
Abstract
Background
Children with complete DiGeorge anomaly (cDGA) have congenital athymia plus a myriad of other challenging clinical conditions. The term cDGA encompasses children with congenital athymia secondary to 22q11.2DS, CHARGE syndrome (coloboma, heart defects, choanal atresia, growth or mental retardation, genital abnormalities, and ear abnormalities and/or deafness), and other genetic abnormalities. Some children have no known genetic defects. Since 1993, more than 100 children with congenital athymia have been treated with cultured thymus tissue implantation (CTTI). Naïve T cells develop approximately 6 to 12 months after CTTI. Most of the children had significant comorbidities such as heart disease, hypoparathyroidism, and infections requiring complex clinical care post cultured thymus tissue implantation (CTTI).
Objective
The purpose of this guidance is to assist multidisciplinary teams in caring for children with cDGA both before and after CTTI.
Methods
Thirty-one specialists, in addition to the authors, were asked to share their experience in caring for children with cDGA at Duke University Health System, before and after CTTI. These specialists included physicians, nurses, dentists, therapists, and dieticians.
Results
The goal of a multidisciplinary approach is to have children in the best possible condition for receiving CTTI and provide optimal care post CTTI through development of naïve T cells and beyond. The CTT (cultured thymus tissue) must be protected from high doses of steroids which can damage CTT. Organs must be protected from adverse effects of immunosuppression.
Conclusion
Creating a multidisciplinary team and a detailed plan of care for children with cDGA is important for optimal outcomes.
Similar content being viewed by others

Introduction
Cultured thymus tissue implantation (CTTI) is an investigational procedure that has been used at Duke University Health System since 1993 to treat more than 100 children with congenital athymia, most of whom had complete DiGeorge anomaly (cDGA) [1,2,3]. These children presented with a myriad of medical issues, the most common of which were heart defects and hypoparathyroidism [2]. The purpose of this guidance is to provide best practices for management of these children both before and after CTTI.
Methods
All children received CTTI under Institutional Review Board–approved protocols after parental consent. Because these children have many clinical issues, this guidance is presented based on organ systems. The immunology section includes both pre- and post-CTTI-specific care as immunologic outcomes are directly impacted by CTTI. Thirty-one clinical specialists were consulted for contributions to this guidance document.
Results
Immunology
Diagnosis: In the USA, newborn screening of T cell receptor rearrangement excision circles (TRECs) identifies children with congenital athymia [4, 5]. Upon receiving the report of low TRECs on a newborn screen, an immunologist should be consulted, protective isolation initiated, and breast-feeding stopped until the diagnosis of athymia is ruled out. No vaccines should be given. If needed, use only leukoreduced, CMV-negative, irradiated blood products. For the diagnosis of cDGA, a lymphocyte enumeration must show very low (< 50/mm3) naïve T cells with B and NK cells present. Patients with severe combined immunodeficiency (SCID) can be identified by a SCID genetic panel or whole exome sequencing (WES). Complete DiGeorge anomaly can also be distinguished from SCID using thymic organoid cultures, currently available in two research laboratories [6, 7]. The criteria for diagnosing congenital athymia are in Table 1.
Pre-CTTI management: Children identified with congenital athymia should receive immunoglobulin replacement to maintain immunoglobulin G (IgG) trough levels of at least 800 mg/dl [8,9,10]. Subcutaneous immunoglobulin is often preferred over intravenous immunoglobulin (IVIG) to avoid complications associated with central lines. Prophylactic medications should be started to prevent infections (see infectious disease section). Lymphocyte enumerations should be obtained every 6 months prior to CTTI.
Hospitalized children with congenital athymia should remain in protective isolation. Once discharged, minimize exposure to people other than immediate family by using excellent hand hygiene, limiting visitors, homeschooling other young children, and family members showering/changing clothes upon reentry to the home from work.
Autologous graft versus host disease (aGVHD) with immune dysregulation: From birth to 6 to 12 months post CTTI, children with congenital athymia may develop aGVHD from uneducated recipient T cells that did not develop in a thymus [2]. The clinical presentation of aGVHD varies but usually involves the skin and gut. Skin rashes are often accompanied by lymphadenopathy [11]. Skin biopsy is necessary to confirm the diagnosis of aGVHD (see the “Dermatology’ section). For persistent diarrhea, consider gut biopsies. Total parenteral nutrition (TPN) may be required.
For suspected aGVHD, a complete blood count, differential, and lymphocyte enumeration are indicated. T cell counts can increase to over 27,000/mm3. These T cells do not express naïve T cell or recent thymus emigrant (RTE) markers [11].
The liver and lungs are infrequent targets of uneducated T cells of aGVHD. However, if serum aminotransferases increase to over 150 U/L, liver biopsy should be considered to confirm the diagnosis before starting immunosuppression [12]. If lungs are involved, infection should be ruled out before starting immunosuppression.
The first-line treatments of the rash in children with aGVHD are topical moisturizers, steroid creams, and/or calcineurin inhibitor creams. If topical treatments are ineffective, low doses of a systemic calcineurin inhibitor (cyclosporine, tacrolimus) may be needed.
In severe cases, cyclosporine trough levels of 180 to 220 ng/ml or tacrolimus trough levels of 7 to 10 ng/ml may be required. In rare cases, such as recurrence of previously controlled aGVHD, trough levels may be increased to 250 to 300 ng/ml for cyclosporine and 10 to 15 ng/ml for tacrolimus.
If calcineurin inhibitors are not tolerated, e.g., resulting in hypertension and/or low glomerular filtration rate (GFR), steroids can be used at the lowest effective dose. In severe cases, 1 to 2 mg/kg/day of methylprednisolone may be needed to decrease symptoms. Pulse steroids (methylprednisolone of 30 to 40 mg/kg/day for 3 days) should never be given post CTTI as the pulse can permanently damage the CTT [13].
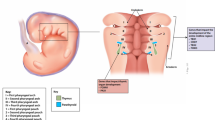
CTTI: CTTI procedures have been reported in multiple publications [14, 15]. Rice et al. (2004) describe the surgical procedure [14]. Briefly, an incision is made in the skin and the surface of the quadriceps is exposed. CTT is placed in “pockets” made in the quadriceps muscle, chosen for its good blood supply to provide oxygen and nutrients to CTT.
Post CTTI: Naïve T cells usually develop 6 to 12 months post CTTI [1]. Thus, upon return to the referring center, clinical care is essentially identical to that prior to CTTI.
Children can develop aGVHD at any time prior to naïve T cell development. They should be evaluated weekly for the first 2 months and at least monthly for the remainder of the first year. Lymphocyte enumerations should be done every 3 months until naïve T cells are greater than 10% of the total T cells and at 12 months post CTTI. When naïve T cells are over 10% of total T cells, the calcineurin inhibitor is weaned over approximately 10 weeks. Prophylactic medications, immunoglobulin replacement, and protective isolation can usually be stopped by 1 year if all criteria in Table 2 are met.
After CTTI, children with congenital athymia develop improved T cell numbers although usually less than the 10th percentile for age [1]. The children do well and are immunologically similar to the 10% of the general population and children with partial DGA (pDGA) who have counts below the 10th percentile for age.
After the first year post CTTI, lifetime annual evaluations should include assessment of T cell counts and monitoring for long-term adverse events including autoimmune disease, osteopenia, renal damage, and scoliosis. Recommended annual tests post CTTI are in Table 3.
Live vaccines were not given early in the CTTI program but more recently have been given once patients are off immunosuppression and demonstrate the ability to mount an immune response to inactivated vaccines. Live vaccines have been well tolerated [3]. Check titers to vaccines to insure that the patient is protected, especially to varicella and measles. Guidances for administration and timing of immunizations are in Tables 4 and 5.
Cardiology
A baseline echocardiogram is recommended when there are signs of cardiac involvement [2]. Blood pressures should be maintained below the 90th percentile for age. Cardiac surgery, if indicated, should not be delayed while awaiting CTTI. Of note, wound healing is normal in children with congenital athymia/cDGA. Generally, CTTI is not performed in children with unrepaired cyanotic congenital heart disease due to the risk of hemodynamic instability and need for surgical intervention requiring high dose steroids in the immediate pre-/post-CTTI period. High-dose steroids are contraindicated in the immediate post CTTI period as they can damage the CTT. CTTI should not be performed in children with anticipated cardiac intervention within 2 months of CTTI.
Dentistry
Excellent oral hygiene is needed to prevent dental or gingival problems. In children with concern for cavities, a dental consult should be obtained prior to CTTI. Confirm with the cardiologist if bacterial endocarditis prophylaxis is indicated. The dentist will provide guidance about side effects of medications, such as cyclosporine, that can cause gingival hyperplasia.
Dermatology
A new rash in a child with congenital athymia/cDGA can be indicative of aGVHD. A skin biopsy, with T cells present, is necessary to confirm the diagnosis [11]. If topical steroids were applied prior to the biopsy, this can alter the histologic findings. If no T cells are present in the initial biopsy, allow the rash to evolve and repeat the biopsy. Classically there is exocytosis, parakeratosis and spongiosis identified by pathology. Dyskeratosis and satellite cell necrosis can be seen [17]. It is important to start immunosuppression for severe rashes associated with aGVHD. Topical steroids or calcineurin inhibitors can be used as a first-line treatment. In many cases, a systemic calcineurin inhibitor is required to control the rash.
Endocrinology
Many children with cDGA have persistent hypoparathyroidism [1, 2]. Hypoparathyroidism can be present at birth but often develops around 5 weeks after birth. Unlike many children with pDGA, hypoparathyroidism in children with cDGA is usually a lifelong problem [18,19,20,21,22,23]. With stressors such as acute illness, infections, or surgery, calcium requirements can increase dramatically. Children are usually treated with calcitriol and calcium (see Table 6). Some centers may use synthetic parathyroid hormone [24].
Thyroid disease is present in approximately 30% of children with cDGA and can occur before or after CTTI [2]. Arrest of linear growth can be an early sign of hypothyroidism; therefore, length should be followed regularly. Hyperthyroidism is less frequent. A thyroid panel is recommended prior to CTTI and then every 6 months through 12 months post CTTI and then annually to assess for thyroid disease.
Gastroenterology/Hepatology/Pancreas
Children with cDGA may present with suboptimal growth and nutritional status related to feeding difficulties such as dysphagia, enteropathy, gastrointestinal dysmotility (constipation), and/or other medical comorbidities [18, 21, 23, 25,26,27,28,29,30,31,32,33,34]. The majority of children have involvement of the gastrointestinal tract and require enteral nutritional support via tube feeding [35, 36]. It is important to avoid obesity from overfeeding and maintain a healthy body mass (weight for length).
Dysphagia with gagging/regurgitation of nipple feeds and risk of aspiration has been described on videofluoroscopic swallow studies in affected children independent of palatal anatomy [25, 26, 37]. Speech-language pathologists should evaluate efficiency and safety of swallow when indicated.
Diarrhea may be associated with enteropathy, infection, or may be a secondary effect of medications/antibiotics. Biopsies of the upper and lower GI tract should be considered to assess diarrhea and also hypoalbuminemia that is present without a cause. Mucosal apoptosis can be seen with aGVHD. Constipation is common requiring appropriate laxative regimens.
Elevated serum aminotransferase levels may indicate T cell–mediated hepatitis, and levels should be performed monthly for 3 months and then every 3 months through 12 months post CTTI. When other etiologies for elevated enzymes are not found (hepatotoxic medications or infection), liver biopsy may be necessary to identify T cells in the parenchyma [12] or evidence of autoimmune hepatitis prior to starting immunosuppression.
For children on calcineurin inhibitors who develop new gastrointestinal symptoms (especially abdominal pain), pancreatitis must be considered. Serum lipase should be monitored.
Genetics
It is important to rule out SCID using a genetic SCID panel or whole-exome sequencing as coexistent SCID and DiGeorge anomaly have been reported. Multiple genes are implicated in congenital athymia as found in cDGA or Foxn1 deficiency (see Table 7).
Growth and Development
Children with cDGA, as those with pDGA, usually have developmental delay though the severity can vary widely [18, 22, 28, 30, 31]. Early intervention with physical, occupational, and speech therapies is critical to optimizing development. Scoliosis can start at a very young age in these children, so monitoring is essential to identify the curve early so that bracing can be initiated [55]. Behavioral and psychiatric problems are common [22, 23, 26, 55,56,57,58,59,60,61]. Therapies should not be delayed while waiting for CTTI.
Hematology
Cytopenias (thrombocytopenia and anemia) due to immune dysregulation are common before and in the first year after CTTI as in pDGA patients [62, 63]. Neutropenia is less frequent. Cytopenias may also be due to marrow suppression from intercurrent infections or medications.
Some children develop coagulopathies related to infections, nutritional deficiencies, or liver dysfunction. They may require central venous access and prolonged hospitalizations which increase risk of thrombosis.
Infectious Disease
Prophylactic antibiotics and antifungals should be started as described below. As soon as the diagnosis of congenital athymia is made, fluconazole (3 to 6 mg/kg/day) and azithromycin (20 mg/kg weekly) should be started. Azithromycin given weekly can cause significant increases in calcineurin inhibitor levels leading to kidney and liver damage. Once daily dosing (5 mg/kg/dose) can be considered in patients on calcineurin inhibitors. After 1 month of life, trimethoprim-sulfamethoxazole should be started at prophylaxis dosing. Family members should be up to date on vaccinations including seasonal vaccines, as age appropriate, with the exception of rotavirus and varicella vaccines, which should not be administered in siblings of immunocompromised patients.
Mycobacterial infections (Mycobacterium avium complex and M. kansasii) have been identified in a small number of children prior to development of naïve T cells. Mycobacterial infections require multiple anti-microbials and a prolonged treatment course for resolution. Children with mycobacterial infection should not be treated with steroids prior to the development of naïve T cells.
Vaccines should not be given prior to development of naïve T cells. See Tables 4 and 5 for guidelines. See Table 8 for treatment after exposure to varicella or measles.
Invasive Procedures
Careful preoperative preparation is essential and should involve surgeons, anesthesiologists and radiologists as indicated. Complete review of medical conditions, medications, electrolyte disturbances, cardiac function, and pulmonary status is critical. A comprehensive anesthesia plan should be made prior to any procedure, especially CTTI, to ensure the anesthesiologist, surgeon, and medical staff are aware of the underlying medical issues (in particular congenital heart disease and airway anomalies) and medications are administered or discontinued as appropriate prior to the procedure. The anesthesiologist should be aware that steroids should be avoided, if possible, because of the potential harm to the CTT.
It is important to keep the calcium stable during surgeries. An ionized calcium should be obtained 4 h prior to surgery to ensure adequate calcium levels. Even in children who normally require ionized calcium levels < 1.0 mmol/L to prevent nephrocalcinosis, a higher level of 1.5 to 2 mmol/L is desirable during operative procedures.
Peripherally inserted central catheters (PICC) are preferred for children needing short-term central venous access around the time of implantation. For children requiring central lines for clinical care such as TPN, frequent blood draws or longer term intravenous immunosuppression, the type of access should be based on the needs and characteristics of the patient (i.e., age, weight, length of time line is needed, potential for infection). Wound healing can be suboptimal in children who are on steroids; special attention should be given to securing external lines. The central line should be looped under a secure dressing to decrease the risk of the line being dislodged and may be tunneled out the back in an active child.
Nephrology/Urology
Congenital kidney and urinary tract anomalies exist in 30% of children with cDGA as in pDGA [64, 65]. These anomalies include renal hypoplasia or dysplasia, obstruction, reflux, and unilateral renal agenesis. Children with cDGA with renal hypoplasia, dysplasia, or unilateral renal agenesis are especially vulnerable to renal insufficiency if on calcineurin inhibitors. For certain anomalies, surgical procedures such as re-implantation of ureters or repair of fistulas between the rectum and the bladder may be necessary [66]. Timing of these repairs should be per urologic surgical standards.
Nephrology consultation is often indicated. The glomerular filtration rate (GFR) should be assessed with an initial nuclear medicine study. Renal monitoring should include BUN, creatinine, cystatin-C, and proteinuria (urine protein to creatinine ratio). Proteinuria is a marker of renal disease. Children with underlying renal hypoplasia/dysplasia can have proteinuria prior to CTTI. After CTTI, common causes for proteinuria are calcineurin inhibitors and other renal toxic medications such as vancomycin, aminoglycosides, and cidofovir. Avoid nephrotoxic medications and iodinated contrast when possible. Dosing and/or medication adjustments may be required to protect the kidneys, for instance, using linezolid instead of vancomycin. Occasionally, proteinuria associated with nephrotic syndrome can be caused by auto-immune-associated glomerular disease and may require a kidney biopsy [67]. Serum creatinine and urinalysis should be performed monthly for 3 months and then every 3 months through 12 months post CTTI. Lastly, nephrocalcinosis can cause more renal damage and may occur due to medical therapies for hypocalcemia. Monitoring for hypercalciuria and, when appropriate, obtaining a renal ultrasound to evaluate/monitor nephrocalcinosis may be indicated.
Neurology/Psychiatry
Neurologists and psychiatrists are important for care of children with cDGA. Brain magnetic resonance imaging may be useful in evaluating children with CHARGE and 22q11.2DS for significant abnormalities [68]. Occasionally children with cDGA have seizures. Neurologists can help determine the etiology for the seizures and manage the seizure disorder. Seizures can also be related to hypocalcemia especially in infancy. Psychiatric disease such as schizophrenia and bipolar disorder can develop in the teen years or later [69].
Ophthalmology
An ophthalmology examination should be done to rule out coloboma in children with CHARGE [31,32,33]. Microphthalmos is common in CHARGE syndrome [26, 28, 30]. Hooded eyelids and hypertelorism are seen in children with 22q11.2DS [61].
Otolaryngology (ENT)
Children with cDGA can have multiple problems involving the head and neck. A hearing evaluation should be obtained early (often under anesthesia) in children with CHARGE. Hearing aids should be ordered as soon as possible to facilitate speech and language development. Children with middle ear effusions may benefit from myringotomy tubes. For severe sensorineural hearing loss, cochlear implants can be considered, preferably once T cells develop post CTTI.
Children with CHARGE are often born with choanal atresia, a challenging condition that frequently recurs after the initial dilation surgery necessitating additional dilation procedures [26, 30, 32, 33].
Children may have recurrent or chronic sinus infections due to immune deficiencies. These may be refractory to standard medical management and require surgical intervention. In those instances, a computed tomography (CT) scan of the sinuses can help assess the degree and severity of the underlying sinus condition.
A CT assessment of the semicircular canals is important for children with CHARGE [30]. This allows the physicians to provide anticipatory guidance related to problems with balance in the child as they reach the age to sit, stand and walk.
Children with structural airway abnormalities may have obstructive sleep apnea. Adenoidectomies and tonsillectomies are sometimes recommended for management. It is important to note that there can be medial displacement of the carotid artery in children with cDGA. This can increase the risk of life-threatening bleeding with tonsillectomy or adenoidectomy; therefore, when the anatomy is in question, computerized tomography angiography should be performed to determine the location of the carotid arteries prior to surgery [70]. Complete DGA associated with certain genetic defects can include velopharyngeal insufficiency which can be worsened by adenoidectomy [23].
Children with 22q11.2DS and CHARGE often present with cleft lip and/or palate [18, 22, 23, 26, 28, 30,31,32,33]. The cleft lip and cleft palate repair may be delayed until after CTTI. Children with velopharyngeal insufficiency without overt cleft palate (common among 22q11.2DS) require a multidisciplinary workup including speech pathology, plastic surgery, and otolaryngology often in context of a cleft team. Many will require surgical intervention in addition to speech therapy.
Pulmonary
Children with cDGA can have bronchomalacia, atelectasis, recurrent pneumonia and occasionally pulmonary fibrosis [71,72,73]. If a child develops increased work of breathing, hypoxia, or a chronic cough, a chest radiograph should be done to rule out pneumonia or atelectasis. A chest CT scan is often necessary if the etiology is not apparent on a chest radiograph. Persistent atelectasis or recurrent pneumonia may require bronchoscopy to diagnose bronchomalacia. A broncho-alveolar lavage can be performed during the bronchoscopy to identify infectious organisms. If the chest CT shows evidence of interstitial lung disease, a lung biopsy may be necessary.
For children requiring long-term mechanical ventilation, tracheostomy should be considered as it allows for a stable airway without sedation and may allow for successful ventilator weaning. Without sedation, these children can have better developmental outcomes.
Psychosocial/Family
Caring for a child with disabilities can be very stressful. The parents/family may need psychosocial support and mental health services. Referrals to mental health providers that utilize resilience-based interventions provide opportunities for strategies to improve coping and family functioning. Psychosocial support from child life specialists can help reduce the stress associated with hospitalization.
Having a family member with a chronic illness can have a significant negative impact on the entire family unit [74]. Involving siblings in the care of the child can help affirm that they are important members of the family.
Discussion
Children with congenital athymia should have a medical home, preferably in a health system with specialists knowledgeable in the underlying problems of the children both before and after CTTI. Physicians, nurses, and therapists need to be aware of the multiple medical issues to provide comprehensive care for these children. Currently there are many 22q11.2DS and CHARGE clinics in the USA. After CTTI and development of naïve T cells, these clinics are a good resource for the families.
After the children develop normal T cells and complete their immunizations, families may require reminders about annual evaluations. These evaluations are important to detect new issues allowing referral to the appropriate specialists. Children with cDGA have developed hypocalcemia, arthritis, thyroid disease, and psychiatric disease later in childhood similar to patients with pDGA who also can develop these conditions later in life [55].
The immunologist should continue to follow the patient for life. Although the first patient who received CTTI is now over 20 years old, it is important to realize that the stability of T cell counts after this time period is not yet known.
Data Availability
Not applicable.
Code Availability
Not applicable.
Abbreviations
- aGVHD:
-
Autologous graft versus host disease
- CHARGE syndrome:
-
Coloboma, heart defects, choanal atresia, growth or mental retardation, genital abnormalities, and ear abnormalities and/or deafness
- cDGA:
-
Complete DiGeorge anomaly
- CT:
-
Computed tomography
- cpm:
-
Counts per minute
- CTTI:
-
Cultured thymus tissue implantation
- CTT:
-
Cultured thymus tissue
- GFR:
-
Glomerular filtration rate
- IgG:
-
Immunoglobulin G
- IVIG:
-
Intravenous immunoglobulin
- MMR:
-
Measles/mumps/rubella
- PTH:
-
Parathyroid hormone
- pDGA:
-
Partial DiGeorge anomaly
- PHA:
-
Phytohemagglutinin
- PJP:
-
Pneumocystis jiroveci pneumonia
- SCID:
-
Severe combined immunodeficiency
- TRECs:
-
T cell receptor rearrangement excision circles
- TPN:
-
Total parenteral nutrition
References
Markert ML, Devlin BH, McCarthy EA. Thymus transplantation. Clin Immunol. 2010;135(2):236–46.
Markert ML, Devlin BH, Alexieff MJ, Li J, McCarthy EA, Gupton SE, et al. Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: outcome of 44 consecutive transplants. Blood. 2007;109(10):4539–47.
Markert ML, Marques JG, Neven B, Devlin BH, McCarthy EA, Chinn IK, et al. First use of thymus transplantation therapy for FOXN1 deficiency (nude/SCID): a report of 2 cases. Blood. 2011;117(2):688–96.
Puck JM. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia. Immunol Rev. 2019;287(1):241–52.
Amatuni GS, Currier RJ, Church JA, Bishop T, Grimbacher E, Nguyen AA, et al. Newborn Screening for Severe Combined Immunodeficiency and T-cell Lymphopenia in California, 2010-2017. Pediatrics. 2019;143(2).
Bifsha P, Leiding JW, Pai SY, Colamartino ABL, Hartog N, Church JA, et al. Diagnostic assay to assist clinical decisions for unclassified severe combined immune deficiency. Blood Adv. 2020;4(12):2606–10.
Bosticardo M, Pala F, Calzoni E, Delmonte OM, Dobbs K, Gardner CL, et al. Artificial thymic organoids represent a reliable tool to study T-cell differentiation in patients with severe T-cell lymphopenia. Blood Adv. 2020;4(12):2611–2616. Blood Adv. 2020;4(15):3507.
Orange JS, Belohradsky BH, Berger M, Borte M, Hagan J, Jolles S, et al. Evaluation of correlation between dose and clinical outcomes in subcutaneous immunoglobulin replacement therapy. Clin Exp Immunol. 2012;169(2):172–81.
Orange JS, Grossman WJ, Navickis RJ, Wilkes MM. Impact of trough IgG on pneumonia incidence in primary immunodeficiency: a meta-analysis of clinical studies. Clin Immunol. 2010;137(1):21–30.
Perez EE, Orange JS, Bonilla F, Chinen J, Chinn IK, Dorsey M, et al. Update on the use of immunoglobulin in human disease: a review of evidence. J Allergy Clin Immunol. 2017;139(3s):S1–s46.
Markert ML, Alexieff MJ, Li J, Sarzotti M, Ozaki DA, Devlin BH, et al. Complete DiGeorge syndrome: development of rash, lymphadenopathy, and oligoclonal T cells in 5 cases. J Allergy Clin Immunol. 2004;113(4):734–41.
Markert ML, Devlin BH, Chinn IK, McCarthy EA. Thymus transplantation in complete DiGeorge anomaly. Immunol Res. 2009;44(1–3):61–70.
Markert ML, Li J, Devlin BH, Hoehner JC, Rice HE, Skinner MA, et al. Use of allograft biopsies to assess thymopoiesis after thymus transplantation. J Immunol. 2008;180(9):6354–64.
Rice HE, Skinner MA, Mahaffey SM, Oldham KT, Ing RJ, Hale LP, et al. Thymic transplantation for complete DiGeorge syndrome: medical and surgical considerations. J Pediatr Surg. 2004;39(11):1607–15.
Markert, M.L., Devlin, B. H., McCarthy, E. A., Chinn, I. K., Hale, L. P., Thymus Transplantation, in Thymus Gland Pathology: Clinical, Diagnostic, and Therapeutic Features, M.C. Lavinin C, Morandi U, Schoenhuber R. , Editor. 2008, Springer-Verlag Italia: Milan p 255-267.
Rager-Zisman B, Bazarsky E, Skibin A, Chamney S, Belmaker I, Shai I, et al. The effect of measles-mumps-rubella (MMR) immunization on the immune responses of previously immunized primary school children. Vaccine. 2003;21(19–20):2580–8.
Selim MA, Markert ML, Burchette JL, Herman CM, Turner JW. The cutaneous manifestations of atypical complete DiGeorge syndrome: a histopathologic and immunohistochemical study. J Cutan Pathol. 2008;35(4):380–5.
Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34(10):798–804.
Ferry BL, Jones J, Bateman EA, Woodham N, Warnatz K, Schlesier M, et al. Measurement of peripheral B cell subpopulations in common variable immunodeficiency (CVID) using a whole blood method. Clin Exp Immunol. 2005;140(3):532–9.
Chinen J, Rosenblatt HM, Smith EO'B, Shearer WT, Noroski LM. Long-term assessment of T-cell populations in DiGeorge syndrome. J Allergy Clin Immunol. 2003;111(3):573–9.
Choi JH, Shin YL, Kim GH, Seo EJ, Kim Y, Park IS, et al. Endocrine manifestations of chromosome 22q11.2 microdeletion syndrome. Horm Res. 2005;63(6):294–9.
Sullivan KE. Chromosome 22q11.2 deletion syndrome: DiGeorge syndrome/velocardiofacial syndrome. Immunol Allergy Clin N Am. 2008;28(2):353–66.
McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine (Baltimore). 2011;90(1):1–18.
Winer KK, Fulton KA, Albert PS, Cutler GB, Jr. Effects of pump versus twice-daily injection delivery of synthetic parathyroid hormone 1-34 in children with severe congenital hypoparathyroidism. J Pediatr. 2014;165(3):556–63.e1.
White DR, Giambra BK, Hopkin RJ, Daines CL, Rutter MJ. Aspiration in children with CHARGE syndrome. Int J Pediatr Otorhinolaryngol. 2005;69(9):1205–9.
Stromland K, Sjogreen L, Johansson M, Ekman Joelsson BM, Miller M, Danielsson S, et al. CHARGE association in Sweden: malformations and functional deficits. Am J Med Genet A. 2005;133(3):331–9.
Digilio MC, Angioni A, de Santis M, Lombardo A, Giannotti A, Dallapiccola B, et al. Spectrum of clinical variability in familial deletion 22q11.2: from full manifestation to extremely mild clinical anomalies. Clin Genet. 2003;63(4):308–13.
Aramaki M, Udaka T, Kosaki R, Makita Y, Okamoto N, Yoshihashi H, et al. Phenotypic spectrum of CHARGE syndrome with CHD7 mutations. J Pediatr. 2006;148(3):410–4.
Digilio MC, Marino B, Giannotti A, Castro M, Colistro F, Ferretti F, et al. Screening for celiac disease in patients with deletion 22q11.2 (DiGeorge/velo-cardio-facial syndrome). Am J Med Genet A. 2003;121A(3):286–8.
Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, van Ravenswaaij-Arts CM. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet 2011;48(5):334–42.
Blake KD, Salem-Hartshorne N, Daoud MA, Gradstein J. Adolescent and adult issues in CHARGE syndrome. Clin Pediatr (Phila). 2005;44(2):151–9.
Blake KD, Davenport SLH, Hall BD, Hefner MA, Pagon RA, Williams MS, et al. CHARGE association: an update and review for the primary pediatrician. Clin Pediatr (Phila). 1998;37(3):159–73.
Issekutz KA, Graham JM, Prasad C, Smith IM, Blake KD. An epidemiological analysis of CHARGE syndrome: preliminary results from a Canadian study. Am J Med Genet A. 2005;133A(3):309–17.
Macdonald M, Hudson A, Bladon A, Ratcliffe E, Blake K. Experiences in feeding and gastrointestinal dysfunction in children with CHARGE syndrome. Am J Med Genet A. 2017;173(11):2947–53.
Giardino G, Cirillo E, Maio F, Gallo V, Esposito T, Naddei R, et al. Gastrointestinal involvement in patients affected with 22q11.2 deletion syndrome. Scand J Gastroenterol. 2014;49(3):274–9.
Campbell IM, Sheppard SE, Crowley TB, McGinn DE, Bailey A, McGinn MJ, et al. What is new with 22q? An update from the 22q and You Center at the Children's Hospital of Philadelphia. Am J Med Genet A. 2018;176(10):2058–69.
Eicher PS, McDonald-McGinn DM, Fox CA, Driscoll DA, Emanuel BS, Zackai EH. Dysphagia in children with a 22q11.2 deletion: unusual pattern found on modified barium swallow. J Pediatr. 2000;137(2):158–64.
Devriendt K, Fryns JP, Mortier G, van Thienen MN, Keymolen K. The annual incidence of DiGeorge/velocardiofacial syndrome. J Med Genet. 1998;35(9):789–90.
Goodship J, Cross I, LiLing J, Wren C. A population study of chromosome 22q11 deletions in infancy. Arch Dis Child. 1998;79(4):348–51.
Tezenas Du Montcel S, Mendizabai H, Ayme S, Levy A, Philip N. Prevalence of 22q11 microdeletion. J Med Genet. 1996;33(8):719.
Shprintzen RJ. Velo-cardio-facial syndrome: 30 years of study. Dev Disabil Res Rev. 2008;14(1):3–10.
Bernstock JD, Totten AH, Elkahloun AG, Johnson KR, Hurst AC, Goldman F, et al.Recurrent microdeletions at chromosome 2p11.2 are associated with thymic hypoplasia and features resembling DiGeorge syndrome. J Allergy Clin Immunol. 2020;145(1):358–367.e2.
Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A. 2005;133A(3):306–8.
Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36(9):955–7.
Janssen N, Bergman JEH, Swertz MA, Tranebjaerg L, Lodahl M, Schoots J, et al. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat. 2012;33(8):1149–60.
Frank J, Pignata C, Panteleyev AA, Prowse DM, Baden H, Weiner L, et al. Exposing the human nude phenotype. Nature. 1999;398(6727):473–4.
Pignata C, Gaetaniello L, Masci AM, Frank J, Christiano A, Matrecano E, et al. Human equivalent of the mouse nude/SCID phenotype: long-term evaluation of immunologic reconstitution after bone marrow transplantation. Blood. 2001;97(4):880–5.
Giardino G, Sharapova SO, Ciznar P, Dhalla F, Maragliano L, Radha Rama Devi A, et al. Expanding the nude SCID/CID phenotype associated with FOXN1 homozygous, compound heterozygous, or heterozygous mutations. J Clin Immunol. 2021;41:756–68.
Hasegawa K, Tanaka H, Higuchi Y, Hayashi Y, Kobayashi K, Tsukahara H. Novel heterozygous mutation in TBX1 in an infant with hypocalcemic seizures. Clin Pediatr Endocrinol. 2018;27(3):159–64.
Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362(9393):1366–73.
Liu N, Schoch K, Luo X, Pena LDM, Bhavana VH, Kukolich MK, et al. Functional variants in TBX2 are associated with a syndromic cardiovascular and skeletal developmental disorder. Hum Mol Genet. 2018;27(14):2454–65.
Paganini I, Sestini R, Capone GL, Putignano AL, Contini E, Giotti I, et al. A novel PAX1 null homozygous mutation in autosomal recessive otofaciocervical syndrome associated with severe combined immunodeficiency. Clin Genet. 2017;92(6):664–8.
Lalani SR, Safiullah AM, Molinari LM, Fernbach SD, Martin DM, Belmont JW. SEMA3E mutation in a patient with CHARGE syndrome. J Med Genet. 2004;41(7):e94.
Schuffenhauer S, Lichtner P, Peykar-Derakhshandeh P, Murken J, Haas OA, Back E, et al. Deletion mapping on chromosome 10p and definition of a critical region for the second DiGeorge syndrome locus (DGS2). Eur J Hum Genet. 1998;6(3):213–25.
Bassett AS, Chow EW, Husted J, Weksberg R, Caluseriu O, Webb GD, et al. Clinical features of 78 adults with 22q11 deletion syndrome. Am J Med Genet A. 2005;138(4):307–13.
Fine SE, Weissman A, Gerdes M, Pinto-Martin J, Zackai EH, McDonald-McGinn DM, et al. Autism spectrum disorders and symptoms in children with molecularly confirmed 22q11.2 deletion syndrome. J Autism Dev Disord. 2005;35(4):461–70.
Hartshorne TS, Grialou TL, Parker KR. Autistic-like behavior in CHARGE syndrome. Am J Med Genet A. 2005;133A(3):257–61.
Vorstman JA, Morcus ME, Duijff SN, Klaassen PW, Heineman-de Boer JA, Beemer FA, et al. The 22q11.2 deletion in children: high rate of autistic disorders and early onset of psychotic symptoms. J Am Acad Child Adolesc Psychiatry. 2006;45(9):1104–13.
Eliez S. Autism in children with 22q11.2 deletion syndrome. J Am Acad Child Adolesc Psychiatry. 2007;46(4):433–4 author reply 434-4.
Drew LJ, Crabtree GW, Markx S, Stark KL, Chaverneff F, Xu B, et al. The 22q11.2 microdeletion: fifteen years of insights into the genetic and neural complexity of psychiatric disorders. Int J Dev Neurosci. 2011;29(3):259–81.
Monteiro FP, Vieira TP, Sgardioli IC, Molck MC, Damiano AP, Souza J, et al. Defining new guidelines for screening the 22q11.2 deletion based on a clinical and dysmorphologic evaluation of 194 individuals and review of the literature. Eur J Pediatr, 2013.
Jawad AF, McDonald-McGinn DM, Zackai E, Sullivan KE. Immunologic features of chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). J Pediatr. 2001;139(5):715–23.
Lawrence S, McDonald-McGinn DM, Zackai E, Sullivan KE. Thrombocytopenia in patients with chromosome 22q11.2 deletion syndrome. J Pediatr. 2003;143(2):277–8.
Lopez-Rivera E, Liu YP, Verbitsky M, Anderson BR, Capone VP, Otto EA, et al. Genetic drivers of kidney defects in the DiGeorge syndrome. N Engl J Med. 2017;376(8):742–54.
Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370(9596):1443–52.
Hall G, Routh JC, Gbadegesin RA. Urinary anomalies in 22q11.2 deletion (DiGeorge syndrome): from copy number variations to single-gene determinants of phenotype. Am J Kidney Dis. 2017;70(1):8–10.
McCloskey O, Maxwell AP. Diagnosis and management of nephrotic syndrome. Practitioner. 2017;261(1801):11–5.
de Geus CM, Free RH, Verbist BM, Sival DA, Blake KD, Meiners LC, et al. Guidelines in CHARGE syndrome and the missing link: cranial imaging. Am J Med Genet C Semin Med Genet. 2017;175(4):450–64.
Olsen L, Sparsø T, Weinsheimer SM, Dos Santos MBQ, Mazin W, Rosengren A, et al. Prevalence of rearrangements in the 22q11.2 region and population-based risk of neuropsychiatric and developmental disorders in a Danish population: a case-cohort study. Lancet Psychiatry. 2018;5(7):573–80.
Dyce O, McDonald-McGinn D, Kirschner RE, Zackai E, Young K, Jacobs IN. Otolaryngologic manifestations of the 22q11.2 deletion syndrome. Arch Otolaryngol Head Neck Surg. 2002;128(12):1408–12.
Deerojanawong J, Chang AB, Eng PA, Robertson CF, Kemp AS. Pulmonary diseases in children with severe combined immune deficiency and DiGeorge syndrome. Pediatr Pulmonol. 1997;24(5):324–30.
Markert ML, Majure M, Harville TO, Hulka G, Oldham K. Severe laryngomalacia and bronchomalacia in DiGeorge syndrome and CHARGE association. Pediatr Pulmonol. 1997;24(5):364–9.
Wong NS, Feng Z, Rappazzo C, Turk C, Randall C, Ongkasuwan J. Patterns of Dysphagia and Airway Protection in Infants with 22q11.2-Deletion Syndrome. Laryngoscope, 2019.
Yoo J, Halley MC, Lown EA, Yank V, Ort K, Cowan MJ, et al. Supporting caregivers during hematopoietic cell transplantation for children with primary immunodeficiency disorders. J Allergy Clin Immunol. 2019;143(6):2271–8.
Acknowledgments
Multiple pediatric specialists at Duke University Medical Center contributed to this publication including Cristina Aluri, PT, DPT, PCS, occupational therapy; Jane S. Bellet, MD, dermatologist; Susan Budziszewski, child life specialist; Michael G. W. Camitta MD, cardiology; Eileen T. Chambers, MD, nephrology; Jeanette Dickson, CCC-SLP, speech pathologist; Teisha Lightbourne Douglass, MPH, RD, LDN, CNSC, ICLC, bone and marrow transplant dietitian; John B. Eck, MD, anesthesiology; Jennifer Edelschick, DPT, C/NDT, PCS, PT, physical therapy, Mai ElMallah MBBCh, MD, pulmonologist; Sharon F. Freedman, MD, ophthalmology; Michael Freemark MD, endocrinology and diabetes; Lindsay Gallo, MSW, LCSW, clinical social worker; Sheryl L. Jewett, OTR/L, occupational therapy; Sameer Kamath, MBBS, MD, critical Care; Martha Ann Keels, DDS, PhD, dentistry; Robert K. Lark, MD, orthopedic surgeon; Debra Lugo, MD, infectious disease; Jeffrey R. Marcus, MD, plastic and reconstructive surgery; Richard J. Noel, MD, gastroenterology; Eileen M. Raynor, MD, head and neck surgery; Henry E. Rice, MD, general surgery, Jennifer A. Rothman, MD, hematology-oncology; Jonathan C. Routh MD, urology; Vandana Shashi, MBBS, MD, medical genetics; Muhammad Zafar, MBBS, MD, neurology.
Funding
The funding of the writing effort by Markert was covered by the Pediatric Allergy and Immunology Division at Duke University Medical Center. Funding of the writing efforts of Gupton and McCarthy was from private donations made to the Thymus Program.
Author information
Authors and Affiliations
Contributions
All authors contributed to this guidance based on our clinical experience caring for and following thymus implantation recipients over many years. The manuscript was written and edited by all authors and all read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Cultured thymus tissue implantation (CTTI) is an investigational product implanted into patients under an Investigational New Drug (IND) application with the Food and Drug Administration (FDA). Dr. Markert is the “sponsor” of the investigations. Dr. Markert developed the technology for CTTI. Duke has licensed the technology to Enzyvant Therapeutics GmbH. Dr. Markert and Duke have received royalties from Enzyvant. Portions of Dr. Markert’s and her research team’s salaries are being paid by funding from Enzyvant. If the technology is commercially successful in the future, Dr. Markert and Duke may benefit financially. The salary and other items needed to create CTTI are paid at cost by insurance.
There is no conflict of interest to disclose by Gupton and McCarthy.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gupton, S.E., McCarthy, E.A. & Markert, M.L. Care of Children with DiGeorge Before and After Cultured Thymus Tissue Implantation. J Clin Immunol 41, 896–905 (2021). https://doi.org/10.1007/s10875-021-01044-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-021-01044-0