
Abstract
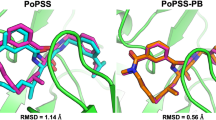
To extend the utility of ligand 3D shape similarity into pose prediction and virtual screening, we have previously developed CDVS and PoPSS methods. Both of them utilize ligand 3D shape similarity with the crystallographic ligands to improve pose prediction. While CDVS utilizes shape similarity to select suitable receptor structures for molecular docking, PoPSS places a ligand conformation of the highest shape similarity with crystal ligands into the target protein binding pocket which is then refined by side-chain repacking and Monte Carlo energy minimization. Analyses of PoPSS revealed some drawbacks in ligand conformation generation and the scoring scheme used. Moreover, as PoPSS does not sample the ligand conformation after placing it in the binding pocket, it relies solely on conformation generation methods to produce native like conformations. To address these limitations of PoPSS method, we report here a modified approach named as PoPSS-Lite, where side-chain repacking was replaced by a simple grid-based energy minimization. This modification also allowed the sampling of terminal functional groups while keeping the core scaffold fixed. Furthermore, shape similarity calculations were improved by increasing the number of ligand conformations and using a different similarity metric. The performance of PoPSS-Lite was prospectively evaluated in D3R GC3. Comparison of PoPSS-Lite demonstrated superior performance over PoPSS and CDVS with lower mean and median RMSDs. Furthermore, comparison with other D3R GC3 pose prediction submissions revealed top performance for PoPSS-Lite. Our D3R GC3 result extends our perspective that ligand 3D shape similarity with known crystallographic information can be successfully used to predict the binding pose of ligands with unknown binding modes. Our D3R GC3 results further highlight the necessity for improvement in conformer generation methods in order to improve shape similarity guided pose prediction.
Graphical abstract









Similar content being viewed by others
References
Maggiora G, Vogt M, Stumpfe D, Bajorath J (2014) Molecular similarity in medicinal chemistry. J Med Chem 57(8):3186–3204. https://doi.org/10.1021/jm401411z
Zauhar RJ, Moyna G, Tian L, Li Z, Welsh WJ (2003) Shape signatures: a new approach to computer-aided ligand- and receptor-based drug design. J Med Chem 46(26):5674–5690. https://doi.org/10.1021/jm030242k
Rush TS 3rd, Grant JA, Mosyak L, Nicholls A (2005) A shape-based 3-D scaffold hopping method and its application to a bacterial protein-protein interaction. J Med Chem 48(5):1489–1495. https://doi.org/10.1021/jm040163o
Kortagere S, Krasowski MD, Ekins S (2009) The importance of discerning shape in molecular pharmacology. Trends Pharmacol Sci 30(3):138–147. https://doi.org/10.1016/j.tips.2008.12.001
Schnecke V, Bostrom J (2006) Computational chemistry-driven decision making in lead generation. Drug Discov Today 11(1–2):43–50. https://doi.org/10.1016/S1359-6446(05)03703-7
Ballester PJ, Westwood I, Laurieri N, Sim E, Richards WG (2010) Prospective virtual screening with Ultrafast Shape Recognition: the identification of novel inhibitors of arylamine N-acetyltransferases. J R Soc Interface 7(43):335–342. https://doi.org/10.1098/rsif.2009.0170
Hoeger B, Diether M, Ballester PJ, Kohn M (2014) Biochemical evaluation of virtual screening methods reveals a cell-active inhibitor of the cancer-promoting phosphatases of regenerating liver. Eur J Med Chem 88:89–100. https://doi.org/10.1016/j.ejmech.2014.08.060
Patil SP, Ballester PJ, Kerezsi CR (2014) Prospective virtual screening for novel p53-MDM2 inhibitors using ultrafast shape recognition. J Comput Aided Mol Des 28(2):89–97. https://doi.org/10.1007/s10822-014-9732-4
Boström J, Berggren K, Elebring T, Greasley PJ, Wilstermann M (2007) Scaffold hopping, synthesis and structure–activity relationships of 5,6-diaryl-pyrazine-2-amide derivatives: a novel series of CB1 receptor antagonists. Bioorg Med Chem 15(12):4077–4084. https://doi.org/10.1016/j.bmc.2007.03.075
Freitas RF, Oprea TI, Montanari CA (2008) 2D QSAR and similarity studies on cruzain inhibitors aimed at improving selectivity over cathepsin L. Bioorg Med Chem 16(2):838–853. https://doi.org/10.1016/j.bmc.2007.10.048
Temml V, Voss CV, Dirsch VM, Schuster D (2014) Discovery of new liver X receptor agonists by pharmacophore modeling and shape-based virtual screening. J Chem Inf Model 54(2):367–371. https://doi.org/10.1021/ci400682b
Kumar A, Parkesh R, Sznajder LJ, Childs-Disney JL, Sobczak K, Disney MD (2012) Chemical correction of pre-mRNA splicing defects associated with sequestration of muscleblind-like 1 protein by expanded r(CAG)-containing transcripts. ACS Chem Biol 7(3):496–505. https://doi.org/10.1021/cb200413a
Vasudevan SR, Moore JB, Schymura Y, Churchill GC (2012) Shape-based reprofiling of FDA-approved drugs for the H1 histamine receptor. J Med Chem 55(16):7054–7060. https://doi.org/10.1021/jm300671m
Sun H, Xu X, Wu X, Zhang X, Liu F, Jia J, Guo X, Huang J, Jiang Z, Feng T, Chu H, Zhou Y, Zhang S, Liu Z, You Q (2013) Discovery and design of tricyclic scaffolds as protein kinase CK2 (CK2) inhibitors through a combination of shape-based virtual screening and structure-based molecular modification. J Chem Inf Model 53(8):2093–2102. https://doi.org/10.1021/ci400114f
Chen W-L, Wang Z-H, Feng T-T, Li D-D, Wang C-H, Xu X-L, Zhang X-J, You Q-D, Guo X-K (2016) Discovery, design and synthesis of 6H-anthra[1,9-cd]isoxazol-6-one scaffold as G9a inhibitor through a combination of shape-based virtual screening and structure-based molecular modification. Bioorg Med Chem 24(22):6102–6108. https://doi.org/10.1016/j.bmc.2016.09.071
Bassetto M, Leyssen P, Neyts J, Yerukhimovich MM, Frick DN, Brancale A (2017) Shape-based virtual screening, synthesis and evaluation of novel pyrrolone derivatives as antiviral agents against HCV. Bioorg Med Chem Lett 27(4):936–940. https://doi.org/10.1016/j.bmcl.2016.12.087
Hevener KE, Mehboob S, Su P-C, Truong K, Boci T, Deng J, Ghassemi M, Cook JL, Johnson ME (2011) Discovery of a novel and potent class of F. tularensis enoyl-reductase (FabI) inhibitors by molecular shape and electrostatic matching. J Med Chem 55(1):268–279. https://doi.org/10.1021/jm201168g
Kaoud TS, Yan C, Mitra S, Tseng C-C, Jose J, Taliaferro JM, Tuohetahuntila M, Devkota A, Sammons R, Park J, Park H, Shi Y, Hong J, Ren P, Dalby KN (2012) From in silico discovery to intracellular activity: targeting JNK–protein interactions with small molecules. ACS Med Chem Lett 3(9):721–725. https://doi.org/10.1021/ml300129b
Naylor E, Arredouani A, Vasudevan SR, Lewis AM, Parkesh R, Mizote A, Rosen D, Thomas JM, Izumi M, Ganesan A, Galione A, Churchill GC (2009) Identification of a chemical probe for NAADP by virtual screening. Nat Chem Biol 5(4):220–226. https://doi.org/10.1038/nchembio.150
Bostrom J, Grant JA, Fjellstrom O, Thelin A, Gustafsson D (2013) Potent fibrinolysis inhibitor discovered by shape and electrostatic complementarity to the drug tranexamic acid. J Med Chem 56(8):3273–3280. https://doi.org/10.1021/jm301818g
Kumar A, Ito A, Hirohama M, Yoshida M, Zhang KYJ (2016) Identification of new SUMO activating enzyme 1 inhibitors using virtual screening and scaffold hopping. Bioorg Med Chem Lett 26(4):1218–1223. https://doi.org/10.1016/j.bmcl.2016.01.030
Kong Y, Bender A, Yan A (2018) Identification of novel aurora kinase A (AURKA) inhibitors via hierarchical ligand-based virtual screening. J Chem Inf Model 58(1):36–47. https://doi.org/10.1021/acs.jcim.7b00300
Wu G, Vieth M (2004) SDOCKER: a method utilizing existing X-ray structures to improve docking accuracy. J Med Chem 47(12):3142–3148. https://doi.org/10.1021/jm040015y
Fukunishi Y, Nakamura H (2008) Prediction of protein-ligand complex structure by docking software guided by other complex structures. J Mol Graph Model 26(6):1030–1033. https://doi.org/10.1016/j.jmgm.2007.07.001
Fukunishi Y, Nakamura H (2012) Integration of ligand-based drug screening with structure-based drug screening by combining maximum volume overlapping score with ligand docking. Pharmaceuticals 5(12):1332–1345. https://doi.org/10.3390/ph5121332
Huang SY, Li M, Wang J, Pan Y (2015) HybridDock: a hybrid protein-ligand docking protocol integrating protein- and ligand-based approaches. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.5b00275
Fleishman SJ, Leaver-Fay A, Corn JE, Strauch EM, Khare SD, Koga N, Ashworth J, Murphy P, Richter F, Lemmon G, Meiler J, Baker D (2011) RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS ONE 6(6):e20161. https://doi.org/10.1371/journal.pone.0020161
McGann M (2012) FRED and HYBRID docking performance on standardized datasets. J Comput Aided Mol Des 26(8):897–906. https://doi.org/10.1007/s10822-012-9584-8
Davis IW, Baker D (2009) RosettaLigand docking with full ligand and receptor flexibility. J Mol Biol 385(2):381–392. https://doi.org/10.1016/j.jmb.2008.11.010
Kelley BP, Brown SP, Warren GL, Muchmore SW (2015) POSIT: flexible shape-guided docking for pose prediction. J Chem Inf Model 55(8):1771–1780. https://doi.org/10.1021/acs.jcim.5b00142
Roy A, Srinivasan B, Skolnick J (2015) PoLi: a virtual screening pipeline based on template pocket and ligand similarity. J Chem Inf Model 55(8):1757–1770. https://doi.org/10.1021/acs.jcim.5b00232
Kumar A, Zhang KYJ (2015) Application of shape similarity in pose selection and virtual screening in CSARdock2014 exercise. J Chem Inf Model. https://doi.org/10.1021/acs.jcim.5b00279
Kumar A, Zhang KYJ (2018) A cross docking pipeline for improving pose prediction and virtual screening performance. J Comput Aided Mol Des 32(1):163–173. https://doi.org/10.1007/s10822-017-0048-z
Kumar A, Zhang KYJ (2016) A pose prediction approach based on ligand 3D shape similarity. J Comput Aided Mol Des 30(6):457–469. https://doi.org/10.1007/s10822-016-9923-2
Kumar A, Zhang KYJ (2016) Prospective evaluation of shape similarity based pose prediction method in D3R Grand Challenge 2015. J Comput Aided Mol Des 30(9):685–693. https://doi.org/10.1007/s10822-016-9931-2
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28(1):235–242
Bower MJ, Cohen FE, Dunbrack RL Jr (1997) Prediction of protein side-chain rotamers from a backbone-dependent rotamer library: a new homology modeling tool. J Mol Biol 267(5):1268–1282. https://doi.org/10.1006/jmbi.1997.0926
Dunbrack RL Jr, Karplus M (1993) Backbone-dependent rotamer library for proteins. Application to side-chain prediction. J Mol Biol 230(2):543–574. https://doi.org/10.1006/jmbi.1993.1170
Li Z, Scheraga HA (1987) Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc Natl Acad Sci USA 84(19):6611–6615
Alford RF, Leaver-Fay A, Jeliazkov JR, O’Meara MJ, DiMaio FP, Park H, Shapovalov MV, Renfrew PD, Mulligan VK, Kappel K, Labonte JW, Pacella MS, Bonneau R, Bradley P, Dunbrack RL, Das R, Baker D, Kuhlman B, Kortemme T, Gray JJ (2017) The Rosetta all-atom energy function for macromolecular modeling and design. J Chem Theory Comput 13(6):3031–3048. https://doi.org/10.1021/acs.jctc.7b00125
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47(7):1739–1749. https://doi.org/10.1021/jm0306430
Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49(21):6177–6196. https://doi.org/10.1021/jm051256o
Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL (2004) Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 47(7):1750–1759. https://doi.org/10.1021/jm030644s
Schrödinger Release 2015-3: LigPrep, version 3.5, Schrödinger, LLC, New York, NY, 2015
Schrödinger Release 2015-3: Maestro, version 10.3, Schrödinger, LLC, New York, NY, 2015
Hawkins PC, Nicholls A (2012) Conformer generation with OMEGA: learning from the data set and the analysis of failures. J Chem Inf Model 52(11):2919–2936. https://doi.org/10.1021/ci300314k
Hawkins PC, Skillman AG, Warren GL, Ellingson BA, Stahl MT (2010) Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model 50(4):572–584. https://doi.org/10.1021/ci100031x
OMEGA 2.5.1.4: OpenEye Scientific Software, Santa Fe, NM. http://www.eyesopen.com/
Hawkins PCD, Skillman AG, Nicholls A (2006) Comparison of shape-matching and docking as virtual screening tools. J Med Chem 50(1):74–82. https://doi.org/10.1021/jm0603365
Krissinel E, Henrick K (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D 60(12-1):2256–2268. https://doi.org/10.1107/S0907444904026460
Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D 67(4):235–242. https://doi.org/10.1107/S0907444910045749
Tversky A (1977) Features of similarity. Psychol Rev 84(4):327–352. https://doi.org/10.1037/0033-295X.84.4.327
Warren GL, Andrews CW, Capelli A-M, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS (2006) A critical assessment of docking programs and scoring functions. J Med Chem 49(20):5912–5931. https://doi.org/10.1021/jm050362n
Plewczynski D, Łaźniewski M, Augustyniak R, Ginalski K (2011) Can we trust docking results? Evaluation of seven commonly used programs on PDBbind database. J Comput Chem 32(4):742–755. https://doi.org/10.1002/jcc.21643
Hawkins PCD (2017) Conformation generation: the state of the art. J Chem Inf Model 57(8):1747–1756. https://doi.org/10.1021/acs.jcim.7b00221
Watts KS, Dalal P, Murphy RB, Sherman W, Friesner RA, Shelley JC (2010) ConfGen: a conformational search method for efficient generation of bioactive conformers. J Chem Inf Model 50(4):534–546. https://doi.org/10.1021/ci100015j
RDKit: Open-source cheminformatics; http://www.rdkit.org/
Vainio MJ, Johnson MS (2007) Generating conformer ensembles using a multiobjective genetic algorithm. J Chem Inf Model 47(6):2462–2474. https://doi.org/10.1021/ci6005646
Santeri PJ, VM J, S. JM (2010) Accurate conformation-dependent molecular electrostatic potentials for high-throughput in silico drug discovery. J Comput Chem 31(8):1722–1732. https://doi.org/10.1002/jcc.21460
Rogers DJ, Tanimoto TT (1960) A computer program for classifying plants. Science 132(3434):1115–1118. https://doi.org/10.1126/science.132.3434.1115
O’Hagan S, Kell DB (2016) MetMaxStruct: a Tversky-similarity-based strategy for analysing the (sub)structural similarities of drugs and endogenous metabolites. Front Pharmacol. https://doi.org/10.3389/fphar.2016.00266
Horvath D, Marcou G, Varnek A (2013) Do not hesitate to use Tversky—and other hints for successful active analogue searches with feature count descriptors. J Chem Inf Model 53(7):1543–1562. https://doi.org/10.1021/ci400106g
Bender A, Mussa HY, Glen RC, Reiling S (2004) Similarity searching of chemical databases using atom environment descriptors (MOLPRINT 2D): evaluation of performance. J Chem Inf Comput Sci 44(5):1708–1718. https://doi.org/10.1021/ci0498719
Acknowledgements
We acknowledge RIKEN ACCC for the supercomputing resources at the Hokusai BigWaterfall supercomputer used in this study. This research was supported by Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number JP18am0101082. We thank members of our lab for help and discussions.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kumar, A., Zhang, K.Y.J. Shape similarity guided pose prediction: lessons from D3R Grand Challenge 3. J Comput Aided Mol Des 33, 47–59 (2019). https://doi.org/10.1007/s10822-018-0142-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-018-0142-x