
Abstract
We and others have been aiming at modifications to maintain or to enhance solubility while enabling permeability for cyclic hexapeptides. Especially, the 2-pyridyl-Ala modification was investigated, since in this case, the pyridyl-nitrogen is able to form an H-bond to the NH of the same residue. The hypothesis of a backbone side-chain interaction was demonstrated by NMR experiments, and further results obtained on a variety of pyridyl-Ala derivatives, studied systematically in the context of permeability, are presented in this contribution. Thus, this study sheds some more light on the pyridyl-Ala modification, which had been reported earlier. In addition to the in vitro profiling, the extent of oral bioavailability was assessed in rats. In principle, the pyridyl-Ala residue can be considered as an amino acid supporting oral uptake.
Graphic Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Discovering a strategy to rationally enhance the permeability and ultimately the oral bioavailability of cyclic peptides remains a major challenge in bio-organic and medicinal chemistry. This is intertwined with scientific endeavours to harness the therapeutic potential of “beyond-the-rule-of-five” molecular entities for targets that have proven undruggable by conventional LMW approaches. An initial approach to improve permeability involved N-methylation of amide bonds within a cyclic peptide structure. While this approach did improve permeability, this was typically at a cost of very much reduced solubility (Lewis et al. 2015). For further improvement of absorption, distribution, metabolism, and excretion properties (ADME) for peptides, e.g. to enable delivery by the oral route, we are interested in identifying modifications in the backbone and the side chain to build a tool box for a more straight-forward optimization in the drug discovery process. In an earlier study, we realized beneficial effects on solubility, permeability and metabolism of cyclic hexapeptides carrying only two N-methylated residues and an aliphatic side chain. Instead of an N-methylated Tyr as in the original experiments, we were able to shield an exposed amide group by these side chains (Vorherr et al. 2018). This strategy aimed at unlocking the door to enhanced permeability, with maintained good solubility, is termed “masking of polarity”. In this study, we decided to focus on shielding by backbone—side chain interactions involving an H-bond, which in turn can provide simultaneously improved solubility and improved permeability. As a consequence, we decided to study the pyridyl-Ala (Pal) side chain enabling hydrogen bonding between the pyridyl-nitrogen and the backbone for the 2-Pal residue. In a more systematic approach, we desired to better understand the beneficial role of these modifications in terms of hydrogen bonding and conformation, and the influence on permeability, metabolism and oral bioavailability. The effect of various side chains on permeability was reported from the Lokey group earlier (Rand et al. 2012). However, the Pal building block was not integrated in this study, but a more detailed investigation with respect to the beneficial H-bonding of the 2-Pal versus 3- and 4-Pals has been published earlier (Rafi et al. 2012). This study, including transcellular experiments on small model compounds, showed a positive effect regarding the ability to form an H-bond between the NH and the 2-Pal side chain in a cellular assessment, suggesting that the pyridyl nitrogen acts as an intramolecular H-bond acceptor. On the other hand, a publication appeared, which according to cellular tests did not support the 2-Pal residue as a suitable modification, but in this case no in vivo studies were performed (Thansandote et al. 2015). While this more systematic work was in progress, our colleagues published on positive effects of 2-Pal modified cyclic decamers with respects to oral uptake (Fouche et al. 2016). Since fine-tuning of properties is of particular importance for macromolecules designed for oral bioavailability, we decided to study various Pal residues incorporated in the same cyclic hexapeptide scaffold in more detail. In order to compare the outcome to our previous study, we have chosen the original NMe-Tyr position for insertion of the Pal variations (Fig. 1). The focus of this study is to evaluate this modification in a series of cyclic peptides. Following up on our previous investigations, we assessed the 3D polar surface area called SAPSA (solvent-accessible polar surface area). In addition, we investigated in vitro permeability parameters for these peptides and generated in vivo oral bioavailability data (%BAV) in rats.

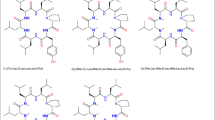
Cyclohexapeptides (1)–(6)
Standard abbreviations for amino acids and peptide derivatives are according to the suggestion of the IUPAC-IUB Joint commission on Biochemistry Nomenclature: Eur J Biochem 138:9-37 (1984) (see "Abbreviations").
Methods
Synthesis
Amino acids, reagents and solvents were purchased from Bachem or Aldrich. Cyclic peptides were synthesized with the first Fmoc amino acid linked to a chloro-trityl polystyrene resin (TentaGel S Trt Cl from RAPP Polymere GmbH) or alternatively, 2-Chlorotrityl chloride resin (100–200 mesh, 1% DVB, 1.0–1.6 mmol Cl/g, from ABCR). Syntheses were carried out manually or alternatively automatic syntheses were carried out using an Initiator + Alstra microwave peptide synthesizer from Biotage or a Symphony synthesizer from PTI.
Peptides were prepared utilizing a synthetic strategy involving attachment of the Fmoc protected C-terminal amino acid e.g. Fmoc-d-Pro residue via the C-terminal carboxy group to the 2-Cl-Trt-resin and subsequent assembly of the linear peptide on the resin. Peptides were prepared by automated peptide synthesis, cleaved from the resin, subsequently cyclized in solution and purified by preparative HPLC. Typically, peptides were synthesized as follows: Fmoc removal was performed using 4-methylpiperidine/DMF 1:4 (2 × 5 min, 5 mL). The resin was washed with DMF (× 3) and DCM (× 4) for 10 min each. Coupling of AAs was performed using 2 eq. of Fmoc-AA-OH, 4.0 eq. of HATU and 8.0 eq. of DIPEA at room temperature. Subsequently, the resin was washed with DMF (× 3) and DCM (× 4). The linear peptide resin was cleaved from the resin by shaking at room temperature for 3 × 10 min then 1 × 5 min with HFIP/DCM (25:75) (6 mL each time). The resin was then washed with DCM (× 2) and the cleavage and washing solutions were filtered off, then concentrated to dryness in-vacuo. Subsequent to cleavage, cyclization was carried out in solution. Typically a 500 mL round-bottomed flask was equipped with a magnetic stirrer and stirring was carried out at 45 °C for several hours. Preparative reversed phase purification was carried out as follows: Column: Waters XBridge Prep C18 5 μm OBD, 30 × 250 mm; Waters Part No 186004025; Eluent A: 0.1% TFA in water; Eluent B:AcN; Flow: 30 mL/min; Gradient: 30% B linearly increasing in 30 min to 100% B, fractions collected with a Waters 2767 Sample Manager and UV detection with a Waters 2996 Photo Diode Array Detector linked to a Waters 2525 Binary Gradient Module Pump. Analytical UPLC-MS was carried out as follows: Peptide standard method: Waters Synapt G2; column: Acquity UPLC CSH C18, 1.7 μm, 2.1x100 mm at 80 °C, Eluent A: H2O + 0.05% TFA, B:AcN + 0.05% TFA, Gradient: hold 5% B for 0.2 min; from 5 to 98% B in 9.2 min, flow: 0.5 mL/min. Peptide non-polar method: Waters Synapt G2; column: Acquity UPLC BEH C4, 1.7 μm, 2.1 × 100 mm at 80 °C, Eluent A: H2O + 0.05% TFA, B:AcN + 0.05% TFA, Gradient: hold 5% B for 0.2 min; from 5 to 98% B in 4.8 min, flow: 0.5 mL/min.
In case of further MPLC purification, the peptide was typically dissolved in 6 mL water and purified on a medium pressure MPLC-C18 Column [Merck Lichroprep (15–25 µm) 26 × 100 mm] with 20 mL/min flow-rate, eluent A:Water, eluent B:AcN, gradient 100% A 2 min isocratic, 100% A 4 min increasing linearly to 70% A:30% B, 4 min isocratic 70% A:30% B, UV Detection: 225 nm providing the purified peptide as a white lyophilisate after lyophilization. For desalting, preparative HPLC fractions containing the peptide as a TFA salt are typically mixed with 0.6 g NaHCO3, concentrated to 50 mL and pumped over the water equilibrated MPLC-C18 column (Merck Lichroprep (15–25 µm) 26 × 100 mm) with 50 mL/min flow-rate, eluent A: Water, eluent B:AcN, gradient 100%A—2 min—> 100%A—30 s—10%A—3 min—> 0%A—4 min—> 0%A, UV Detection: 225 nm providing the de-salted peptide as a white lyophilisate after lyophilization.
Synthesis of Compound (1): [Phe-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro]: Cyclization Procedure
Crude H2N-Phe-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH (216 mg, 0.291 mmol) was dissolved in DCM (290 mL). HOAt (59.4 mg, 0.436 mmol), HATU (442 mg, 1.163 mmol) and 2,6-lutidine (1.016 mL, 8.72 mmol) were subsequently added and the reaction mixture was vigorously stirred. HPLC and UPLC-MS analysis after 1 h showed complete conversion. Solvent was removed in-vacuo and the crude product was dissolved in 2 mL NMP and 2 mL H2O. The crude product was purified by preparative HPLC providing pure cyclic peptide (110 mg, 152 mmol, 52% yield, > 99% % purity, (M + H)+ = 725.5, tR = 3.67 min) as a white lyophilisate.
Synthesis of Precursor of Compound (2): H2N-2-L-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH
Coupling was carried out as follows: TPTU (202 mg, 0.680 mmol) dissolved in 6 mL of NMP/DCE (1/1) to provide solution 1. (S)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-3-(pyridin-2-yl)propanoic acid, Fmoc-2-Pal-OH (264 mg, 0.680 mmol) was dissolved in 8 mL of NMP/DCE (1/1) and 479 μL of collidine was added. The solution was added to the resin and heated to 45 °C while stirring to provide solution 2. Then, solution 1 was added slowly (60 mL/h) in 10 min to the solubilized resin in solution 2. After the additon, the black reaction mixture was stirred for 5 min at RT. The cleavage of the resin was carried out for 2x15 min with a solution of 40% HFIP in DCM. All the fractions were collected together and volatiles were fully evaporated with high vacuum (DCM and HFIP). The crude linear peptide was then purified prior to cyclization by preparative HPLC (45% B for 7 min, 60% B for 7 min, 70% B for 2 min). The linear peptide H2N-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH (270 mg, 363 mmol, 61.3% yield, 100% purity, (M + H)+ = 743.5, tR = 1.579) was isolated as a white lyophilisate and the diastereomeric linear peptide H2N-D-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH (87 mg, 117 mmol, 19.7% yield, 100% purity, (M + H)+ = 743.5, tR = 1.604) was isolated additionally as a white lyophilisate.
Synthesis of Compound (2): [2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro]: Cyclization Procedure
The linear peptide H2N-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH was dissolved in 25 mL AcN, with DIPEA (2 eq) to provide solution 1. In 25 mL AcN, HATU (3 eq) and DIPEA (4 eq) were dissolved (this addition has to be done at the last minute) and this solution was heated to 45 °C while stirring to provide solution 2. Solution 1 was then added slowly (100 mL/h) in 15 min to the solution 2. After the additon, the orange reaction mixture was stirred for 5 min at RT. The crude cyclized peptide was purified by preparative HPLC to provide the cyclic peptide (132.5 mg, 183 mmol, 48.5% yield, 97% purity UPLC-MS-Non-Polar method, (M + H)+ = 726.5, tR = 3.92 min, 96% purity, UPLC-MS-Standard method, tR = 4.91 min) as a white lyophilisate. Chiral integrity of compound (2) was investigated by C.A.T. Tuebingen, verifying 98.82% 2-Pal and 1.18% undesired D-Pal enantiomer.
Synthesis of Building Block for Compound (3): Boc-NMe-2-Pal-OH
Boc-2-Pal-OH (500 mg, 1.784 mmol) was dissolved in 10 mL dry THF. The solution was cooled to 0 °C under vigorous stirring, then 4 eq. MeI (0.451 mL, 7.13 mmol) was added. Subsequently 5 eq of NaH (~ 356 mg, dispersion in oil) was added and after 30 min, a further 5 eq of NaH (~ 356 mg, dispersion in oil) was added to the solution at 0 °C. The solution was then warmed up to 45 °C and after 60 min, full conversion was observed. The solution was then cooled to 0 °C and 12 mL of AcOH (dilute) were added, then the solution was evaporated to dryness. The residue was then dissolved in 6 mL water and purified on a medium pressure MPLC-C18 Column (Merck Lichroprep (15–25 µm) 26 × 100 mm) with 20 mL/min flow-rate, eluent A:Water, eluent B:AcN, 100% A 2 min isocratic, 100% A 4 min increasing linearly to 70% A:30% B, 4 min isocratic 70% A:30% B. UV Detection: 225 nm providing the Boc-2-NMe-Pal)-OH as a white lyophilisate (320 mg, 1.139 mmol, 63.4% yield, 99% purity, (M + H)+ = 281.1, tR = 2.80 min), 1H NMR (400 MHz, DMSO-d6) δ 12.84 (s br, 0H), 8.53–8.44 (m, 1H), 7.73–7.69 (m, 1H), 7.29–7.16 (m, 2H), 4.97, 4.83 (1H) (dd, J = 10.4, 5.2 Hz) (dd, J = 10.7, 4.6 Hz), 3.30–3.13 (m, 2H), 2.69, 2.64 (s, 3H), 1.29, 1.24 (s, 9H).
Synthesis of Precursor of Compound (3): H2N-NMe-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH
The peptide-resin Fmoc-HN-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-Cl-Trt-Resin was swollen in DCM (+ 0.3% of DIPEA) for 15 min. Subsequently Fmoc was removed using 4-methylpiperidine/DMF 1:4 (2 × 5 min, 5 mL) and then the resin was washed. Then Boc-2-NMe-Pal-OH dissolved in 3 mL NMP/DCM (1/1), and 240 μL of collidine was added, and then this solution was combined with the suspended peptide resin. Subsequently, TPTU (1.5 eq) was dissolved in 5 mL NMP/DCM (1:1) and this solution was added slowly over 15 min to the peptide resin. After the addition, the reaction mixture was black colored. A test cleavage was carried out with a 40% solution of HFIP in DCM. UPLC-MS of the peptide identified the desired Boc protected linear peptide as a diastereomeric mixture, tR = 1.35 min, purity 30%, (M + H)+ = 858.7; tR = 1.38 min, purity 49%, (M + H)+ = 858.7. Then, the peptide resin was cleaved with a 40% solution of HFIP in DCM with shaking for 2 × 15 min. The volatiles were fully evaporated with high vacuum (DCM and HFIP) providing 395 mg of crude Boc protected linear peptide. The Boc protecting group was removed using 4 mL of a solution of TFA:H2O:AcN (50:25:25) which was added to the peptide and the mixture was heated at 50 °C. The reaction mixture was then stirred for 30 min and UPLC-MS showed completion of the reaction. Two peaks could be identified in UPLC, H2N-2-L-NMe-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH (tR = 1.673 min, 60.4% purity by UV) and H2N-2-D-NMe-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH (tR = 1.729 min, 28.4% UV). The diastereomeric linear peptide was purified by preparative HPLC providing the desired product H2N-2-L-NMe-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH, (70 mg, 92 mmol, 40.7% yield, > 99% purity, (M + H)+ = 758.1, tR = 1.69 min) and the diastereomer H2N-2-D-NMe-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH, (30 mg, 39.6 mmol, 17.4% yield, > 99% purity, (M + H)+ = 758.6, tR = 1.75 min) as white lyophilisates.
Synthesis of Compound (3): [NMe-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro]: Cyclization Procedure
The linear peptide (H2N-NMe-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH) was dissolved in 10 mL AcN, with DIPEA (2 eq) to provide solution 1. In 10 mL AcN, HATU (3 eq) and DIPEA (4 eq) were freshly dissolved and the solution was heated to 45 °C while stirring to provide solution 2. Then, solution 1 was added slowly (60 mL/h) during 10 min to solution 2. After the addition, the orange reaction mixture was stirred for 15 min at RT. The reaction was monitored by UPLC-MS after 15 min, tR = 2.00 min, purity 82.8% UV, UPLC-MS (Standard Peptide method): tR = 1.41 min, (M + H)+ = 740.5. The crude cyclized peptide was purified by preparative HPLC. UPLC analysis was carried out (tR = 1.99 min, purity > 99% UV) on the purified cyclic peptide obtained as a white lyophilisate (20.5 mg, 28 mmol, 29.8% yield, > 99% purity UPLC-MS (Non polar method), (M + H)+ = 740.5, tR = 4.05 min, 99% purity; UPLC-MS (standard method), tR = 5.03 min). Chiral integrity was successfully determined by analytical UPLC using a Luna C8 column, eluent A: water + 5 mM HCl, eluent B:AcN; gradient 22%B linearly increasing to 80% B in 2.5 min at 75 °C indicating 96.4% of the target cyclic peptide [2-NMe-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro] and 3.6% of the diastereomeric cyclic peptide [2-D-NMe-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro]. Interestingly, for compound (3) the GC method of determining chiral integrity was not successful.
Synthesis of Precursor of Compound (4): H2N-D-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH
The peptide-resin was first swollen in NMP (10 mL). The Fmoc-D-2-Pal-OH (2 eq) was dissolved in 10 mL NMP with HOBT (3 eq). DIC was added in 2 portions, at 0 min and after 30 min. The resin was washed and stored in the fridge overnight. A double coupling was made, with 1 eq Fmoc-D-2-Pal-OH and 1.5 eq DIC/HOBT. After 7 h, the resin was washed and a test cleavage was carried out. UPLC-MS analysis of the test cleavage showed two diastereomeric peaks at tR = 2.93 min and 2.95 min, 43% purity and 23% purity, (M + H)+ = 966. The Fmoc deprotection was performed using 10 mL 20% 4-methylpiperidine in NMP for 3 × 15 min, and the resin was washed with 2x NMP and 1x iPrOH. The cleavage of the peptide-resin was made 3 × 15 min with a 1% TFA solution in DCM (10 mL), to provide crude linear peptide (121 mg, 160 mmol, 46% yield, 27% purity, (M + H)+ = 744.5, tR = 2.44 min).
Synthesis of Compound (4): [D-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro]: Cyclization Procedure
The crude linear peptide H2N-D-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH was dissolved in 25 mL DCM with DIPEA (2 eq) to provide solution 1. In 80 mL MeCN, HATU (3 eq) and DIPEA (4 eq) were dissolved and the solution was warmed up to 45 °C while stirring to provide solution 2. Then, solution 1 was added slowly (60 mL/h) in 17 min to the warm solution 2. After the addition, the orange reaction mixture was stirred for 30 min at 45 °C. Subsequently, the volatiles were evaporated after 30 min to provide the crude cyclic peptide, UPLC-MS tR = 2.57 min, 27% purity UV, (M + H)+ = 726. The crude cyclic peptide was purified by preparative HPLC. Fractions were compared to the 2-Pal diastereomer previously synthesized by UPLC. The results confirmed that peptide in fractions 19 and 28 were the D-2-Pal diastereomer and peptide in fraction 21 is the 2-Pal diastereomer. The fractions were directly frozen and lyophilized, yielding [D-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro] as a white lyophilisate (15.2 mg, 21 mmol, 10.8% yield, > 99% purity, (M + H)+ = 726.5, UPLC tR = 1.455 min, UPLC-MS (standard method) 98% purity, tR = 4.85 min) and the diastereomeric product [2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro] as a white lyophilisate (3.3 mg, 4 mmol, 2.3% yield, > 99% purity, (M + H)+ = 726.5, tR = 1.460 min). Chiral integrity of compound (4) obtained by this route was investigated by C.A.T. Tuebingen GmbH, verifying 95.96% D-2-Pal and chiral integrity of compound (2) obtained by this route as a byproduct was additionally investigated verifying 89.85% 2-Pal enantiomer.
Synthesis of Compound (5): [3-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro]: Cyclization Procedure
A solution of H2N-3-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH (149 mg, 0.200 mmol), HOAt (40.8 mg, 0.300 mmol) and HATU (304 mg, 0.800 mmol) in DCM (200 mL) and 2,6-lutidine (0.699 mL, 6.00 mmol) was stirred at RT for 16 h then concentrated to dryness in-vacuo. The product was isolated by preparative HPLC. The pure fractions were combined and lyophilised, providing the cyclic peptide as a white lyophilisate (46.7 mg, 56 mmol, 27.8% yield, 98% purity UPLC-MS Standard method, (M + H)+ = 726.5, tR = 2.89 min, > 99% purity HPLC, tR = 6.17 min).
Synthesis of Compound (6): [4-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro]: Cyclization Procedure
The linear peptide H2N-4-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH was dissolved in AcN (20 mL) with DIPEA (2 eq) to provide solution 1. HATU (3 eq) and DIPEA (4 eq) were dissolved in 120 mL AcN and the fresh solution was warmed up to 45 °C while stirring to provide solution 2. Then solution 1 was added slowly (60 mL/h) in 15 min to the warm solution 2. After the additon, the yellow reaction mixture was cooled to RT and stirred for 18 h. The crude cyclic peptide was concentrated in-vacuo and UPLC-MS of the crude cyclized peptide was carried out (tR = 2.44 min, 50% purity UV, (M + H)+ = 726). The crude cyclic peptide was purified and UPLC of combined fractions showed a purity of 98.3% (tR = 1.66 min). The volatiles were evaporated in-vacuo and the mixture was frozen and lyophilized providing the cyclic peptide as a white lyophilisate (145.1 mg, 199 mmol, 74.7% yield, > 99% purity UPLC-MS Standard method, (M + H)+ = 726.5, tR = 2.55 min).
Analysis for Optical Purity
The degree of chiral integrity was determined by two different methods. As a first test analytical GC after deuteration of the hydrolyzed peptide product, as carried out at C.A.T. GmbH (Tuebingen, Germany), was employed. This method was successful in determining the % presence of the epimeric D-2-Pal-OH in the [2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro] cyclic peptide, and similarly, the % of the epimeric 2-Pal-OH in the [D-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro] sequence.
Concerning the synthesis of cyclic peptide (3), preparative HPLC separation of the protected linear precursor H-NMe-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH from the undesired H–D-2-NMe-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH was identified as being the superior approach. Quantification of the chiral integrity of this cyclic peptide was successfully achieved by an alternative method using an optimized UPLC analysis, where the two diastereomeric peptides [NMe-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro] and [NMe-D-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro] could be separated. In order to validate and optimize the UPLC method, a 1:1 mixture of [NMe-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro] and [NMe-D-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro] was prepared and analyzed using different conditions. An UPLC method using a Luna C8 column, combined with eluent A:water + 5 mM HCl, eluent B:AcN; gradient 22%B linearly increasing to 80%B in 2.5 min at 75 °C resulted as the optimal method illustrating 96.4% purity of the target cyclic peptide (3) [NMe-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro] and only 3.6% of the [NMe-D-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro] diastereoisomer.
Assays for Permeability and Solubility
Protocols for high-throughput equilibrium solubility, PAMPA and cellular permeability by the MDCK-LE and MDCK-MDR1 assays were followed as reported earlier (Lewis et al. 2015; Tatrai et al. 2019). The values relate to single experiments run in triplicates. For the MDCK assay, the LLOQ (lower limit of quantification) was 5 nM, and for PAMPA 2 nM, respectively. For both PAMPA and MDCK, the standard deviation in log units for those assays is 0.2 (1.6 fold on a linear scale). This has been estimated by looking at independent replicates across the assay dynamic range. For the determination of the apparent passive permeability, Papp, apical to basolateral was determined using MDCK-Low efflux cells (Sigma #8412903) and for the determination of efflux ratios Papp, apical to basolateral, basolateral to apical was determined using MDCK-MDR1 cells (Netherland Cancer Institute).
SAPSA
Molecular dynamics simulations were performed over 20 ns for every peptide using an implicit solvent model of water (Schaefer and Karplus 1996) generating a set of 20000 conformations for each as previously reported (Lewis et al. 2015). The initial 3-dimensional structures were generated using the program Corina (Sadowski et al. 1994). Before starting data collection, the peptides were minimized for 400 steps and heated to 300 K by performing 10 ps Langevin dynamics. A water model was applied for the dynamics, rather than a model representing the membrane interior (e.g., chloroform), since this leads to faster transitions between conformations in the simulations and better sampling of conformation space (Chen et al. 2008). For every structure thus obtained, we computed the 3-dimensional solvent-accessible polar surface area (SAPSA). By averaging over conformations observed between 10 and 20 ns, we determined the time-averaged SAPSA for the various peptides. Conformational changes and the dynamic formation of hydrogen bonds were facilitated by a high dielectric constant of 80 representing water as the solvent, and a low friction constant of 1/ps for performing constant temperature Langevin dynamics (Brooks et al. 1983). The SAPSA for each frame is determined as the area covered by the center of a solvent probe sphere with a radius of 1.4 Å in contact with oxygen, nitrogen, and their bonded hydrogens (Richmond 1984). For the solute atoms, we employ the same Bondi radii as are used to compute the conventional polar surface area (PSA) (Ertl et al. 2000).
SFC
Conditions for analytical supercritical fluid chromatography (SFC) were taken from previous publications (Goetz et al. 2014a, b). The Pirkle chiral stationary phase Chirex 3014, a silica bonded (S)-valine and (R)-1-(α-naphthyl)ethylamine with a urea linkage, was selected. A mobile phase of 15% methanol in supercritical CO2 enabled adequate separation in combination with a low-slope gradient. High retention times were mitigated by increasing the percentage of methanol in the eluent and the addition of the relatively weak salt pair ammonium formate. In the resulting chromatographic system, differences in retention depend upon hydrophobic interactions, the ability for hydrogen bonding, the presence of dipoles, and van der Waals interactions of the solutes with the stationary phase (Goetz et al. 2014a, b).
Potentiometric pKa Determination
Potentiometric ionization constants were determined on the commercial SiriusT3 instruments (Pion-inc.com) as described by Völgyi et al. (2007). Briefly, 0.3–1 mM of test solutions were titrated from pH 2 to 12 for bases or 12 to 2 for acids. Titrations were conducted at 25 °C and in 0.15 M ionic strength. Aqueous titrations were performed in triplicate in 0.15 M KCl, while sparingly soluble test compounds were titrated in 10–60 %wt methanol, 1,4-dioxane, or dimethyl sulfoxide co-solvent. A minimum of three titrations in varying amounts of co-solvent were performed for extrapolation to the aqueous pKa. For each titration, initial estimates of pKa values were obtained from Bjerrum difference plots (number of bound protons versus pH) and then were refined by a weighted non-linear least-squares procedure (Avdeef 1992, 1993) available in the instrument software. Experimental variability was determined from 389 duplicate measurements from different days and experimentalists, with a standard deviation of 0.28.
NMR Experiments
Solid peptide samples (2–3 mg) were dissolved in 600 μL CDCl3 and treated with dried aluminum oxide. All NMR data were collected on a Bruker Avance 600 MHz NMR instrument with a 5 mm TCI cryoprobe at 298 K. Titration experiments were carried out by serial addition of 10 to 50 μL of DMSO-d6 to 500 μL of CDCl3 samples with 10 μL increments. Chemical shift changes of amide protons were analyzed against the volume of DMSO-d6 added.
In-Vivo Studies
For rat PK, blood concentration versus time profiles were obtained from 3 male Sprague–Dawley rats. For intravenous PK, the compound was administered intravenously by bolus injection (0.5 mL/kg) at a dose of 1 mg/kg, solubilized in NMP (30%) and PEG200 (70%). For oral PK, after a washout period of 48 h, an oral gavage was administered to the same animals at a dose of 3 mg/kg at a dosing volume of 2.5 mL/kg. The oral dose was dispersed in water (99%), Tween80 (0.5%) and methylcellulose (0.5%), forming a homogenous suspension. For both rodents, blood samples (10–50 µL) were collected at 0.08 (i.v. only), 0.25, 0.5, 1, 2, 4, 7, and 24 h after dosing. Analyses of parent compound concentrations were carried out in blood using LC–MS/MS. An aliquot of 30 μL was taken and added to 200 μL AcN (including Glyburide (c = 50 ng/mL) as internal standard) for protein precipitation. Sample analysis was performed on a LC–MS/MS system consisting of an AB SCIEX API 5500 QTrap mass spectrometer equipped with a TurboIon SprayTM interface (Framingham, USA). The MS system was connected to a HTS CTC PAL auto-sampler (Zwingen, Switzerland) and to a Flux Rheos 2200 pump system (Reinach, Switzerland). The supernatants (2 µl) were injected directly onto the LC–MS/MS system for analysis. The test articles and its internal standard were separated with a Phenomenex Kinetex C18 (50 x 2 mm ID, 2.7 µm pore size). A binary gradient with a mobile phase consisting of water (A) and AcN (B) was used for the LC-separation. Both mobile phases (A) and (B) were acidified with 0.1% formic acid. The elution gradient program was as follows: [time (min), (% mobile phase B): (0, 95) (5.8, 85) (5.81, 99) (6.5, 99) (6.51, 95) (8, 95]. The column temperature was maintained at 50 °C using a column heater. Under these experimental conditions, the LLOQ was about 0.7 ng/mL.
All animals were maintained under standard housing conditions with access to standard pelleted food and water ad libitum, including throughout experiments. Animal experiments were conducted in accordance with Swiss national animal welfare regulations, under the ethically approved animal experimentation licenses authorized by the Cantonal Veterinary Authority of Basel city and the Federal Veterinary Office of Switzerland.
Results
Design and Synthesis
The cyclic peptides, as illustrated in Fig. 1, were primarily designed to compare solubility, permeability and oral uptake in order to check for scope and limitations of the various Pal modifications. In the first instance, we ran MD simulations. In particular for the 2-Pal modified peptide (peptide 2) in a low dielectric environment, we could clearly identify the intramolecular H-bond between the pyridyl-nitrogen and the NH of this residue (Fig. 2a). However under these conditions, the 3-Pal residue was not able to form an H-bond from the backbone to the side chain (Fig. 2b). Interestingly, the canonical intramolecular H-bonding pattern and the side chain interaction could not be observed for peptide 2, if the simulation was carried out in a more polar environment. Next, we prepared the derivatives as indicated in Fig. 1. Although we originally did not plan to investigate peptide 4, however due to the epimerization of the 2-Pal residue during coupling, we obtained this peptide and thought of studying it as a negative control. In addition, we prepared peptide 3, the N-methylated-2-Pal derivative, since we thought of this peptide as a suitable positive control, which can be compared to the corresponding NMe-Tyr cyclic hexapeptide reported earlier (White et al. 2011).

Results of simulations on peptides 2 and 6
As outlined in the introduction, the 2-Pal moiety is known to be able to form an intramolecular hydrogen bond between its pyridyl side chain and the hydrogen atom of its N-α amide group. While this is advantageous in terms of design, masking polarity and potentially providing improved permeability, this interesting structural feature presents challenges in terms of maintaining chiral integrity during the synthesis process. In our hands, the coupling of Fmoc-2-Pal-OH was especially prone to epimerization. This can be mitigated by the judicious choice of coupling reagent, careful addition of the pre-activated Fmoc-2-Pal-OH to the peptide resin, and optimization of temperature to ensure rapid completion of the coupling (see “Methods”). UPLC monitoring of the reaction enables observation of the desired product versus the undesired epimeric side product. Furthermore, separation of the Fmoc deprotected linear peptide, prior to cyclization, utilizing preparative HPLC purification is a successful strategy to obtain the desired cyclic product with high chiral integrity. Indeed, the synthesis and optical integrity determination of (2) [2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro] and (3) [NMe-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro] can be compared and contrasted (see “Methods”). The linear sequence precursor of compound (2) H2N-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH could be prepared after preparative HPLC purification providing pure 2-Pal desired linear peptide (270 mg, 80.1% yield, purity > 99%) and the separated pure diastereomeric D-2-Pal linear peptide (80 mg, 23.7% yield, purity > 99%). The target 2-Pal linear sequence could then be cyclized to provide the cyclic peptide (2) (132.5 mg, 183 mmol, 48.5% yield, 97% purity) as a white lyophilisate. Chiral integrity of compound (2) was determined by C.A.T. GmbH (Tuebingen), using deuteration followed by GC–MS verifying 98.82% 2-Pal and 1.18% undesired D-2-Pal enantiomer.
In contrast, the N-methylated linear sequence precursor of compound (3) H2N-NMe-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH could be prepared after preparative HPLC purification providing the desired product H2N-2-NMe-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro-OH, (70 mg, 92 mmol, 40.7% yield, > 99% purity) and the diastereomer H2N-2-D-NMe-Pal-Leu-NMe-D-Leu-NMe-LeuLeu-D-Pro-OH, (30 mg, 39.6 mmol, 17.4% yield, > 99% purity) as white lyophilisates. The target NMe-2-Pal linear sequence could then be cyclized and purified by preparative HPLC to provide the cyclic peptide (20.5 mg, 28 mmol, 29.8% yield, 100% purity). For this NMe-2-Pal cyclic peptide (3), the GC–MS method for determining chiral integrity was not successful, because the NMe-2-Pal enantiomers could not be separated after hydrolysis. As an alternative, an UPLC method was developed using a Luna C8 column at 75 °C indicating 96.4% of the target cyclic peptide [2-NMe-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro] and 3.6% of the diastereomeric cyclic peptide [2-D-NMe-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro]. Epimerization for incorporation of 3- or 4-Pal residues was not observed. In the next step, the physico-chemical parameters like solubility, permeability, and pharmacokinetic properties (PK) were investigated for this set of peptides (Fig. 1).
In-Silico and In Vitro Profiling
We calculated the SAPSA following MD-simulations to compare polar surface area, averaging over an ensemble of 3-dimensional structures, for the various peptides (Table 1). This method assesses in silico the solvent-accessibility of polar atoms. As expected for the 2-Pal peptide, the SAPSA was slightly higher for the Phe analogue (1), and considerably lower than the SAPSA determined for the 3- and 4-Pal cyclic peptides (Table 1). The lower SAPSA for the N-methylated 2-Pal peptide compared to the compound without N-methylation in this position was also expected. Since the D-2-Pal residue in peptide (4) was expected to disrupt the geometry of the scaffold, a similar SAPSA value for this compound did come as a surprise. SFC-analyses, indicating the exposure of polar groups in the liquid CO2-medium, was then compared to the outcome of the SAPSA calculations (Goetz et al. 2014a, b). In this case, the N-methylated 2-Pal peptide showed the shortest retention time, and thus interacts less with H-bond acceptors and donors on the stationary phase. The 2-Pal peptide had a shorter retention than the Phe control, and the 3- and 4-Pal peptides as well as the D-2-Pal peptide clearly showed a longer retention, and thus, exhibited more polar groups. Results from the PAMPA assay qualitatively agreed with the SFC measurements in the sense of showing better permeability for the Phe-peptide. All 2-Pal peptides were rated highly permeable, whereas the 3- and 4-Pal modifications were ranked only medium permeable (Table 1). In terms of solubility, only the 2-Pal peptides showed significant increases compared to peptide (1). Although as expected, the 3- and 4-Pal derivatives are associated with higher basicity as measured by the pKa (Table 1), the solubility was surprisingly low. For the time being, this finding cannot be rationalized. MDCK (low efflux) measurements were performed on a cell line expressing only minor amounts of the multidrug-resistance-protein-1 (mdr1). The transport measured across this cell monolayer is consistent with a lower permeability for peptides (5) and (6), and showed a surprisingly high value for the D-2-Pal peptide (Table 2). In the MDCK mdr1 cell line, which overexpresses the multidrug resistance protein (mdr1), fluxes in both directions have been measured and the ratios in the column (BA/AB) are discussed in the first instance. While all peptides are exported to a certain degree, according to this criterion the peptides (5) and (6) are considered the best substrates for the efflux pump, although similar to the low efflux experiments the transport rates for peptides (5) and (6) are lower. These results clearly demonstrate the influence of minor modifications on cellular uptake and of course, the outcome of these kind of experiments is dependent on the cell line employed.
Intramolecular H-Bond Investigation by NMR
In order to correlate the strength of intramolecular H-bonds to results from the permeability assessment, we carried out DMSO titration experiments starting from peptides dissolved in CDCl3. As a hydrogen bond acceptor, DMSO will form H-bond with exposed amide protons, resulting in downfield chemical shift changes; whereas chemical shifts of the amide protons involved in intramolecular H-bonds remain relatively stable upon DMSO addition. Chemical shift changes of amide protons were then analyzed against the volume of DMSO-d6 added to these solutions (Fig. 3). For peptide (1), the NH of the Phe residue is not involved in an H-bond, while the two transannular H-bonds involving amide protons of Leu3 and Leu6 are quite stable as shown in Fig. 3a. In the case of the 2-Pal peptide (2), the DMSO titration result indicates that the 2-Pal NH is involved in an intramolecular H-bond accepted by the pyridyl nitrogen (Fig. 3b). Surprisingly, we have also observed an H-bond in case of the D-2-Pal peptide (4) (Fig. 3d), which could be understood only when analyzing the MD trajectory for this peptide in more detail. Indeed, as shown in Fig. 4, the amide bond of the D-2-Pal residue is thought to invert, and thus favors an orientation enabling this backone-side chain contact. As expected the 3- and 4-Pal residues cannot form an intramolecular H-bond as indicated by the chemical shift changes of the corresponding NH protons upon DMSO titration. The result for the two transannular H-bonds of the NMe-2-Pal peptide was anticipated, although in this case the NH of Leu3 shows a stronger intramolecular hydrogen bond than those observed for the other peptides. The unexpected conformational behavior as indicated for the D-2-Pal amino acid (peptide 4) may explain the superiority of this modification regarding permeability when compared to the 3- and 4-Pal incorporation.

Results of NMR studies on peptides (1)–(6)

MD in low dielectric medium peptide (4) [D-2-Pal-Leu-NMe-D-Leu-NMe-Leu-Leu-D-Pro]
i.v. and per os (p.o.) Study in Rats: Pharmacokinetics of the Cyclohexapeptides
The results of the in vivo p.o. study in rats led to some surprising results (Table 3). As discussed earlier in the context of metabolism, a distinct difference in clearance was observed (Vorherr et al. 2018). Although peptide 2 is able to form an H-bond between the 2-Pal residue and the backbone, this positive effect on permeability does not translate to high oral absorption (Fig. 5a). Only the Phe control, the N-methylated 2-Pal and the 3-Pal peptide showed a low clearance. Both of these Pal sequences exhibited an astonishingly high exposure upon oral administration. As a consequence, for the N-methylated 2-Pal peptide (3) an oral bioavailability (BAV) of 88% was observed, which is significantly higher than reported for other cyclic variants carrying 3 N-methylated amide groups (Fig. 5b). For the 3-Pal sequence, an oral BAV of 41% points to the idea that the pyridyl-nitrogen, although not involved in a H-bond, is not completely exposed and the ring system is able to shield the free NH of this residue. Although both 2-Pal peptides, the l- and d-form [(2) and (4)], are able to form a side chain—backbone H-bond, this conformational feature does not translate in either high exposures or a significant fraction absorbed. However for the latter, the high metabolic turnover, and in addition the increased efflux compared to peptide (2), are believed to outweigh the permeability component. Furthermore, considering the variations in clearance and solubility, the outcome of the p.o. studies currently cannot be rationalized. Furthermore, it is astonishing to observe an oral BAV of 13%, for the 4-Pal peptide (6) although the clearance is quite high and the exposed pyridyl-nitrogen clearly has a higher basicity. Overall, the original hypothesis predicting an increased oral uptake by shielding in addition to what is achieved by the Phe residue (peptide 1) could not be simply translated into an effective medicinal chemistry practice due to multiple intertwining factors. However, it seems the different Pal variants could be applied to retain intestinal permeability. Due to the large differences in clearance, the contribution of conformational features, which influence in vivo oral exposure, e.g. by fixation of the side chain by an H-bond, is difficult to assess independently. In these cases, subtle differences in conformational flexibility depending on the nature of the side chain, and thus, a slightly altered scaffold geometry are thought to have a profound effect on the in vivo pharmacokinetics of these compounds.

a Blood concentration i.v. and p.o. from rat PK study on peptide (2). Data are mean ± standard deviation (n = 3 animals). b Blood concentration i.v. and p.o. from rat PK study on peptide (3). Data are mean ± standard deviation (n = 3 animals)
Discussion
As explained in the introduction, we were building on previous results to introduce more hydrophilic groups without compromising permeability in the sense of enabling cyclic peptides for oral uptake in a more straight-forward drug discovery process. In this context, we would like to validate appropriate side-chain and backbone modifications to assemble a toolbox, which can be used to fine-tune properties, e.g. permeability. Starting out from in silico considerations, options regarding the backbone—side chain H-bonding interaction, e.g. like the 2-Pal modification as reported earlier, have been suggested for the cyclic hexapeptide scaffold to enable shielding of the pyridyl-nitrogen and a non N-methylated amide bond at the same time. These theoretical findings on the basis of MD simulations were confirmed by NMR measurements using DMSO-titrations to determine the strength of intramolecular H-bonds. Accordingly, the hypothesis of bringing in additional polarity without limiting the transition through membranes could be verified, in particular for the 2-Pal modification, as shown in Fig. 2a. In the introduction, we described a mixed outcome regarding the Pal-modification. The study by Thansandote et al. cannot be readily compared to this work due to a slight but important variation in the scaffold (no N-methylation) and the choice for positioning the 2-Pal residue. In addition, the artificial membrane assay employed for this study has not been widely applied to draw conclusions regarding the correlation with PAMPA or cellular assays. In this study, the SFC and PAMPA values clearly predict the N-methylated 2-Pal peptide (3) as the most permeable sequence. For the 3- and 4-Pal modifications the in vitro results obtained with these assays are in line with the expectation, that more exposed basic groups will compromise permeability to some extent. These latter findings are also reflected in the two different MDCK assays. In light of the variation of fluxes A → B and B → A in the two MDCK assays employed, we believe there is a need to perform more independent permeability assessments to understand, e.g. the contributions of efflux pumps. In fact, we tried to shed some more light on the role of exporters by employing the MDCK mdr1 cell line, and could confirm quite a substantial effect of closely related Pal-modifications on the export. However, we did not determine the role of efflux systems with respect to intestinal permeability. Nevertheless, this small collection, when compared to the Phe residue (peptide 1), offers some insights to what extent properties can be modulated. Interestingly, it seems different Pal variants, maybe with the exception of the 4-Pal moiety, could be applied to retain a reasonable percentage of fraction absorbed. On the basis of cyclic hexapeptides, a potent CXCR7 modulator has been developed (Boehm et al. 2017). This peptide has a Ki of 9 nM, and shows an oral bioavailability of 18% in rats despite hepatic-blood flow limited clearance. The polar surface area could be considerably reduced by employing one peptoidic structure, but no N-methylation was present in the structure selected for in vivo experiments. Interestingly, Pal residues were also studied during this optimization, and to some extent, these modifications were tolerated by the target. As outlined in the introduction, the positive effects of the N-methylated 2-Pal and the 2-Pal modification have been demonstrated in the context of multi N-methylated cyclic decamers (Fouché et al. 2016). In this case an oral BAV of 33% (N-methylation at 2-Pal) and 40% (2-Pal) were reported, while clearance was low and good exposures upon p.o. administration were achieved. However, for molecules containing two N-methylated 2-Pal or simply 2-Pal residues a low exposure i.v. and no oral uptake was observed.
Conclusions
As indicated above, the contribution of conformational features, which influence in vivo pharmacokinetic studies, e.g. by fixation of the side chain by an H-bond, is difficult to assess independently. In these cases, subtle differences in conformational flexibility depending on the nature of the side chain, and thus, a slightly altered scaffold geometry is thought to have a profound effect on the in vivo pharmacokinetics of these compounds. Nevertheless, the permeability assessment can help to filter out the decoration, which is best in terms of permeability. Shortcomings of this type of assays have been discussed recently (Vorherr et al. 2018), and any new formats to be used in addition or tests on intestinal tissue, e.g. of rodent or other species, will help to better qualify cyclic peptides for their ability to be taken up by the oral route. However in this study, the influence of export phenomena for the in vivo oral uptake could not be readily assessed, since the clearance values also differed quite significantly. This means, the overall oral bioavailability is observed as a result of potentially counterbalancing properties like intestinal permeability, contributions of importers and exporters, and the liver metabolism, not to mention other PK parameters. As published earlier (Vorherr et al. 2018), it seems that minor changes in the structure account for large differences observed for the clearance, which we also attribute to liver metabolism for the peptides studied in this contribution. Considering the in vivo outcome, the original hypothesis of predicting the translation of conformational features, e.g. internalized H-bond interactions of polar groups, to an enhanced oral bioavailability proved to be difficult due to multiple parameters, which have an influence on exposure. In a recent review on macrocyclic peptides, similar issues related to drug development have been discussed (Vinogradov et al. 2019). However, despite the progress achieved, the development of orally active therapeutic peptides continues to be considered as a challenging task.
Abbreviations
- ADME:
-
Absorption, distribution, metabolism, and excretion
- AUC:
-
Area under the curve
- %BAV:
-
Bioavailability on p.o. administration
- CL:
-
In-vitro or in vivo clearance
- Cmax :
-
Observed maximum concentration in blood
- d.n.:
-
Dose normalized
- i.v.:
-
Intravenous
- LLOQ:
-
Lower limit of quantification
- MDCK:
-
Madin Darby canine kidney cells
- mdr1:
-
Multidrug resistance-protein 1
- MRT:
-
Mean residence time
- Pal:
-
Pyridyl-Ala
- PAMPA:
-
Parallel artificial membrane permeability assay
- PK:
-
Pharmacokinetics
- rmsd:
-
Root mean squared deviation
- p.o.:
-
Per os
- PSA:
-
Polar surface area
- SAPSA:
-
Solvent-accessible polar surface area
- SFC:
-
Supercritical fluid chromatography
- T1/2 term:
-
Terminal half-life for elimination
- Tmax :
-
Time of observed maximal blood concentration
- Vss :
-
Apparent volume of distribution at steady state
References
Avdeef A (1992) pH-Metric log P. Part 1. Difference plots for determining ion-pair octanol-water partition coefficients of multiprotic substances. Mol Inform 11(4):510–517
Avdeef A, Comer JEA, Thomson SJ (1993) pH-Metric log P. 3. Glass electrode calibration in methanol-water, applied to pKa determination of water-insoluble substances. Anal. Chem. 65(1):42–49
Boehm M, Beaumon K, Jones R, Kalgutkar AS, Zhang L, Atkinson K, Bai G, Brown JA, Eng H, Goetz GH et al (2017) Discovery of potent and orally bioavailable macrocyclic peptide-peptoid hybrid CXCR3 modulators. J Med Chem 60:9653–9663
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comp Chem 4:87–217
Chen J, Brooks CL III, Khandogin J (2008) Recent advances in implicit solvent-based methods for biomolecular simulations. Curr Opin Struct Biol 18(2):140–148
Ertl P, Rohde B, Selzer P (2000) Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport. J Med Chem 43:3714–3717
Fouche M, Schaefer M, Blatter M, Berghausen J, Desrayaud S, Roth HJ (2016) Pharmacokinetic studies around the mono- and difunctionalization of a bioavailable cyclic decapeptide scaffold. Chem Med Chem 11:1060–1068
Goetz GH, Farrell W, Shalaeva M, Sciabola S, Anderson Dennis, Yan Jiangli, Philippe L, Shapiro MJ (2014a) High throughput method for the indirect detection of intramolecular hydrogen bonding. J Med Chem 57:2920–2929
Goetz GH, Philippe L, Shapiro MJ (2014b) EPSA: a novel supercritical fluid chromatography technique enabling the design of permeable cyclic peptides. ACS Med Chem Lett 5:1167–1172
Lewis I, Schaefer M, Wagner T, Oberer L, Sager E, Wipfli P, Vorherr T (2015) A detailed investigation on conformation, permeability and PK properties of two related cyclohexapeptides. Int J Pept Prot Ther 21:205–221
Rafi SB, Hearn BR, Vedantham P, Jacobson MP, Renslo AR (2012) Predicting and improving the membrane permeability of peptidic small molecules. J Med Chem 55(7):3163–3169
Rand AC, Leung SSF, Eng H, Rotter CJ, Sharma R, Kalgutkar AS, Zhang Y, Varma MV, Farley KA, Khunte B, Limberakis C, Price DA, Liras S, Mathiowetz AM, Jacobson MP, Lokey RS (2012) Optimizing PK properties of cyclic peptides: the effect of side chain substitutions on permeability and clearance. Med Chem Commun 3(10):1282–1289
Richmond TJ (1984) Solvent accessible surface area and excluded volume in proteins. J Mol Biol 178(1):63–89
Sadowski J, Gasteiger J, Klebe G (1994) Comparison of automatic three-dimensional model builders using 639 X-ray structures. J Chem Inf Comput Sci 34:1000–1008
Schaefer M, Karplus M (1996) A comprehensive analytical treatment of continuum electrostatics. J Phys Chem B 100:1578–1599
Tatrai P, Schweigler P, Poller B, Domange N, de Wilde R, Hanna I, Gaborik Z, Huth F (2019) A systematic in vitro investigation of the inhibitor preincubation effect on multiple classes of clinically relevant transporters. Submitted to Drug Metab and Disposition in March 2019
Thansandote P, Harris RM, Dexter HL, Simpson GL, Pal S, Upton RJ, Valko K (2015) Improving the passive permeability of macrocyclic peptides: balancing permeability with other physicochemical properties. Bioorg Med Chem 23:322–327
Vinogradov AA, Yin Y, Hiroaki Suga H (2019) Macrocyclic peptides as drug candidates: recent progress and remaining challenges. JACS 141(10):4167–4181
Völgyi G, Ruiz R, Box K, Comer J, Bosch E, Takács-Novák K (2007) Potentiometric and spectrophotometric pKa determination of water-insoluble compounds: validation study in a new cosolvent system. Anal Chim Acta 583(2):418–428
Vorherr T, Lewis I, Berghausen J, Desrayaud S, Schaefer M (2018) Modulation of oral bioavailability and metabolism for closely related cyclic hexapeptides. Int J Pept Res Ther 24:35–48
White TR, Renzelman CM, Rand AC, Rezai T, McEwen CM, Gelev VM, Turner RA, Linington RG, Leung SSF, Kalgutkar AS, Bauman JN, Zhang Y, Liras S, Price DA, Mathiowetz AM, Jacobsen MP, Lokey S (2011) On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat Chem Biol 7:810–817
Acknowledgements
The contributions of Isabelle Adam, Jerome Andre, Norbert Domange, Stefan Grueninger, Pauline Heymes, Alexander Marziale, Stephane Rodde, Franziska Schneider, Mina Sellak, and Peter Wipfli are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors Thomas Vorherr, Ian Lewis, Joerg Berghausen, Felix Huth, Michael Schaefer, Roman Wille, Jinhai Gao, and Bing Wang declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Vorherr, T., Lewis, I., Berghausen, J. et al. Pyridyl-Ala Modified Cyclic Hexapeptides: In-Vitro and In-Vivo Profiling for Oral Bioavailability. Int J Pept Res Ther 26, 1383–1397 (2020). https://doi.org/10.1007/s10989-019-09935-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-019-09935-y