after relapse was not related to biological factors and was very poor despite several patients receiving intensive treatments. Exploration of new drugs is warranted, preferably based on tumour biology from biopsy of the relapsed tumour.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The 5-year progression free survival for medulloblastoma (MB), the most common malignant brain tumour in childhood, is now expected to be 70–80 % in the ‘standard’ or ‘average’ risk subgroup [1]. Between 2001 and 2006, the International Society of Paediatric Oncology (SIOP)-Europe Brain Tumour Group conducted a multicentre Phase III trial, the HIT-SIOP-PNET4 study, for children and young people with ‘standard-risk’ MB, with >100 participating centres. After clinical staging, patients were randomised to receive either conventional/standard radiotherapy (STRT) or hyperfractionated radiotherapy (HFRT) followed by chemotherapy. In addition, tumour biomarkers were examined [2]. We previously reported no significant difference in the probability of event free survival (EFS) between the two treatment groups after a median follow-up of 4.8 years [2]. The outcome after a median follow-up of 7.8 years is included in this report.
In standard risk MB, the major cause of death is due to relapse that occurs in 20−30 % of patients. Several reports have demonstrated that the prognosis at relapse is poor, with generally less than 10 % survival [3–5]. This has caused uncertainty whether intensive second line treatment including re-irradiation or myeloablative chemotherapy is appropriate. In addition, there are increasing reports on the occurrence of second malignant neoplasms (SMN) as a consequence of MB therapy [1]. We therefore describe here the patterns of relapsed disease, treatments received at relapse as chosen by various institutions across Europe, and associated outcomes, for patients enrolled on the HIT-SIOP-PNET4 study.
Patients and methods
Patients (n = 338) from Germany, France, Italy, UK, Austria, Spain, The Netherlands, Sweden, Norway and Denmark were included in the HIT-SIOP-PNET4 trial. Patients were 4–21 years of age, with a MB without metastases on craniospinal MRI or cerebrospinal fluid (CSF) cytology. All patients were randomised to receive either STRT (1.8 Gy daily) with 23.4 Gy to the craniospinal axis (CSA) and a 30 Gy boost to the posterior fossa (PF) or HFRT (1 Gy twice daily) with 36 Gy CSA, a 24 Gy boost to the PF and another 8 Gy boost to the tumour bed. During RT, a weekly dose of vincristine was given. RT was followed by eight cycles of chemotherapy with cisplatin, vincristine and CCNU, given at 6 week intervals [2]. The study was approved by each national/institutional review board and all patients/parents/guardians had consented to participate.
Until June 30th 2013, when the database was frozen, patients were followed to time of relapse/progression (hereafter called relapse) or SMN as primary event. Time of relapse was defined as the date of radiological examination confirming the relapse. In Germany, MRI scans were centrally reviewed at relapse, in all other countries radiological relapse was diagnosed at the treating centres. For non-German patients, a case report form specifically designed for this analysis was sent to the treating institutions asking for more detailed information on relapse site, symptoms and treatment of relapses and the date of death. For German patients, this information was collected from the HIT Group Relapsed MB database.
At primary diagnosis, tumour material was centrally reviewed by appointed study pathologists, confirming the diagnosis and the histological MB variant. In addition, a series of MB biomarkers were investigated as part of the HIT-SIOP-PNET4 study [6]; (i) β-catenin nuclear accumulation by immunohistochemistry (IHC) and CTNNB1 exon 3 mutation (DNA sequencing), which both defined the favourable-risk wingless (WNT)-activated MB subgroup [6], (ii) MYC and MYCN gene amplification status (by FISH), associated with a poor prognosis in previous studies [7], and (iii) Chromosome 17 alterations (Chr 17(im)/diploid background; by FISH), which were previously associated with a poor prognosis in the HIT-SIOP-PNET4 cohort [6].
Statistical methods
SPSS software was used for the statistical analyses (IBM SPSS Statistics, version 22, 2013, IBM Corp., Armonk, NY, USA). Relapse, SMN, death in remission and death after relapse were defined as events. The EFS and overall survival (OS) after relapse were estimated using the Kaplan–Meier method and differences in outcome between patients groups were tested using the Log Rank method. Comparisons of patient characteristics between subgroups were performed using Fisher’s exact test, Chi square test or ANOVA where appropriate. Prognostic factors after relapse were evaluated by using the Cox multivariate, proportional regression analysis. The significance level was set to p = 0.05.
Results
Long term survival of the patients included in the HIT-SIOP-PNET4 study
At 10 years from diagnosis, EFS and OS for all the 338 patients were 76 ± 2 % and 78 ± 2 % respectively. The EFS for the two treatment arms continued to show no significant difference; HFRT 78 ± 3 %, STRT 76 ± 3 % (Fig. 1a). At the time when data collection was halted, 72/338 patients (21 %) had suffered a relapse and three patients a SMN (see below). One patient died in CCR1. Four patients were lost to follow up.

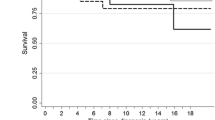
a Long-term probability of event free survival (EFS) of the two randomised treatment arms of HIT-SIOP-PNET4 at 5 and 10 years after primary diagnosis (Kaplan–Meier, Log Rank test). b Probability of overall survival (OS) after first relapse in the HIT-SIOP-PNET4 cohort (Kaplan–Meier). c Probability of overall survival (OS) after first relapse in relation to site of relapse (Kaplan–Meier, Log Rank test)
In order to make our long term results comparable with the COG A9961 study [1], and only for this particular calculation, two adjustments were made: 31 patients with large residual tumours >1.5 cm2 at diagnosis were excluded as well as another 21 patients who had local but not central review of postoperative MRI performed which could allow for disseminated disease being missed. The adjusted EFS and OS at 10 years, were 79 ± 3 % and 81 ± 2 % (n = 286). All other results below are based on the total patient group.
Diagnosis of relapse
MRI scans diagnosing relapse were centrally reviewed in 29/72 cases (40 %). Information on the reason for performing the MRI that demonstrated relapse was available in 65 patients (90 %). Forty-five relapses (69 %) were detected on surveillance MRI. For the remaining 20 patients whose relapse presented symptomatically, the most common symptoms were headache and/or vomiting (n = 11), followed by back pain (n = 5) and loss of specific neurological function (n = 3). The patients presenting with symptoms had a significantly shorter survival after relapse than those detected by surveillance MRI p < 0.01, but the OS (after primary diagnosis) did not differ p = 0.21 (Log Rank test).
Relapse site and timing
The 72 relapses occurred at a mean/median of 30/26 months from diagnosis; range 2–95 months. All patients had complete radiological staging with cranial and spinal MRI at relapse. Metastatic CNS relapse with or without involvement of the PF was the most common relapse site; 59/72 patients (82 %), at a mean/median of 28/26 months after diagnosis. The majority of metastatic relapses, 47/59 (80 %), were multifocal, including cases with leptomeningeal dissemination. In the remaining 20 % there was a solitary metastatic relapse in the spine (n = 7) or brain (n = 5) (Table 1). Eighteen relapses (25 %) were confined exclusively to the spine, and occurred at a mean/median of 30/26 months from diagnosis; range 9–65 months. Twelve of these were diagnosed by surveillance MRI. Eight (44 %) of the MRI scans showing only spinal recurrence were centrally reviewed. There was information on CSF cytology in 61/73 patients. Twenty-one (34 %) had malignant cells in CSF at relapse, always in combination with other tumour manifestations. An isolated local relapse in PF was seen in 13 patients (18 %) after a mean/median of 38/35 months. Late relapse i.e. >5 years from initial diagnosis, occurred in 6 patients (8 %). Five of these were isolated relapses in PF (n = 3) or spine (n = 2). There was no difference in relapse distribution (isolated PF vs not isolated PF) between early and late relapses, although there was a trend for more late, isolated PF relapses (p = 0.07, Fisher’s exact test). Late relapses were diagnosed by surveillance MRI in 2/6 and due to symptoms in 2/6, (2/6 no data). Neither the timing nor the site of relapse differed between the two randomised treatment arms. Furthermore, we compared patients who received ≤4 chemotherapy courses vs patients who received >4 (max 8) courses in the primary tumour treatment, and found no significant difference in the number of relapses between these two groups, (p = 0.398, Fisher’s exact test).
Relapse in relation to histology and biology at primary diagnosis
Histology at primary diagnosis (classic 57, desmoplastic 10, large cell/anaplastic 5) was not related to time in CCR1, (p = 0.56, ANOVA), localisation, (p = 0.97, Chi square) or survival after relapse, (overall p = 0.32, Log Rank test). Nuclear β-catenin IHC status at diagnosis was known in 58/72 patients (14 % positive, 8/58 tumours assessed) and CTNNB1 mutation status in 55 (5 % mutated, 3/55), MYC/MYCN status in 33/72 patients (9 % MYCN amplified, 3/33, no MYC amplified), and chromosome 17 imbalances/diploid background in 32/72 patients (ch17(im)/diploid(cen); 31 %, 10/32) [6]. Neither nuclear β-catenin status nor Chr 17 status had any significant impact on time in CCR1, (p = 0.50/0.51 respectively, ANOVA), localisation (p = 0.61/0.07, Chi square) or survival after relapse, (p = 0.60/0.46, Log Rank test). Eight tumours showing nuclear beta-catenin accumulation relapsed, and in 4/8 of these other unfavourable risk factors were present (Table 2). Six of these relapses were previously reported [6], two more patients have relapsed since that report. Notably, none of eight relapsed WNT-activated MBs is alive in remission (7 DOD). Only seventeen patients underwent surgery at relapse. Therefore, an analysis of outcome with respect to histology and biology on tumour material taken at relapse was not feasible.
Relapse treatment
In four patients no relapse treatment was given, (no treatment data available in one patient). All remaining 67 patients received treatment. The combination of treatments comparing those given to isolated PF vs all other relapses is shown in Table 1. Surgery was performed in 25 % with radical surgery in less than half of the procedures, RT in 22 %, conventional chemotherapy in 90 % and HDSCR in 21 % of all relapsed patients (Table 1). Surgery and HDSCR were significantly more often used (p = 0.027) in isolated PF relapses. Fifteen patients received re-treatment with focal RT (20-45 Gy), most often to spinal metastases (n = 10), supratentorial localised relapse (n = 3) and PF relapse (n = 2). The most common chemotherapy drug was temozolomide alone or in combination with other drugs. Carboplatin-based combinations were also common (Table 3).
Information on the use of intrathecal chemotherapy was available in 66 patients with 17 (26 %) receiving either etoposide (n = 7), methotrexate (n = 6), Depocyte™ (liposomal cytarabine) (n = 3) and one unknown. Concordant information on CSF cytology and intrathecal chemotherapy in 53 patients showed that 9/21 (43 %) patients with CSF positivity received intrathecal treatment as did 5/32 (16 %) patients without malignant cells in CSF.
Survival after relapse
The mean/median survival time after relapse was 23/18 months (Fig. 1b). The OS after relapse, was 20 % (± 5 %) at 3 years, and 6 % (± 4 %) at 5 years after relapse. The OS after isolated PF relapse was significantly higher compared to all other relapses (p = 0.02) (Fig. 1c). In Cox multivariate analysis, surgery (p < 0.01) and isolated PF relapse (p < 0.01) were associated with longer survival after relapse unlike treatment with RT (p = 0.10) or HDSCR (p = 0.44). When the data base was frozen, nine patients were still alive. A detailed description of the survivors is shown in Table 4.
Second malignant neoplasms (SMN)
Three SMNs were reported as primary events namely: a pontine anaplastic astrocytoma, a PF glioblastoma, and an abdominal rhabdoid tumour in a child with Li-Fraumeni syndrome occurring 61, 55, and 35 months after primary diagnosis respectively. All three patients have died. Review of the Li-Fraumeni case confirmed the original (desmoplastic) MB diagnosis (INI-1 positive, SHH-activated with p53 alteration).
Discussion
The primary aim of the HIT-SIOP-PNET4 study was to compare two radiotherapy protocols. It was hypothesised that the HFRT regimen used would be superior for survival without causing more late effects. With a median follow up time of 7.8 years, the estimated OS at 10 years, remains not significantly different in the two treatment arms. Since some patients with standard risk MB are known to relapse later than 5 years after diagnosis (in this study 8 % of the patients), a long follow up is necessary to definitely accept the null hypothesis.
The Children’s Oncology Group (COG) A9961 RCT Study, published in 2012, comparing two different chemotherapy protocols while giving the same conventional RT as in our study, reported a 10-year OS of 81.3 ± 2.1 % [1] which is very similar to our adjusted OS of 81 ± 2 %. The time to relapse, median 23 months, and the amount of disseminated relapses with a tendency to occur earlier than local PF relapses, is also in line with the COG study [1]. Consequently, the two largest studies of standard risk MB, including almost 700 patients, have produced very similar long-term results albeit using slightly different treatment regimes. It is noteworthy that in both studies, patients who were not optimally staged at primary diagnosis (due to incomplete/poor quality MRIs or no central review of MRIs), or had excess residual tumour (>1.5 cm2) on review, had worse outcome [2, 8].
A relevant question today is whether the MB subgroup could explain the pattern and timing of relapse [1]. Ramaswamy et al. showed that the different MB subgroups had different relapse patterns, with more local relapses in patients with Sonic Hedgehog (SHH) tumours compared to Group 3 and 4 tumours, which tended to relapse with metastases [9]. Furthermore, patients with Group 4 tumours were shown to survive longer after relapse, compared to the other subgroups [9]. In this study, only the WNT-activated subgroup could be prospectively delineated, and insufficient tumour material remained to do further retrospective subgrouping. It is well known that tumours showing β-catenin nuclear accumulation (IHC) are prognostically favourable [10]. Indeed only eight patients with relapse showed this biomarker, however, the timing and pattern of relapse for WNT-activated tumours did not differ from the non-WNT tumours. The prognosis after relapse is poor also for this group, and frontline de-escalation of therapy should only be done within trials, as salvage options are limited.
The majority of relapses were found on surveillance imaging when the patients had not yet developed signs or symptoms of relapse and seemingly these patients had a longer survival after relapse. This confirms previous findings but is probably due to the effect of lead time and length time bias as eventually almost all patients died due to progressive disease [3, 11]. The authors agree with others that with respect to survival, the benefit of early discovery of relapse by surveillance MRI remains unclear [12, 13], but could be justified in the perspective of possible inclusion in a relapse study, e.g. an early drug development trial. In that context, a spinal MRI should be considered, since 17 % of relapses were both asymptomatic and confined only to the spine. The high frequency of exclusively spinal recurrences (25 %) contrasts to a smaller study by Bartels et al., where 12/24 relapses had a spinal component but no exclusively spinal relapses were found [12]. Admittedly, this discrepancy could to some extent be explained by the lack of consistent central review of MRI scans in the present study, but since 8/18 (44 %) of cases with exclusive spinal relapse had a central review of the MRI scan, it seems that spinal relapses (leptomeningeal or solid) without concurrent cranial involvement do occur.
We found a relatively low incidence of SMNs, with only three cases reported compared to the COG A9961 study from which fifteen cases were reported albeit with a few years longer follow up. We should thus expect more SMNs and it appears that this is becoming a major problem affecting outcome as MB patients survive longer [1]. However, some SMNs may have gone undetected due to the low number of tumour biopsies at relapse 17/72 (24 %), in the present study.
The poor outcome after MB relapse for patients previously treated with radiotherapy is in line with other reports [3, 4, 11, 14, 15]. With regard to the use of HDSCR, there was hope that this approach would be of significant benefit in relapsed malignant brain tumours, and single institution reports showed encouraging results [16]. However, some studies that reported survival from the time of HDSCR may particularly have over-estimated the benefit of HDSCR-based strategies to the total population of relapsing patients. As noted in both the UK and German studies discussed below, a significant proportion of patients with chemo-resistant disease may not reach HDSCR despite an initial treatment plan to include HDSCR with curative intent.
There have been only limited national studies specifically for relapsed MB that aimed to investigate treatment that may provide long-term disease control. In the UK CCLG relapsed PNET study (2000–2007), at a median follow-up of 7.4 years, only three MB patients were alive, (5 year OS of 8.2 %) [15]. Similarly in 2014, Bode et al. published the results of the German HIT-REZ-97 trial [4]. This national study tested a non-randomised but stratified relapse protocol using either intensive chemotherapy +/- HDSCR as a potentially curative therapy or oral chemotherapy as a palliative option. Survival was poor with only 2/72 patients alive and in CCR at the time of the report. As of September 2015, these two patients are alive and in CCR 155 and 145 months after first relapse (G Fleischhack—personal communication). HDSCR was associated with severe toxicity with a treatment related mortality rate of 7.4 %. The poor results of a HDSCR-based approach have also been shown in recent small institutionally based studies [17, 18]. To date there has been no randomised trial evaluating the role of HDSCR in MB relapse in a multimodal therapy approach and in comparison with conventional chemotherapy, metronomic chemotherapy, targeted therapy or anti-angiogenic therapy.
As noted in this study, oral palliative chemotherapy with, for example, temozolomide or etoposide was frequently employed and may provide time-limited disease control [14]. There has been recent interest in low intensity multi-agent drug combinations with antiangiogenic effect often referred to as ‘metronomic chemotherapy’. In Europe, such therapy is being investigated in studies such as MEMMAT and COMBAT [19, 20], with reports showing a degree of promise with respect to disease control.
The role of intrathecal therapy at relapse is difficult to evaluate. It was not used to its full extent in this study considering that less than half of the patients with malignant cells in CSF at relapse received this treatment. However, the non-randomised German HIT relapse study which involved the use of intraventricular etoposide prior and simultaneously to the conventional systemic chemotherapy showed short-term disease stabilisation in a relevant number of patients but did not achieve higher survival [21]. Re-irradiation, although not a factor for prolonged survival in the present study, has previously been shown to be of benefit, and considering the high frequency of metastatic relapses in MB, craniospinal re-irradiation has been suggested as a therapeutic option worth exploring, although this requires careful balancing against the risk of side effects [22].
A limitation of this study is the lack of information on tumour biology at relapse. This area is now being actively investigated. For example, Wang et al. demonstrated subgroup stability across the primary and the metastatic compartments in newly diagnosed MB [23]. Ramaswamy et al. showed subgroup stability between tumour at diagnosis and relapse in MB [9] and Hill et al. recently undertook a comprehensive investigation of 29 relapsed MB that confirmed this finding [5]. Importantly, Hill et al. also demonstrated significant changes in biological characteristics of tissue at relapse compared to diagnosis. In particular, MYC family (MYC, MYCN) gene amplifications and TP53 pathway defects commonly emerged in combination at relapse and predicted rapid progression to death [5]. Another limitation is that only 40 % of the relapses were confirmed by central review of MRI scans, and also the diversity of relapse treatments.
The strength of this study is the long-time follow up of standard risk MB in children treated according to the same protocol in over one hundred European institutions. This would indicate an outcome not biased by treatment at few or tertiary centres. The report shows the outcome of relapse treatment without the use of a common protocol and gives a survival curve based on a variety of treatments to which further studies could be compared.
The optimal approach to treating relapsed MB in previously irradiated children remains in doubt. The question is still whether it is possible to identify a subgroup of patients in whom a curative approach is justified, and if this is possible, what therapy should be applied. Certainly, in cases where relapse is localised, surgical resection is appropriate. Surgery has added value with regard to provide material to enhance our understanding of this disease and to exclude a SMN. It appears clear however, that the vast majority of relapsed patients cannot be cured and in most patients exploration of new drugs in early phase trials is appropriate, preferably guided by biopsies of the relapsed tumour, and not only the primary tumour.
References
Packer RJ, Zhou T, Holmes E, Vezina G, Gajjar A (2012) Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: results of Children’s Oncology Group trial A9961. Neuro Oncol 15:97–103. doi:10.1093/neuonc/nos267
Lannering B, Rutkowski S, Doz F, Pizer B, Gustafsson G, Navajas A, Massimino M, Reddingius R, Benesch M, Carrie C, Taylor R, Gandola L, Bjork-Eriksson T, Giralt J, Oldenburger F, Pietsch T, Figarella-Branger D, Robson K, Forni M, Clifford SC, Warmuth-Metz M, von Hoff K, Faldum A, Mosseri V, Kortmann R (2012) Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: results from the randomized multicenter HIT-SIOP PNET 4 trial. J Clin Oncol 30:3187–3193. doi:10.1200/jco.2011.39.8719
Bouffet E, Doz F, Demaille MC, Tron P, Roche H, Plantaz D, Thyss A, Stephan JL, Lejars O, Sariban E, Buclon M, Zucker JM, Brunat-Mentigny M, Bernard JL, Gentet JC (1998) Improving survival in recurrent medulloblastoma: earlier detection, better treatment or still an impasse? Br J Cancer 77:1321–1326
Bode U, Zimmermann M, Moser O, Rutkowski S, Warmuth-Metz M, Pietsch T, Kortmann RD, Faldum A, Fleischhack G (2014) Treatment of recurrent primitive neuroectodermal tumors (PNET) in children and adolescents with high-dose chemotherapy (HDC) and stem cell support: results of the HITREZ 97 multicentre trial. J Neurooncol 120:635–642. doi:10.1007/s11060-014-1598-8
Hill RM, Kuijper S, Lindsey JC, Petrie K, Schwalbe EC, Barker K, Boult JK, Williamson D, Ahmad Z, Hallsworth A, Ryan SL, Poon E, Robinson SP, Ruddle R, Raynaud FI, Howell L, Kwok C, Joshi A, Nicholson SL, Crosier S, Ellison DW, Wharton SB, Robson K, Michalski A, Hargrave D, Jacques TS, Pizer B, Bailey S, Swartling FJ, Weiss WA, Chesler L, Clifford SC (2015) Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell 27:72–84. doi:10.1016/j.ccell.2014.11.002
Clifford SC, Lannering B, Schwalbe EC, Hicks D, O’Toole K, Nicholson SL, Goschzik T, Zur Muhlen A, Figarella-Branger D, Doz F, Rutkowski S, Gustafsson G, Pietsch T (2015) Biomarker-driven stratification of disease-risk in non-metastatic medulloblastoma: results from the multi-center HIT-SIOP-PNET4 clinical trial. Oncotarget 6:38827–38839. doi:10.18632/oncotarget.5149
Ryan SL, Schwalbe EC, Cole M, Lu Y, Lusher ME, Megahed H, O’Toole K, Nicholson SL, Bognar L, Garami M, Hauser P, Korshunov A, Pfister SM, Williamson D, Taylor RE, Ellison DW, Bailey S, Clifford SC (2012) MYC family amplification and clinical risk-factors interact to predict an extremely poor prognosis in childhood medulloblastoma. Acta Neuropathol 123:501–513. doi:10.1007/s00401-011-0923-y
Packer RJ, Gajjar A, Vezina G, Rorke-Adams L, Burger PC, Robertson PL, Bayer L, LaFond D, Donahue BR, Marymont MH, Muraszko K, Langston J, Sposto R (2006) Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol 24:4202–4208. doi:10.1200/jco.2006.06.4980
Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJ, Shih DJ, Luu B, Dubuc AM, Northcott PA, Schuller U, Gururangan S, McLendon R, Bigner D, Fouladi M, Ligon KL, Pomeroy SL, Dunn S, Triscott J, Jabado N, Fontebasso A, Jones DT, Kool M, Karajannis MA, Gardner SL, Zagzag D, Nunes S, Pimentel J, Mora J, Lipp E, Walter AW, Ryzhova M, Zheludkova O, Kumirova E, Alshami J, Croul SE, Rutka JT, Hawkins C, Tabori U, Codispoti KE, Packer RJ, Pfister SM, Korshunov A, Taylor MD (2013) Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol 14:1200–1207. doi:10.1016/s1470-2045(13)70449-2
Ellison DW, Onilude OE, Lindsey JC, Lusher ME, Weston CL, Taylor RE, Pearson AD, Clifford SC (2005) beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol 23:7951–7957. doi:10.1200/jco.2005.01.5479
Torres CF, Rebsamen S, Silber JH, Sutton LN, Bilaniuk LT, Zimmerman RA, Goldwein JW, Phillips PC, Lange BJ (1994) Surveillance scanning of children with medulloblastoma. N Engl J Med 330:892–895. doi:10.1056/nejm199403313301303
Bartels U, Shroff M, Sung L, Dag-Ellams U, Laperriere N, Rutka J, Bouffet E (2006) Role of spinal MRI in the follow-up of children treated for medulloblastoma. Cancer 107:1340–1347. doi:10.1002/cncr.22129
Saunders DE, Hayward RD, Phipps KP, Chong WK, Wade AM (2003) Surveillance neuroimaging of intracranial medulloblastoma in children: how effective, how often, and for how long? J Neurosurg 99:280–286. doi:10.3171/jns.2003.99.2.0280
Cefalo G, Massimino M, Ruggiero A, Barone G, Ridola V, Spreafico F, Potepan P, Abate ME, Mascarin M, Garre ML, Perilongo G, Madon E, Colosimo C, Riccardi R (2014) Temozolomide is an active agent in children with recurrent medulloblastoma/primitive neuroectodermal tumor: an Italian multi-institutional phase II trial. Neuro Oncol 16:748–753. doi:10.1093/neuonc/not320
Pizer B, Donachie PH, Robinson K, Taylor RE, Michalski A, Punt J, Ellison DW, Picton S (2011) Treatment of recurrent central nervous system primitive neuroectodermal tumours in children and adolescents: results of a Children’s Cancer and Leukaemia Group study. Eur J Cancer 47:1389–1397. doi:10.1016/j.ejca.2011.03.004
Graham ML, Herndon JE 2nd, Casey JR, Chaffee S, Ciocci GH, Krischer JP, Kurtzberg J, Laughlin MJ, Longee DC, Olson JF, Paleologus N, Pennington CN, Friedman HS (1997) High-dose chemotherapy with autologous stem-cell rescue in patients with recurrent and high-risk pediatric brain tumors. J Clin Oncol 15:1814–1823
Gururangan S, Krauser J, Watral MA, Driscoll T, Larrier N, Reardon DA, Rich JN, Quinn JA, Vredenburgh JJ, DesjardinsA, McLendon RE, Fuchs H, Kurtzberg J, Friedman HS (2008) Efficacy of high-dose chemotherapy or standard salvage therapy in patients with recurrent medulloblastoma. Neuro Oncol 10:745–751. doi:10.1215/15228517-2008-044
Massimino M, Gandola L, Spreafico F, Biassoni V, Luksch R, Collini P, Solero CN, Simonetti F, Pignoli E, Cefalo G, Poggi G, Modena P, Mariani L, Potepan P, Podda M, Casanova M, Pecori E, Acerno S, Ferrari A, Terenziani M, Meazza C, Polastri D, Ravagnani F, Fossati-Bellani F (2009) No salvage using high-dose chemotherapy plus/minus reirradiation for relapsing previously irradiated medulloblastoma. Int J Radiat Oncol Biol Phys 73:1358–1363. doi:10.1016/j.ijrobp.2008.06.1930
Sterba J, Pavelka Z, Andre N, Ventruba J, Skotakova J, Bajciova V, Bronisova D, Dubska L, Valik D (2010) Second complete remission of relapsed medulloblastoma induced by metronomic chemotherapy. Pediatr Blood Cancer 54:616–617. doi:10.1002/pbc.22382
Peyrl A, Chocholous M, Kieran MW, Azizi AA, Prucker C, Czech T, Dieckmann K, Schmook MT, Haberler C, Leiss U, Slavc I (2012) Antiangiogenic metronomic therapy for children with recurrent embryonal brain tumors. Pediatr Blood Cancer 59:511–517. doi:10.1002/pbc.24006
Tippelt S, Mikasch R, Warmuth-Metz M, Pietsch T, Hilger RA, Kwiecien R, Faldum A, Rutkowski S, Bode U, Siegler N, Fleischhack G (2014) CT-002. Intraventricular therapy with etoposide in recurrent medulloblastomas, pineoblastomas, CNS-PNETs and ependymomas—final results of a phase II study. Neuro Oncol 16:i10–i13. doi:10.1093/neuonc/nou066
Wetmore C, Herington D, Lin T, Onar-Thomas A, Gajjar A, Merchant TE (2014) Reirradiation of recurrent medulloblastoma: does clinical benefit outweigh risk for toxicity? Cancer 120:3731–3737. doi:10.1002/cncr.28907
Wang X, Dubuc AM, Ramaswamy V, Mack S, Gendoo DM, Remke M, Wu X, Garzia L, Luu B, Cavalli F, Peacock J, Lopez B, Skowron P, Zagzag D, Lyden D, Hoffman C, Cho YJ, Eberhart C, MacDonald T, Li XN, Van Meter T, Northcott PA, Haibe-Kains B, Hawkins C, Rutka JT, Bouffet E, Pfister SM, Korshunov A, Taylor MD (2015) Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol 129:449–457. doi:10.1007/s00401-015-1389-0
Acknowledgments
The contributions to this study of the SIOP-Europe brain tumour group, national study group members, and national data managers are gratefully acknowledged. The study was presented in part at the International Symposium on Paediatric Neuro-Oncology in Singapore 2014.
Funding
Funding for this work was provided by: The Swedish Children’s Cancer Foundation, The German Children’s Cancer Foundation, Cancer Research UK, The French Ministry of Health, The French National Cancer Institute (INCa) and Associazione Bianca Garavaglia onlus (B. Arsizio, Milano).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Birgitta Lannering and Barry Pizer have contributed equally.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sabel, M., Fleischhack, G., Tippelt, S. et al. Relapse patterns and outcome after relapse in standard risk medulloblastoma: a report from the HIT-SIOP-PNET4 study. J Neurooncol 129, 515–524 (2016). https://doi.org/10.1007/s11060-016-2202-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-016-2202-1