
Abstract
A second polymorph of 4-aminobenzyl alcohol [orthorhombic, a = 8.95051(15), b = 5.8248(1), c = 12.1645(2) Å, space group Pna21] shows a “herringbone” structure with stacks of hydrogen-bonded molecules when viewed down the b-axis.
Graphical Abstract

A new polymorph of 4-aminobenzyl alcohol adopts a “herringbone” structure with stacks of hydrogen bonded molecules viewed down the b-axis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In connection with current studies on functionalised analogues of azophenine, we recently prepared 4-aminobenzyl alcohol 1. Although this compound was first reported in 1895 [1], we initially believed that its X-ray structure had not hitherto been determined. The combination of amino and hydroxyl groups with a benzene ring gives interesting opportunities for the formation of hydrogen-bonded networks and such features have already been noted for both the meta-isomer 2 [2] and the ortho-isomer 3 [3]. In addition, hydrogen-bonding patterns in structures of the closely related ortho-, meta- and para-hydroxybenzyl alcohols 4–6 have been examined in detail [4, 5] (Scheme 1). We determined the X-ray structure and, at that stage, found a previously published structure [6] but the two were markedly different. In this paper, we describe the crystal and molecular structure of the new polymorph of 1 and compare the observed hydrogen-bonding patterns with those for the originally reported polymorph and related compounds.

Synthetic route used to compound 1, and structures of related compounds 2–6 with CCDC Ref Codes
Experimental
4-Nitrobenzyl alcohol (20 g, 0.12 mol) was added to a boiling suspension of zinc dust (80 g, 1.22 mol) and calcium chloride (8 g, 0.07 mol) in water (400 mL). The resulting solution was boiled for 30 min and then filtered hot. On cooling the filtrate, a reddish solid appeared and this was filtered off and discarded. Sodium carbonate was then added to the filtrate to precipitate the calcium salts, which were then filtered off. The filtrate was evaporated down to dryness. The resulting oil was then recrystallized from toluene to afford a pale yellow solid (3.69 g, 23%) m.p. 60–61 °C, (lit. [7] 58–61 °C).
δH (Bruker AV400, 400 MHz, CDCl3) 7.18 (2H, d, J = 8.5 Hz, ArH) 6.69 (2H, d, J = 8.5 Hz, ArH), 4.57 (2H, s, CH2OH) and 3.65 (2H, s, NH2) (agreement with lit. values [8]).
Data were collected using a Rigaku XtalLAB P200 and a Mercury CCD (confocal optics Cu-Kα radiation) at 125 K. Intensity data were collected using ω/ϕ steps accumulating area detector frames spanning at least a hemisphere of reciprocal space (data were integrated using CrystalClear). All data were corrected for Lorentz, polarisation and long-term intensity fluctuations. Absorption effects were corrected on the basis of multiple equivalent reflections. The structure was solved by direct methods and refined by full-matrix least-squares against F2 (SHELXTL [9]). Carbon-bound hydrogen atoms were assigned riding isotropic displacement parameters and constrained to idealised geometries while OH and NH hydrogen atoms were refined freely. Table 1 summarises the X-ray data.
Results and Discussion
We first attempted to prepare 1 by reduction of the readily available ethyl 4-aminobenzoate (benzocaine) using lithium aluminium hydride. Although this reaction does not appear to have been reported, the closely related reduction of methyl 4-aminotetradeuteriobenzoate with LiAlD4 is reported to afford the hexadeuteriated 4-aminobenzyl alcohol in 47% yield [10]. However in our hands the ethyl ester was recovered completely unchanged after prolonged boiling with a large excess of LiAlH4 in either diethyl ether or THF. We deduce that binding of the amino group to aluminium occurs and inhibits reduction. We therefore reverted to the original 1895 synthesis [1], and found that treatment of 4-nitrobenzyl alcohol with zinc dust in the presence of calcium chloride in boiling water gave the product in acceptable yield and in good purity after recrystallization from toluene. The crystals obtained were directly suitable for X-ray diffraction and the molecular structure of 1 is shown in Fig. 1.

The molecular structure of the new polymorph of 1 showing numbering system used
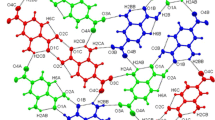
The molecular structure has bond lengths (Table 2) and angles that are unremarkable but the crystal structure is much more interesting with a regular “herringbone” pattern featuring stacks of hydrogen-bonded molecules. A section of the structure viewed along the b-axis is shown in Fig. 2 and the equivalent schematic representation is in Fig. 3. Each OH and NH2 group is involved in one donor and one acceptor interaction, with the oxygen lone pair interacting with an NH and the nitrogen lone pair interacting with an OH. One NH in each molecule is not involved in hydrogen bonding but appears to interact with the π-electrons of a benzene ring. The geometric parameters for this pattern are given in Table 3.

Hydrogen bonding pattern in the crystal structure of 1 viewed along the b-axis

Schematic hydrogen bonding pattern in the crystal structure of 1
The hydrogen-bonding pattern is remarkably different from those involved for the previously reported structure of 1 [6], and the other isomers 2 and 3. We attribute the isolation of a second form of 1 to recrystallization of our sample from toluene which apparently gives a new polymorph. The literature structure used commercially available material with no indication of the recrystallization solvent (if any) in its manufacture. The two polymorphs of 1 differ most obviously in that the previous structure is centrosymmetric with head-to-tail hydrogen bonded dimers which are then linked by further hydrogen bonding interactions in chains (Fig. 4). As is the case for our structure, each OH and NH is involved in one donor and one acceptor interaction with one NH not involved in hydrogen bonding. For the meta-isomer 2 [2], each OH and NH2 group is again involved in one donor and one acceptor interaction, with the oxygen lone pair interacting with an NH and the nitrogen lone pair interacting with an OH, but the molecules are all aligned in the same orientation (Fig. 5). In the ortho-isomer 3 [3], the molecules are arranged in antiparallel lines with an oxygen lone pair to NH interaction linking adjacent molecules within the line and one nitrogen lone pair to OH interaction per pair of molecules linking the two lines in centrosymmetric dimers (Fig. 6). The remaining nitrogen lone pair to OH interactions then link to further lines of molecules behind and in front of the plane giving an infinite layer with a hydrogen bonded interior and hydrophobic exterior.

Hydrogen bonding pattern in the previously reported polymorph of 1 viewed along the c axis [6]

Hydrogen bonding pattern in the crystal structure of 2 [2]

Hydrogen bonding pattern in the crystal structure of 3 [3]
Comparison with the structures adopted by the isomeric hydroxybenzyl alcohols 4–6 [4] is also interesting. The structures adopted by the two ortho-isomers 3 and 6 are closely analogous with centro-symmetric dimers involved in further hydrogen bonding in each case, while for the two meta-isomers, 2 and 5, there is again a good degree of similarity, with molecules all aligned in the same way and connected in a two-dimensional hydrogen bonded network. When we come to the para-isomers 1 and 4 however, the situation differs markedly. Although each hydrogen bonded group is involved in one acceptor and one donor interaction in both structures, the herringbone pattern of 1 is replaced in 4 by a three-dimensional hydrogen bonded network featuring spirals of molecules along the c-axis with a four molecule repeat unit.
Conclusion
The crystal structure for this new polymorph of 1 is quite different from the previous polymorph and those previously reported for the isomers 2 and 3. It also differs markedly from that of the closely similar hydroxy analogue 4. Despite significant progress in being able to predict the X-ray structures of simple aromatic compounds containing OH and NH2 groups [11], it is clear that further work is required to fully understand the forces that determine the structure adopted in a given case.
References
Fischer O, Fischer G (1895) Ber Dtsch Chem Ges 28:879–881
Betz R, Gerber T, Hosten E (2011) Acta Crystallogr Sect E 67:o2118
Zipp CF, Fernandez MA, Marques HM, Michael JP (2012) Acta Crystallogr Sect E 68:o174
Lemmerer A, Esterhuysen C (2011) CrystEngComm 13:5773–5782
Liu W-S, Wei R-P, Tang X-L, Wang W-H, Ju Z-H (2009) Acta Crystallogr Sect E 65:o1689
Bacchi A, Carcelli M, Chiodo T, Cantoni G, De Filippo C, Pipolo S (2009) CrystEngComm 11:1433–1441
Grice R, Owen LN (1963) J Chem Soc 1963:1947–1954
Lawson T, Gannett PM, Yau W-M, Dalal NS, Toth B (1995) J Agric Food Chem 43:2627–2635
Sheldrick GM (2014) Acta Crystallogr Sect A 64:112–122
Pfister R, Ihalainen J, Hamm P, Kolano C (2008) Org Biomol Chem 6:3508–3517
Dey A, Kirchner MT, Vangala VR, Desiraju GR, Mondal R, Howard JAK (2005) J Am Chem Soc 127:10545–10559
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Aitken, R.A., Davidson, L. & Slawin, A.M.Z. The X-ray Structure of 4-Aminobenzyl alcohol (4-Aminophenylmethanol). J Chem Crystallogr 50, 8–13 (2020). https://doi.org/10.1007/s10870-018-0748-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-018-0748-9