
Abstract
Novel coronavirus disease 2019 (COVID-19) has resulted in a global pandemic and is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Several studies have suggested that a precise disulfide-thiol balance is crucial for viral entry and fusion into the host cell and that oxidative stress generated from free radicals can affect this balance. Here, we reviewed the current knowledge about the role of oxidative stress on SARS-CoV and SARS-CoV-2 infections. We focused on the impact of antioxidants, like NADPH and glutathione, and redox proteins, such as thioredoxin and protein disulfide isomerase, that maintain the disulfide-thiol balance in the cell. The possible influence of these biomolecules on the binding of viral protein with the host cell angiotensin-converting enzyme II receptor protein as well as on the severity of COVID-19 infection was discussed.
Similar content being viewed by others

1 Introduction
Coronaviruses (CoVs) are members of the Coronaviridae family and are positive-sense, single-strand enveloped RNA viruses. These viruses contain the largest genome among all known RNA viruses, with a normal size ranging from 27 to 32 kb [1, 2]. CoVs are capable of infecting mammalian and avian species and typically cause respiratory, gastrointestinal, and central nervous system diseases [3]. The novel coronavirus known as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the seventh member of the coronavirus family [4]. The other two viruses in this family that infect humans are severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV). The full-length RNA genome of SARS-CoV and SARS-CoV-2 contain 29,727 and 29,811 nucleotides (~ 30 kb); SARS-CoV-2 genome encodes for 29 proteins whereas SARS-CoV genome contains 14 potential open reading frames [1, 2]. There are four subfamilies of coronaviruses, known as alpha, beta, gamma, and delta. The beta-coronaviruses cause severe disease and fatalities as compared to other subfamilies of coronaviruses; SARS-CoV and SARS-CoV-2 belong to beta subfamily [5].
The maintenance of disulfide-thiol equilibrium is an important aspect of viral entry, viral reactivity, and viral fusion, and can be affected by oxidative stress (vide infra) [6]. Recent studies suggest that oxidative stress plays an important role in viral infections such as SARS-CoV and SARS-CoV-2 infections [7,8,9,10,11,12,13]. Oxidative stress is defined as the disproportion between the presence of antioxidants and free radicals, or prooxidants, in a biological system [14]. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are the byproducts of various cellular processes, including aerobic metabolism. For example, the RNS nitric oxide (NO·) is produced from l-arginine by nitric oxide synthase, which then reacts with superoxide (O2·−) to form peroxynitrite (ONOO–) [15, 16]. These reactive oxygen/nitrogen species (RONS), namely, hydroxyl, superoxide anion, nitric oxide, and nitrosyl anion, are highly reactive molecules due to their unpaired valence electrons [7]. Normally, RONSs play important biological roles in cell signaling (redox signaling) pathways, thiol switches, regulation of inflammatory cytokines, growth factors, etc.[16,17,18]. Under normal physiological conditions, the balance between the cellular level of RONS and antioxidant is maintained. However, when the redox balance is disrupted, these powerful oxidants (free radicals) can have detrimental impacts; they are commonly linked to the onset of several lifestyle diseases [12]. These malignant effects are ultimately caused by unregulated oxygen- and nitrogen-containing free radicals that attack different cells and damage DNA, proteins, and lipids. Several studies have suggested that the production of excess RONS and a disproportionate cellular antioxidant-oxidant balance have an important function in the pathogenesis of respiratory viral infections, such as SARS-CoV infections [7, 12, 19]. Also, recent reports suggested that those with pre-existing conditions such as diabetes, hypertension, and pulmonary, cardiac, and kidney diseases are at a higher risk of developing a severe infection [20,21,22]. Also, these pathologies such as cancers, diabetes mellitus, cardiovascular diseases, chronic kidney disease, etc. cause an enhancement of oxidative stress [7 and references therein].
This review focuses on the significance of oxidative stress in SARS-CoV and SARS-CoV-2 infections, particularly as it relates to viral interaction with ACE2, viral infection as a result of disulfide-thiol balance, the role of ACE2 in lowering oxidative stress, the impact of reduced/oxidized nicotinamide dinucleotide phosphate (NADPH/NADP+) equilibrium on ACE2, and how thioredoxin (TRX) and protein disulfide isomerase (PDI) affect the oxidation state of ACE2.
2 Structure of SARS-CoV/SARS-CoV-2 Spike Protein and its Interactions with Human Cell-Surface Receptor ACE2
2.1 Coronavirus Structure
Among the structural proteins of coronaviruses, at least four are encoded in all coronaviruses: the nucleocapsid (N) protein, the membrane (M) protein and the envelope (E) protein, which are involved in viral assembly, and the spike (S) protein, which is involved in viral entry into host cells (Fig. 1) [1]. The SARS-CoV-2 genome contains open reading frames for six accessory proteins (3a, 6, 7a, 7b, 8, and 10), though it is currently unknown which accessory genes are expressed [23]. The remaining proteins are nonstructural and are expressed as two long polypeptides that are cleaved by the SARS-CoV-2 protease [23]. The S protein is the largest structure and makes the distinct spikes on the surface of the virus (Fig. 1). It is heavily glycosylated, utilizes an N-terminal signal sequence to gain access to the endoplasmic reticulum, and mediates attachment to host receptors [24]. Because of its essential role in host binding and viral entry, the S protein is a key target for the treatment of coronavirus-caused infections.

Structure of SARS-CoV virion
2.2 Spike Protein Structure
Coronavirus S proteins consist of three segments: an ectodomain, a single-pass transmembrane anchor, and an intracellular tail [25]. The ectodomain is further divided into two subunits: the receptor-binding subunit S1, and the fusion-machinery-containing subunit S2 [26]. S1 contains domains essential for binding to a host receptor, comprised of an amino-terminal domain, a carboxy-terminal receptor-binding domain (RBD), and sub-domains 1 and 2. S2 is responsible for the fusion of the viral and host cell membranes, consisting of a fusion peptide, heptad repeats, a central helix, and a connector domain [27]. Electron microscopy studies have shown that the S protein itself is a trimer consisting of three S1 heads and a trimeric S2 stalk, which is an essential structural property for the binding and invagination of the target host cell [28, 29]. Overall, the structure of SARS-CoV-2 resembles the structure of the S protein of SARS-CoV. The RMSD of the 959 Cα atoms is 3.8 Å, while the RMSD of the amino-terminal domains, RBDs, sub-domains 1 and 2, and S2 subunits were found to be 2.6 Å, 3.0 Å, 2.7 Å, and 2.0 Å, respectively, indicating a high degree of structural homology, which is consistent with the fact that these two proteins exhibit greater than 70% sequence identity [27, 30].
2.3 Angiotensin-Converting Enzyme 2
ACE2 is a metallopeptidase found in nearly every human organ which serves as a component to counter-regulation of cardiovascular renin–angiotensin–aldosterone system (RAAS) functioning [31, 32]. It activates angiotensin, a regulator of blood pressure, and binds to the cell membranes of tissues in the lungs, heart, kidneys, intestinal cells, brain, and testes [33]. It can also be found free-floating in a soluble form, albeit in much lower concentrations [33]. ACE2 is a dimeric enzyme containing an M2 domain, an N-terminal peptidase, and a C-terminal collectrin renal amino acid transporter domain [34]. Multiple studies have identified ACE2 as the entry site for SARS-CoV and SARS-CoV-2 into human respiratory epithelial cells [31, 35,36,37]. Preliminary findings suggest that genetic variants in the ACE2 protein can prevent the infection of a person exposed to the virus [38].
2.4 Receptor Recognition and Binding
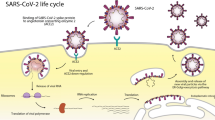
The generalized process for viral fusion is well understood, however, the process becomes more nuanced depending on the specific virus under examination. Overall, when SARS-CoV fuses with ACE2 in the human body, it involves a three-step process. First, the S1 subunit is bound to the peptidase domain of ACE2 [34]. Then, the S protein is cleaved into two parts (S1 and S2) by the receptor transmembrane protease serine 2. Following cleavage, the S2 portion rearranges and completes the fusion between the two membranes. The completed fusion allows the virus to enter the cell, replicate, and continue to infect other cells [33].
The RBD of S1 is responsible for binding to a host cell receptor. The RBD undergoes hinge-like conformational motions to either hide or expose the receptor-binding components. In the “up” or open conformation, the binding constituents are accessible, while in the “down” conformation, the binding constituents are inaccessible. The up conformation is a less stable state and is required in order for binding to occur [27]. Depending on the specific coronavirus, RBDs within the S1 subunit is used to recognize different functional receptors. The coronaviruses Porcine epidemic diarrhea virus and Transmissible gastroenteritis virus recognize aminopeptidase N, while MERS-CoV and HKU4 recognize the serine peptidase dipeptidyl peptidase 4 [1]. Coronaviruses OC43 and HKU1 attach to acetylated sialoside receptors on glycoproteins and glycolipids of the host cell, while MERS-CoV recognizes non-acetylated sialoside receptors [39, 40]. SARS-CoV-2, just like SARS-CoV, interacts with ACE2 to promote cell entry [41, 42]. The structure of SARS-CoV-2 RBD bound to ACE2 was recently determined by Lan et al. and has provided valuable insight into this interaction [43]. The RBD of SARS-CoV-2 contains a twisted five-stranded antiparallel β sheet and forms a core with short connecting helices and loops [43]. An extended insertion known as the receptor-binding motif (RBM) from the core contains two short β strands, two α helices, and loops. The RBM contains most of the residues of SARS-CoV-2 that bind to ACE2. Three disulfide bonds (Cys336–Cys361, Cys379–Cys432, and Cys391–Cys525) stabilize the β sheet structure of the core, while a fourth pair (Cys480–Cys488) connects loops in the distal end of the RBM [43]. The N-terminal peptidase domain of ACE2 forms the peptide substrate binding site between two lobes, while the N-terminal helix contacts a concave outer surface in the RBM. There are 20 residues in ACE2 that interact with both SARS-CoV and SARS-CoV-2 RBDs. 17 of these residues are shared between the two coronaviruses [43]. There are 14 amino acid positions that interact with ACE2 and are shared by RBMs of SARS-CoV and SARS-CoV-2; of these 14, 8 residues are identical and 5 have similar biochemical properties [43]. The RBDs of SARS-CoV and SARS-CoV-2 are very similar, which explains the potent binding of the SARS-CoV antibody, CR3022, with the SARS-CoV-2 RBD [44].
Early investigation of both SARS-CoV and SARS-CoV-2 using HeLa cells suggests that the ACE2 enzyme is necessary for viral infection as HeLa cells without any ACE2 present show no signs of infection [43]. There are several cysteine residues in the RBD of S protein and the peptidase domain of ACE2 (vide supra) that interact with one another (Fig. 2). The disulfide bonds formed at the protein–protein interfaces seem critical for S protein binding to ACE2.

Structures of protein complexes of a SARS-CoV⋯ACE2 (PDB: 3D0G) and b SARS-CoV-2⋯ACE2 (PDB: 6M0J). ACE2 shown in gray and spike glycoprotein in blue. Both visualized using New Cartoon drawing method. All the disulfide bridges between cysteine residues are shown in golden vdW spheres and thiol groups in magenta licorice (Color figure online)
3 Disulfide-Thiol Balance and Viral Infection
3.1 Viral Envelope Proteins
The two main steps of viral entry into the host cell are the binding of the virus particle to the cell-surface receptor and fusion of the viral envelope and cell membrane. A conformational change of viral envelope proteins is a prerequisite for them to undergo fusion to their target cells [45]. There are specific conditions that are necessary for these conformational changes to occur such as acidic pH, enzymatic actions, etc. One of the conditions is the precise disulfide-thiol balance at the viral surface and the cell-surface of the host [45, 46]. A specific balance is necessary to facilitate both, binding of the virus to a cell and for the fusion of the viral and cell membranes [6].
There are many examples of viral infections that display the importance of the native disulfide-thiol balance, where a specific disulfide-thiol balance is required for many different purposes, such as maintaining reactivity, becoming destabilized, aggregating or enabling viral membrane fusion with the target cell [6, 45]. One example is the baculovirus fusion protein, gp64, where changes in the native disulfide-thiol balance cause the protein to aggregate, which drives the fusion process [45, 47]. Another example of a virus displaying the importance of the disulfide-thiol balance is the Sindbis virus, which requires a specific number of disulfide bonds to maintain its stability and function within the protein envelope [45]. Overall, it has been suggested that disulfide-thiol balance is crucial for protein-mediated membrane fusion [47].
Combining the information collected about disulfide-thiol balances for many viruses, including those described above, implies that changes in the native disulfide-thiol balance of viral glycoproteins influence viral entry into a cell [6]. Studies have shown that the ability of a virus and viral envelope glycoproteins to fuse to the surface of a cell membrane depends on the disulfide-thiol balance of the cell [6, 45]. Changes in the native disulfide network at the surface of a target cell have major effects on the ability of many viruses to carry out cell-virus interaction processes [6, 45]. It has also been shown that disulfide-thiol rearrangements within envelopes can cause conformational changes of the peptide, allowing for entry of a virus into a cell [6]. This was determined through the introduction of reducing or alkylating agents, which disrupt the natural disulfide-thiol balance at the surface of a mature viral envelope [45].
3.2 HIV Infection
There have been many studies involving human immunodeficiency virus (HIV), which have resulted in extensive knowledge about how changes in the native disulfide-thiol balance at the surface of a cell has been shown to hinder the ability of a virus to enter its target cells and that HIV infection depends on the presence of free thiol groups [47, 48]. It has been observed that introducing disulfide isomerase inhibitors suppresses HIV infection, suggesting that the disulfide-thiol balance is an important aspect regarding whether the virus can enter its target cell [46]. The reduction of disulfides of the primary receptor, glycoprotein CD4 cells, is required before viral fusion and entry into a cell can occur. Other studies have determined that changes in native disulfide-thiol balance can result in the fusion of a peptide into the cell membrane, ultimately leading to viral entry into the cell [45, 49]. These changes in disulfide-thiol balance trigger a mechanism that activates HIV fusion to its target cell due to protein refolding [45, 50]. Studies have shown that as thiol content in HIV gp120 increases, the ability to bind its respective target cell is decreased [45].
3.3 SARS-CoV and SARS-CoV-2 Infection
Recent studies also highlighted the importance of disulfide-thiol balance in the viral entry of coronaviruses SARS-CoV and SARS-CoV-2. Similar to the HIV gp120 protein, experimental data has shown that as the thiol content in SARS-CoV S1 (the receptor-binding subunit) increased, its ability to bind to its target cells decreased, suggesting that both viruses require a specific thiol content for fusion and entry to occur [45].
In a study conducted by Lavillette, it was found that as SARS-CoV S1 became more reduced, ACE2 binding capacity became more and more reduced until eventually there was minimal binding occurring [6]. It is also possible that the thiol groups and disulfide groups within or between the S complex and proteins on the target cell surface act as electron donors and acceptors, respectively. This, in turn, may activate fusion mechanisms, leading to viral fusion and entry into the cell [6].
Gallagher studied S proteins involved in murine hepatitis virus (MHV), a coronavirus, and found that blocking cysteine residues in the S proteins did not affect cell receptor binding abilities, but did negatively impact the ability for structural changes necessary for fusion to occur [51]. It is known that single residue changes in S proteins can alter protein structure, and from this, it is proposed that even mutations distant from the S1 protein may lead to modifications in the native disulfide-thiol balance [51].
Because of the structural similarities of SARS-CoV to other coronaviruses, including SARS-CoV-2, these coronaviruses function in a manner similar to SARS-CoV, meaning that they depend on a specific disulfide-thiol balance before fusion, and ultimately, viral entry can occur [6]. Recent computational work corroborated with the previous experimental work on SARS-CoV. The complete reduction of disulfide linkages in ACE2 and RBD domain of the S protein resulted in impaired binding between these two proteins [30]. A similar observation was made earlier where the alanine substitution of cysteines disrupted the disulfide bond at the protein–protein interface [45].
4 ACE2 and its Role in Lowering Oxidative Stress
4.1 Vascular Regulation
Vascular regulation is essential for the survival and health of most multicellular organisms. In mammals, this control of the cardiovascular system is exerted in part through the actions of vascular endothelial cells. These cells help to trigger the dilation and constriction of blood vessels and the adjustment of vascular concentrations of numerous chemical species in response to environmental and hormonal stimuli. An important source of this regulation is the renin angiotensin-aldosterone system (RAAS), which utilizes multiple proteins, including ACE 1 and 2. The ACE2 receptor plays a key role in lowering oxidative stress, which in turn plays a key role in modulating the binding affinity of proteins involved in SARS-CoV-2 infection, i.e. S protein and ACE2.
4.2 RAAS Pathway
Classical RAAS starts with the secretion of angiotensinogen from the liver into the bloodstream which then interacts with renin. The signaling molecule that serves to activate the RAAS system is renin, which is produced in kidney cells. Upon being released into the bloodstream, renin interacts with angiotensinogen, which degrades the angiotensinogen into Angiotensin I (Ang 1) (Fig. 3). Inactive Ang I is then converted into Angiotensin II (Ang II) via the cleavage of two amino acids from the C-terminal by ACE1 at the endothelium. Ang II has several important functions and acts as a stimulant for a variety of regulatory systems, in turn playing a pivotal role in the pathophysiological effects of RAAS [52]. Ang II also promotes the presence of superoxide species in a biological system, which gone unchecked, results in oxidative stress. Ang II is regulated by ACE2 which degrades Ang II into Angiotensin 1–7 (Ang 1–7), which counteracts the effects of Ang II via the Mas receptor [52].

Renin angiotensin system. ACE angiotensin-converting enzyme
4.3 Angiotensin-Converting Enzyme 2
ACE2 is a membrane-bound protein and as mentioned above, is responsible for the degradation of Ang II, a vasoconstrictor, to Ang 1–7, a vasodilator. Ang II produces RONS by stimulating membrane-bound NADPH oxidase [53, 54]. The degradation of Ang II into Ang 1–7 by ACE2 mitigates oxidative stress as it inhibits NADPH oxidase, and therefore, Ang II-induced RONS synthesis [55]. This phenomenon is in direct support of studies showing that overexpression of ACE2 can lead to reducing the effects of hypertension in animal models [56,57,58]. Hypertension has been observed to be a side effect directly correlated with oxidative stress, thus supporting the argument that the overexpression of ACE2 leads to a lowering of oxidative stress in a biological system [14]. This is especially important due to the intrinsic function of RAAS in the regulation of pathophysiological pathways. If ACE2 is bound to the S protein, the cellular concentration of Ang II will increase, leading to an increased presence of superoxide species and subsequent cell damage, inevitably creating a cycle of oxidative stress, and ultimately, increasing the risk of severe illness from COVID-19.
Oxidative stress (free RONS) is generated by high concentrations of Ang II and low concentrations of Ang 1–7. These RONS can oxidize the cysteine residues on the peptidase domain of ACE2 receptors and RBD of SARS-CoV and SARS-CoV-2 S proteins, keeping them in oxidized (disulfide) forms, as opposed to reduced (thiol) forms [59]. It seems possible that oxidation of these thiols to disulfides, under a mechanism of oxidative stress, would increase the affinity of SARS-CoV and SARS-CoV-2 S proteins for the ACE2 receptor, and therefore, increase the severity of COVID-19 infection [30, 60].
5 NADPH/NADP+ Equilibrium and its Impact on ACE2
5.1 NADPH Oxidase Promotes Oxidative Stress
NADPH oxidase is a membrane-bound enzymatic complex that reduces O2 to superoxide and is primarily found in phagocytotic cells, endothelial cells, vascular smooth muscle cells, and cardiac myocytes [53]. Binding of the viral S protein with ACE2 increases the concentration of Ang II as ACE2 is no longer available to convert Ang II to Ang 1–7. Under this condition, Ang II binds the angiotensin type 1 receptor (AT1R), which stimulates NADPH activity. This results in increased production of ROS (superoxide) and consumption of NADPH, the electron donor, in the cell.
The relationship between Ang II and NADPH oxidase has been investigated using murine vascular smooth muscle cells [61]. When the cells were exposed to Ang II, researchers observed increased NADPH oxidase activity as well as increased production of superoxide anions. Exact mechanisms for the stimulation of NADPH oxidase are complex but are known to occur at the genetic, transcriptional, and post-transcriptional levels and involve numerous signaling molecules and scaffolding proteins/platforms [62]. Dormant NADPH oxidase contains two subunits: glycoprotein (gp)91phox (for phagocyte oxidase) and p22phox [63]. In the presence of Ang II, NADPH oxidase is activated and the additional subunits p67phox, p47phox, p40phox, and Rac1 shift from the cytosol to the membrane. The activated NADPH oxidase can generate superoxide anions. Xia et al. performed a study in which they removed the ACE2 gene from the brains of mice and then compared the NADPH oxidase activity, among other things, of these mice with non-transgenic littermates after the infusion of Ang II [64]. NADPH oxidase activity was found to increase in the mice without ACE2. A similar study by Wysocki et al. also observed an increase in NADPH oxidase activity [57]. In particular, ACE2 deficiency increases NADPH-mediated oxidative stress in the kidney [57]. Since the binding of SARS-CoV-2 to the ACE2 receptor inhibits the catalytic activity of the enzyme, i.e. conversion of Ang II to Ang 1–7, one can conclude that NADPH oxidase activity might also increase in SARS-CoV-2 patients, subsequently leading to an increase in oxidative stress.
5.2 Glutathione Defends Against Oxidative Stress
Cells maintain a reducing environment through the presence of biomolecules such as glutathione, TRX and glutaredoxin, which reduce ROS to less harmful molecules like water and molecular oxygen. Glutathione (γ-l-glutamyl-l-cysteinyl-glycine) is a biologically abundant tripeptide that plays an important role in cellular detoxification via glutathione S-transferases, antioxidant defense, maintenance of thiol status, and modulation of cell proliferation [65,66,67]. Although not necessary for prokaryotic cells, it has been found essential for eukaryotes [65]. Synthesis of GSH occurs in the cytosol and is closely regulated [66]. Glutathione exists in two states: oxidized (GSSG) and reduced (GSH). It exists predominantly as GSH, with concentrations of 5–10 mM in liver cells; the amount of GSSG, however, is less than 1% that of GSH [68, 69].
As mentioned earlier, oxidative stress occurs when there is a disproportionate amount of ROS, such as H2O2 and superoxide, compared to antioxidants. Oxidative stress is known to stimulate the synthesis of GSH [69, 70]. This GSH is used to reduce H2O2 and organic peroxides, leaving behind the oxidized GSSG (Eq. 1) [71].
The reduction of GSSG utilizes the enzyme glutathione reductase, which requires NADPH to function (Eq. 2).
Thus, the cellular ratio of NADPH to NADP+ has cascade effects on the ability of the glutathione pathway to act as a defense against oxidative stress in a cell.
Lower levels of glutathione increase cellular oxidative stress and are associated with various disease states and immune dysfunctions that lead to a higher susceptibility of viral infections, namely uncontrolled SARS-CoV-2 infection [71]. Uncontrolled replication leads to oxidative damage in the lungs which increases the viral load, thus increasing the severity of infection from the virus [72]. Conversely, high levels of GSH may prevent the virus from replicating efficiently, producing lower viral loads and thus, milder symptoms. A doctor at Kursk State Medical University investigated the effects of GSH levels on an individual’s ability to recover from COVID-19 infection and found that high ROS/GSH ratios seemed to strongly correlate to worsened symptoms and slower recovery times [72].
GSH deficiency can interfere with the body’s ability to detoxify the cellular environment, fold proteins, regenerate antioxidants, maintain healthy immune responses, and even modulate apoptosis events—this promotes suboptimal cellular function and leads to illness [70, 72]. Age has a large impact on the severity of COVID-19 symptoms, where older individuals are at an increased risk compared to younger individuals. Age is also associated with GSH levels, where GSH concentrations decrease as age increases in erythrocytes and mammals [73]. GSH deficiency is known to cause a variety of adverse physiological effects and can alter genes that work together to synthesize vitamin D (VD) and is linked to an increase in oxidative stress (vide infra) [74].
5.3 Vitamin D and Severity of COVID-19 Infection
There is plenty of evidence supporting the relationship between low VD levels and increased severity of COVID-19 symptoms [75, 76]. This relationship is likely attributed to the role of VD in regulating immune function, defense against respiratory illness, and its antioxidant properties [75]. Many diseases including hypertension, diabetes, cardiovascular disease, and metabolic syndrome are associated with low levels of VD. Such diseases are known comorbidities of COVID-19 and may increase the risk of severe infection from SARS-CoV-2 [76]. The significance of VD in protection against SARS-CoV-2 infection is strong, however, it is not known whether GSH deficiency or VD deficiency is more significant in causing severe symptoms from COVID-19 [74].
5.4 NADPH and Severity of COVID-19 Infection
Although NADPH plays an undeniably vital role in the RAAS system, there is currently no literature specifically relating the NADPH/NADP+ equilibrium to ACE2 or SARS-CoV-2 infection. However, it is expected that NADPH has some positive role in reducing the severity of COVID-19 infection. NADPH is important for the regeneration of GSH which is important in reducing ROS (Fig. 4). As discussed earlier, viral infection stimulates the activity of NADPH oxidase, which produces superoxide using NADPH as an electron donor in the cell. Increased activity of NADPH oxidase reduces the concentration of free NADPH that is needed to reduce GSSG to GSH. Less availability of GSH will further increase the oxidative stress in the cell, which in turn will favor S protein-ACE2 interactions and enhance the severity of COVID-19 infection.

NADPH/NADP+ equilibrium and ROS. Glucose 6-phosphate dehydrogenase is an important enzyme in the regeneration of NADPH but was not a focus of this review
6 Thioredoxin, Protein Disulfide Isomerase and, Oxidative State of ACE2
6.1 Thioredoxin System and Transcription Factor Regulation
The thioredoxin system is a selenium-dependent system of enzymes that regulates intracellular ROS concentration and the regeneration of antioxidants [59]. It is comprised of TRX and thioredoxin reductase (TRXR), which work in concert to maintain a redox balance in the cellular environment via disulfide reduction of target proteins. TRX is an oxidoreductase that regulates the activity of multiple redox-sensitive transcription factors (TF) via its disulfide active site [77]. These TFs are necessary for oxidative signaling, hence the importance of the thioredoxin system in many signaling pathways. A notable example is Ref-1, a DNA-repair endonuclease whose activity is dependent on reduction by TRX; when reduced by TRX, Ref-1 increases DNA binding affinity of several TFs, including AP-1, NF-κB, ATF/CREB, Myb, and p53 [78].
6.2 Thioredoxin Structure
The TRX active site contains a conserved sequence (–Cys–Gly–Pro–Cys–) and belongs to a TRX superfamily, in which all members share similar active site sequences –Cys–X–Y–Cys). It has a low redox potential and is known to be a strong reductant under physiological conditions [79]. The reduced dithiol form of thioredoxin (TRX-(SH)2) is the redox-active form that scavenges ROS and reduces disulfides in target proteins. The oxidized disulfide form of thioredoxin (TRX-S2) is inactive and therefore, must be reduced to be functional. The reduction of (TRX-S2) back to its (TRX-(SH)2) form requires flavoenzyme TRXR and NADPH. Formally, TRXR can be described as an oxidoreductase that relies on NADPH and FAD to reduce TRX-S2.
TRX is secreted in response to oxidative stress by multiple cell types, including virally infected cells. Once secreted, TRX can mimic cytokine and chemokine activity to affect the interaction and communication between cells and influence immune response [80, 81]. It is also known that activation of cytoprotective transcription factor NRF2 (nuclear factor-erythroid 2-related factor 2) upregulates TRX and TRXR. Upregulation of these enzymes has been proposed to decrease the oxidative stress impacting the propagation of the COVID-19 disease [82]. The TRX/TRXR system catalyzes the shift of disulfide-thiol reaction equilibrium toward thiolated products, which might facilitate reduction of the disulfide bonds of SARS-CoV-2 as well as ACE2. As predicted from the computational studies, the binding of SARS-CoV S protein to ACE2 will be impaired, ultimately halting the disease propagation [30]. However, there is no experimental proof that either SARS-CoV-2 or ACE2 are targets of TRX/TRXR systems and more experimental work is needed to provide a clear understanding of the interplay of these molecules and COVID-19 infection. In summary, the upregulation of TRX/TRXR could potentially decrease the risk of infection by SARS-CoV-2.
However, a recent study demonstrated that the gold-based drug auranofin, a TRXR inhibitor, also inhibits SARS-CoV-2 replication [79, 83]. An earlier study of HIV suggested that auranofin induces apoptosis in HIV-infected memory T lymphocytes, which are the most important reservoir of latent HIV, under increased cellular oxidative stress due to the inhibition of TRXR [84]. The inhibition of the enzyme TRXR is proposed to be due to the stronger binding of the gold(I) to the enzyme’s selenocysteine, but a theoretical study found the drug could bind favorably to cysteine as well [83, 85]. Therefore, the inhibition of viral replication by auranofin raises more questions about the role of disulfide-thiol exchanges involving TRXR/TRX systems than answers. A thorough study is needed to understand the mechanism of action of TRX and TRXR on SARS-CoV-2 infection, which could be promising targets for developing antiviral drugs against SARS-CoV-2.
6.3 Protein Disulfide Isomerase
PDI is a member of the TRX superfamily that aids in protein folding by regulating and/or catalyzing the formation, isomerization, or reduction of disulfide bonds to regulate their 3-dimensional structure—this activity is dependent on reducing equivalents of glutathione [79, 86]. PDI is found most in the endoplasmic reticulum (ER) but exists in other locations, including endothelial cells [87]. In contrast to its predominant oxidizing activity in the ER, surface-associated PDI acts primarily as a reductant. In the early stages of protein folding, disulfide formation is susceptible to error. PDI acts as a chaperone to prevent aggregation and misfolding of target proteins that have incorrect disulfide pairings or pairing at the wrong residues [88]. At high concentrations, PDI acts as a chaperone to prevent aggregation by facilitating proper folding and production of target proteins, but can promote aggregation at lower concentrations [88, 89].
6.4 Oxidative Stress and PDI
ROS are impossible to eliminate from the cellular environment, as they are necessary for many cellular mechanisms, such as cellular signaling. The human body is a great buffer, and many proteins can maintain function under mild conditions of oxidative stress. However, some proteins may be more susceptible to oxidative damage and subsequent degradation than others.
In one study, PDI was selectively degraded and resynthesized after peroxide exposure [86]. This selective proteolysis is largely catalyzed by the proteasome: a protease complex that carries out selective degradation of proteins [86, 90]. The extent of PDI degradation in response to mild levels of oxidative stress was 90% greater over the span of 24 h than that of other proteins. However, these high levels of degradation were followed by high levels of regeneration, resulting in approximately constant levels of PDI. Because of this caveat, it cannot be said conclusively that PDI is more sensitive to oxidative damage or degradation, however, it is a plausible mechanism. Another possibility is that PDI may be preferentially damaged by the initial peroxide exposure, causing degradation, followed by regeneration from scratch to maintain cellular homeostasis [86].
Other studies show that PDI is inhibited by high levels of oxidative stress. In one study, rats were fed a diet high in fat and ethanol to create an oxidized cellular environment, in which 4-hydroxynonenal (4-HNE) was used as a marker to identify modified proteins. PDI was found to be consistently modified by 4-HNE, specifically at the cysteine residue in its active site. To confirm these observations, purified PDI was treated with different concentrations of 4-HNE in vivo (20–200 µM) to show a 14–56% inhibition, respectively. The same experimental design was used to show inhibition by different lipid peroxidation products (acrolein and 4-oxononenal), resulting in 60% and 100% inhibition, respectively. It was also shown that adding physiological concentrations of GSH partly mitigated this effect by removing 4-HNE from PDI. This compensatory mechanism has a threshold dependent on the redox balance of the cell. Because PDI bound by 4-HNE was identified in vivo, where GSH is present, it suggests that this compensatory mechanism may be limited by high levels of oxidative stress in which GSH concentrations are low [91].
6.5 PDI and Severity of COVID-19 Infection
Through careful analysis of oxidative stress interference on PDI function, predictions about how oxidative stress affects the severity of infection by SARS-CoV-2 can be made. As mentioned, the affinity of SARS-CoV-2 for the ACE2 receptor is intensified by the oxidation of cysteine residues in both proteins. This interaction lowers the concentration of free ACE2 and further enables the generation of oxidative stress by increasing the concentration of Ang 2 and decreasing the concentration of Ang 1–7. Furthermore, the inhibition or reduced activity of PDI due to oxidative stress could have impact on SARS-CoV-2 infection. The role of PDI in SARS-CoV-2 infection could be experimentally verified by performing binding assays between SARS-CoV-2 spike and ACE2 receptor proteins under oxidative stress in the absence and presence of PDI. The binding assays could also be performed in the presence of thiol/disulfide exchange blockers and antibodies against PDI.
7 Potential COVID-19 Treatments that Address Oxidative Stress
7.1 Redox-Modulating Agents in the Treatment of Viral Infections
Imbalance of redox homeostasis in the body is the main aspect of viral infections. The virus manipulates the host cell machinery to throw the cell into a state of oxidative stress to create favorable conditions for viral replication by causing an excess ROS and a deficiency of GSH [92]. Research suggests that oxidative stress may be a viable target in fighting viral infections and that by modulating the sensitive redox pathways, the immune response may be regulated. This was studied in the context of a wide variety of viruses, including SARS-CoV-2. Some of the antioxidant agents of interest are N-acetylcysteine (NAC), GSH, polyphenols, vitamins C, D, and E, and selenium [92, 93]. Although data has been collected, more experimentation is necessary before any treatments are considered effective antiviral treatments.
7.2 N-Acetylcysteine
Oral administration NAC could potentially be a viable drug to treat COVID-19 infection due to its role in the synthesis of glutathione, improving T cell response, and modulating inflammation [10, 94]. L-cysteine is the rate-limiting substrate for the synthesis of glutathione. NAC supplementation provides the cell with an extra source of cysteine to synthesize glutathione, better equipping it to combat oxidative stress and the presence of a thiol group can block ACE2 activity, which would hinder SARS-CoV-2 penetration into target cells [10, 94]. Additionally, SARS-CoV-2 causes the production of even more cytokines than SARS-CoV. These excess cytokines and the viral antigens are the likely causes of T cell exhaustion. However, higher levels of glutathione are known to correlate with lymphocyte proliferation. NAC may be administered to keep glutathione levels high to prevent T cell exhaustion. NAC also inhibits the production of cytokines thereby preventing their inflammatory pathways [10]. Although it proves to be quite promising, further in vitro and in vivo studies are necessary to determine the effectiveness of NAC as a treatment for COVID-19.
Age predisposes individuals to have increased oxidative stress levels leading to the production of inflammatory cytokines, which is only exacerbated by COVID-19 infection. The gradual decrease in the capability of maintaining redox homeostasis amongst the elderly may increase the risk of excessive immune activation and lung damage in response to viral infection [95]. Chronic oxidative stress can lead to lipid peroxidation, a common feature associated with acute respiratory distress syndrome. ROS also impair proteasome and mitochondrial function. NAC is a drug of interest in the early treatment of COVID-19 due to its antioxidant properties, although it has not yet been proven to decrease mortality rates [95].
8 Conclusions
In conclusion, current research findings and reports suggested that oxidative stress could play a major role in viral infections, especially in coronavirus infections such as SARS-CoV and SARS-CoV-2. Understanding the role of oxidative stress, including the roles of antioxidants and redox proteins, in COVID-19 infection may help to direct potential therapeutics. In summary, the present review demonstrates that the absence of or reduced oxidative stress would have a significant beneficial effect during the early stage of viral infection by preventing viral protein binding on the host cells.
Data Availability
This review contains no new data; all data are published elsewhere.
Abbreviations
- 4-HNE:
-
4-Hydroxynonenal
- ACE2:
-
Angiotensin-converting enzyme 2
- Ang 1–7:
-
Angiotensin 1–7
- Ang I:
-
Angiotensin I
- Ang II:
-
Angiotensin II
- COVID-19:
-
Coronavirus disease 2019
- CoVs:
-
Coronaviruses
- GSH:
-
Glutathione (reduced form)
- GSSG:
-
Glutathione (oxidized form)
- HIV:
-
Human immunodeficiency virus
- MERS-CoV:
-
Middle East respiratory syndrome coronavirus
- NAC:
-
N-acetylcysteine
- NADP+ :
-
Nicotinamide dinucleotide phosphate (oxidized form)
- NADPH:
-
Nicotinamide dinucleotide phosphate (reduced form)
- PDI:
-
Protein disulfide isomerase
- RAAS:
-
Renin–angiotensin–aldosterone system
- RBD:
-
Receptor-binding domain
- RBM:
-
Receptor-binding motif
- RONS:
-
Reactive oxygen/nitrogen species
- S protein:
-
Spike protein
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- TF:
-
Transcription factor
- TRX:
-
Thioredoxin
- TRXR:
-
Thioredoxin reductase
- VD:
-
Vitamin D
References
Li F (2016) Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol 3(1):237–261
Ziebuhr J (2004) Molecular biology of severe acute respiratory syndrome coronavirus. Curr Opin Microbiol 7(4):412–419
Li F (2015) Receptor recognition mechanisms of coronaviruses: a decade of structural studies. J Virol 89(4):1954–1964
Denison MR, Graham RL, Donaldson EF, Eckerle LD, Baric RS (2011) Coronaviruses: an RNA proofreading machine regulates replication fidelity and diversity. RNA Biol 8(2):270–279
Velavan TP, Meyer CG (2020) The COVID-19 epidemic. Trop Med Int Health 25(3):278–280
Lavillette D, Barbouche R, Yao Y, Boson B, Cosset F, Jones IM, Fenouillet E (2006) Significant redox insensitivity of the functions of the SARS-CoV spike glycoprotein: comparison with HIV envelope. J Biol Chem 281(14):9200–9204
Ntyonga-Pono MP (2020) COVID-19 infection and oxidative stress: an under-explored approach for prevention and treatment? Pan Afr Med J 35(Suppl 2):12
Derouiche S (2020) Oxidative stress associated with SARS-Cov-2 (COVID-19) increases the severity of the lung disease—a systematic review. J Infect Dis Epidemiol. https://doi.org/10.1096/fj.202001807
Schönrich G, Raftery MJ, Samstag Y (2020) Devilishly radical NETwork in COVID-19: oxidative stress, neutrophil extracellular traps (NETs), and T cell suppression. Adv Biol Regul 77:100741
Poe FL, Corn J (2020) N-Acetylcysteine: a potential therapeutic agent for SARS-CoV-2. Med Hypotheses 143:109862
Laforge M, Elbim C, Frère C, Hémadi M, Massaad C, Nuss P, Benoliel JJ, Becker C (2020) Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat Rev Immunol 20(9):515–516
Delgado-Roche L, Mesta F (2020) Oxidative stress as key player in severe acute respiratory syndrome coronavirus (SARS-CoV) infection. Arch Med Res 51(5):384–387
Cecchini R, Cecchini AL (2020) SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med Hypotheses 143:110102
Yoshikawa T, Naito Y (2000) What is oxidative stress? Jpn Med Assoc J 124(11):1549–1553
Squadrito GL, Pryor WA (1998) Oxidative chemistry of nitric oxide: the roles of superoxide, peroxynitrite, and carbon dioxide. Free Radic Biol Med 25(4–5):392–403
Adams L, Franco MC, Estevez AG (2015) Reactive nitrogen species in cellular signaling. Exp Biol Med (Maywood) 240(6):711–717
Schieber M, Chadnel NS (2014) ROS function in redox signaling and oxidative stress. Curr Biol 24(10):R453-462
Antelmann H, Helmann JD (2011) Thiol-based redox switches and gene regulation. Antioxid Redox Signal 14(6):1049–1063
Camini FC, da Silva Caetano CC, Almeida LT, de Brito Magalhães CL (2017) Implications of oxidative stress on viral pathogenesis. Arch Virol 162(4):907–917
Abdi A, Jalilian M, Sarbarzeh PA, Vlaisavljevic Z (2020) Diabetes and COVID-19: a systematic review on the current evidences. Diabetes Res Clin Pract 166:108347
Mihalopoulos M, Dogra N, Mohamed N, Badani K, Kyprianou N (2020) COVID-19 and kidney disease: molecular determinants and clinical implications in renal cancer. Eur Urol Focus 6(5):1086–1096
Lee LYW, Cazier JB, Starkey T, Briggs SEW, Arnold R, Bisht V, Booth S, Campton NA, Cheng VWT, Collins G, Curley HM, Earwaker P, Fittall MW, Gennatas S, Goel A, Hartley S, Hughes DJ, Kerr D, Lee AJX, Lee RJ, Lee SM, Mckenzie H, Middleton CP, Murugaesu N, Newsom-Davis T, Olsson-Brown AC, Palles C, Powles T, Protheroe EA, Purshouse K, Sharma-Oates A, Sivakumar S, Smith AJ, Topping O, Turnbull CD, Várnai C, Briggs ADM, Middleton G, Kerr R (2020) COVID-19 prevalence and mortality in patients with cancer and the effect of primary tumour subtype and patient demographics: a prospective cohort study. Lancet Oncol 21(10):1309–1316
Kim D, Lee J, Yang J, Kim JW, Kim VN, Chang H (2020) The architecture of SARS-CoV-2 transcriptome. Cell 181(4):914.e10-921.e10
Casalino L, Gaieb Z, Dommer AC, Harbison AM, Fogarty CA, Barros EP, Taylor BC, Fadda E, Amaro RE (2020) Shielding and beyond: the roles of glycans in SARS-CoV-2 spike protein. bioRxiv
Tortorici MA, Veesler D (2019) Structural insights into coronavirus entry. Adv Virus Res 105:93–116
Kirchdoerfer RN, Cottrell CA, Wang N, Pellesen J, Yassine HM, Turner HL, Corbett KS, Graham BS, McLellan JS, Ward AB (2016) Pre-fusion structure of a human coronavirus spike protein. Nature 531(7592):118–121
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C, Abiona O, Graham BS, McLellan JS (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367(6483):1260–1263
Walls AC, Tortorici MA, Bosch BJ, Frenz B, Rottier PJM, DiMaio F, Rey FA, Veesler D (2016) Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 531(7592):114–117
Harrison SC (2008) Viral membrane fusion. Nat Struct Mol Biol 15(7):690–698
Hati S, Bhattacharyya S (2020) Impact of thiol-disulfide balance on the binding of Covid-19 spike protein with angiotensin-converting enzyme 2 receptor. ACS Omega 5(26):16292–16298
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203(2):631–637
Towler P, Staker B, Prasad SG, Menon S, Tang J, Parsons T, Ryan D, Fisher M, Williams D, Dales NA, Patane MA, Pantoliano MW (2004) ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J Biol Chem 279(17):17996–18007
Verdecchia P, Cavallini C, Spanevello A, Angeli F (2020) The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med 76:14–20
Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q (2020) Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367(6485):1444–1448
Wang H, Yang P, Liu K, Guo F, Zhang Y, Zhang G, Jiang C (2008) SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res 18(2):290–301
Millet JK, Whittaker GR (2018) Physiological and molecular triggers for SARS-CoV membrane fusion and entry into host cells. Virology 517:3–8
Cascella M, Rajnik M, Cuomo A, Dulebohn SC, Di Napoli R (2020) Features, evaluation and treatment coronavirus (COVID-19), in StatPearls. 2020, StatPearls Publishing. Copyright © 2020, StatPearls Publishing LLC: Treasure Island
Hussain M, Jabeen N, Raza F, Shabbir S, Baig AA, Amanullah A, Aziz B (2020) Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J Med Virol. https://doi.org/10.1002/jmv.25832
Vlasak R, Luytjes W, Spaan W, Palese P (1988) Human and bovine coronaviruses recognize sialic acid-containing receptors similar to those of influenza C viruses. Proc Natl Acad Sci USA 85(12):4526–4529
Park YJ, Walls AC, Wang Z, Sauer MM, Li W, Tortoriic MA, Bosch B, DiMaio F, Veesler D (2019) Structures of MERS-CoV spike glycoprotein in complex with sialoside attachment receptors. Nat Struct Mol Biol 26(12):1151–1157
Li F, Li W, Farzan M, Harrison SC (2005) Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309(5742):1864–1868
Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D (2020) Structure, Function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181(2):281-292.e6
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X (2020) Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581(7807):215–220
Tian X, Li C, Huang A, Xia S, Lu S, Shi Z, Lu L, Jiang S, Yang Z, Wu Y, Ying T (2020) Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg Microbes Infect 9(1):382–385
Fenouillet E, Barbouche R, Jones IM (2007) Cell entry by enveloped viruses: redox considerations for HIV and SARS-coronavirus. Antioxid Redox Signal 9(8):1009–1034
Stantchev TS, Paciga M, Lankford CR, Schwartzkopff F, Broder CC, Clouse KA (2012) Cell-type specific requirements for thiol/disulfide exchange during HIV-1 entry and infection. Retrovirology 9:97
Markovic I, Stantchev TS, Fields KH, Tiffany LJ, Tomiç M, Weiss CD, Broder CC, Strebel K, Clouse KA (2004) Thiol/disulfide exchange is a prerequisite for CXCR4-tropic HIV-1 envelope-mediated T-cell fusion during viral entry. Blood 103(5):1586–1594
Ryser HJ, Levy EM, Mandel R, DiSciullo GJ (1994) Inhibition of human immunodeficiency virus infection by agents that interfere with thiol-disulfide interchange upon virus-receptor interaction. Proc Natl Acad Sci USA 91(10):4559–4563
Bechtel TJ, Weerapana E (2017) From structure to redox: The diverse functional roles of disulfides and implications in disease. Proteomics. https://doi.org/10.1002/pmic.201600391
Jain S, McGinnes LW, Morrison TG (2007) Thiol/disulfide exchange is required for membrane fusion directed by the Newcastle disease virus fusion protein. J Virol 81(5):2328–2339
Gallagher TM (1996) Murine coronavirus membrane fusion is blocked by modification of thiols buried within the spike protein. J Virol 70(7):4683–4690
Guang C, Phillips RD, Jiang B, Milani F (2012) Three key proteases–angiotensin-I-converting enzyme (ACE), ACE2 and renin—within and beyond the renin-angiotensin system. Arch Cardiovasc Dis 105(6–7):373–385
Wen H, Gwathmey JK, Xie LH (2012) Oxidative stress-mediated effects of angiotensin II in the cardiovascular system. World J Hypertens 2(4):34–44
Touyz RM (2004) Reactive oxygen species and angiotensin II signaling in vascular cells—implications in cardiovascular disease. Braz J Med Biol Res 37(8):1263–1273
Lovren F, Pan Y, Quan A, Teoh H, Wang G, Shukla PC, Levitt KS, Oudit GY, Al-Omran M, Stewart DJ, Slutsky AS, Peterson MD, Backx PH, Penninger JM, Verma S (2008) Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am J Physiol Heart Circ Physiol 295(4):H1377-1384
Wysocki J, Ye M, Rodriguez E, González-Pacheco FR, Barrios C, Evora K, Schuster M, Loibner H, Brosnihan KB, Ferrario CM, Penninger JM, Batlle D (2010) Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension 55(1):90–98
Wysocki J, Ortiz-Melo DI, Mattocks NK, Xu K, Prescott J, Evora K, Ye M, Sparks MA, Haque SK, Batlle D, Gurley SB (2014) ACE2 deficiency increases NADPH-mediated oxidative stress in the kidney. Physiol Rep 2(3):e00264
Rentzsch B, Todiras M, Iliescu R, Popova E, Campos LA, Oliveira ML, Baltatu OC, Santos RA, Bader M (2008) Transgenic angiotensin-converting enzyme 2 overexpression in vessels of SHRSP rats reduces blood pressure and improves endothelial function. Hypertension 52(5):967–973
Nordberg J, Arnér ES (2001) Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med 31(11):1287–1312
Busse LW, Chow JH, McCurdy MT, Khanna AK (2020) COVID-19 and the RAAS-a potential role for angiotensin II? Crit Care 24(1):136
Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW (1994) Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 74(6):1141–1148
Nguyen Dinh Cat A, Montezano AC, Burger D, Touyz RM (2013) Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid Redox Signal 19(10):1110–1120
Sachse A, Wolf G (2007) Angiotensin II-induced reactive oxygen species and the kidney. J Am Soc Nephrol 18(9):2439–2446
Xia H, Suda S, Bindom S, Feng Y, Gurley SB, Seth D, Navar LG, Lazartigues E (2011) ACE2-mediated reduction of oxidative stress in the central nervous system is associated with improvement of autonomic function. PLoS ONE 6(7):e22682
Pallardó FV, Markovic J, García JL, Viña J (2009) Role of nuclear glutathione as a key regulator of cell proliferation. Mol Aspects Med 30(1–2):77–85
Lu SC (2009) Regulation of glutathione synthesis. Mol Aspects Med 30(1–2):42–59
Armstrong RN (1997) Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem Res Toxicol 10(1):2–18
Pramono AA, Rather GM, Herman H, Lestari K, Bertino JR (2020) NAD- and NADPH-contributing enzymes as therapeutic targets in cancer: an overview. Biomolecules 10(3):358
Meister A (1988) Glutathione metabolism and its selective modification. J Biol Chem 263(33):17205–17208
Forman HJ, Zhang H, Rinna A (2009) Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med 30(1–2):1–12
Fraternale A, Paoletti MF, Casabianca A, Oiry J, Clayette P, Vogel JU, Cinatl J Jr, Palamara AT, Sgarbanti R, Garaci E, Millo E, Benatti U, Magnani M (2006) Antiviral and immunomodulatory properties of new pro-glutathione (GSH) molecules. Curr Med Chem 13(15):1749–1755
Polonikov A (2020) Endogenous Deficiency of glutathione as the most likely cause of serious manifestations and death in COVID-19 patients. ACS Infect Dis 6(7):1558–1562
Abraham EC, Taylor JF, Lang CA (1978) Influence of mouse age and erythrocyte age on glutathione metabolism. Biochem J 174(3):819–825
Parsanathan R, Jain SK (2019) Glutathione deficiency induces epigenetic alterations of vitamin D metabolism genes in the livers of high-fat diet-fed obese mice. Sci Rep 9(1):14784
Razdan K, Singh K, Singh D (2020) Vitamin D Levels and COVID-19 Susceptibility: Is there any Correlation? Med Drug Discov. https://doi.org/10.1016/j.medidd.2020.100051
Biesalski H (2020) Vitamin D deficiency and co-morbidities in COVID-19 patients—a fatal relationship? NFS J. https://doi.org/10.1016/j.nfs.2020.06.001
Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE (2014) Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev 94(2):329–354
Nishinaka Y, Masutani H, Nakamura H, Yodoi J (2001) Regulatory roles of thioredoxin in oxidative stress-induced cellular responses. Redox Rep 6(5):289–295
Mathys L, Balzarini J (2016) The role of cellular oxidoreductases in viral entry and virus infection-associated oxidative stress: potential therapeutic applications. Expert Opin Ther Targets 20(1):123–143
Zhang JM, An J (2007) Cytokines, inflammation, and pain. Int Anesthesiol Clin 45(2):27–37
Hughes CE (2018) Nibbs RJB (2018) A guide to chemokines and their receptors. FEBS J 285(16):2944–2971
Cuadrado A, Pajares M, Benito C, Jiménez-Villegas J, Escoll M, Fernández-Ginés R, Garcia Yagüe AJ, Lastra D, Manda G, Rojo AI, Dinkova-Kostova AT (2020) Can Activation of NRF2 Be a Strategy against COVID-19? Trends Pharmacol Sci 41(9):598–610
Rothan HA, Stone S, Natekar J, Kumari P, Arora K, Kumar M (2020) The FDA-approved gold drug auranofin inhibits novel coronavirus (SARS-COV-2) replication and attenuates inflammation in human cells. Virology 547:7–11
Chirullo B, Sgarbanti R, Limongi D, Shytaj IL, Alvarez D, Das B, Boe A, DaFonseca S, Chomont N, Liotta L, Petricoin EI, Norelli S, Pelosi E, Garaci E, Savarino A, Palamara AT (2013) A candidate anti-HIV reservoir compound, auranofin, exerts a selective “anti-memory” effect by exploiting the baseline oxidative status of lymphocytes. Cell Death Dis 4(12):e944
Dos Santos HF (2014) Reactivity of auranofin with S-, Se- and N-containing amino acids. Comput Theor Chem 1048:95–101
Grune T, Reinheckel T, Li R, North JA, Davies KJ (2002) Proteasome-dependent turnover of protein disulfide isomerase in oxidatively stressed cells. Arch Biochem Biophys 397(2):407–413
Turano C, Coppari S, Altieri F, Ferraro A (2002) Proteins of the PDI family: unpredicted non-ER locations and functions. J Cell Physiol 193(2):154–163
Gilbert HF (1997) Protein disulfide isomerase and assisted protein folding. J Biol Chem 272(47):29399–29402
Wilkinson B, Gilbert HF (2004) Protein disulfide isomerase. Biochim Biophys Acta 1699(1–2):35–44
Tanaka K (2009) The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci 85(1):12–36
Carbone DL, Doorn JA, Kiebler Z, Petersen DR (2005) Cysteine modification by lipid peroxidation products inhibits protein disulfide isomerase. Chem Res Toxicol 18(8):1324–1331
Checconi P, DeAngelis M, Marcocci ME, Fraternale A, Magnani M, Palamara AT, Nencioni L (2020) Redox-modulating agents in the treatment of viral infections. Int J Mol Sci. https://doi.org/10.3390/ijms21114084
Bellavite P, Donzelli A (2020) Hesperidin and SARS-CoV-2: new light on the healthy function of citrus fruits. Antioxidants 9:742
De Flora S, Balansky R, La Maestra S (2020) Rationale for the use of N-acetylcysteine in both prevention and adjuvant therapy of COVID-19. FASEB J. https://doi.org/10.1096/fj.202001807
Nasi A, McArdle S, Gaudernack G, Westman G, Melief C, Rockberg J, Arens R, Kouretas D, Sjölin J, Mangsbo S (2020) Reactive oxygen species as an initiator of toxic innate immune responses in retort to SARS-CoV-2 in an ageing population, consider N-acetylcysteine as early therapeutic intervention. Toxicol Rep 7:768–771
Funding
This work was supported in part by National Institute of Health [Grant Number: GM117510-01 (S.B. and S.H.)] and by the Office of Research and Sponsored Programs of the University of Wisconsin-Eau Claire, Eau Claire, WI.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Suhail, S., Zajac, J., Fossum, C. et al. Role of Oxidative Stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) Infection: A Review. Protein J 39, 644–656 (2020). https://doi.org/10.1007/s10930-020-09935-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-020-09935-8