
Abstract
Revealing patterns of genetic diversity and barriers for gene flow are key points for successful conservation in endangered species. Methods based on molecular markers are also often used to delineate conservation units such as evolutionary significant units and management units. Here we combine phylo-geographic analyses (based on mtDNA) with population and landscape genetic analyses (based on microsatellites) for the endangered yellow-bellied toad Bombina variegata over a wide distribution range in Germany. Our analyses show that two genetic clusters are present in the study area, a northern and a southern/central one, but that these clusters are not deeply divergent. The genetic data suggest high fragmentation among toad occurrences and consequently low genetic diversity. Genetic diversity and genetic connectivity showed a negative relationship with road densities and urban areas surrounding toad occurrences, indicating that these landscape features act as barriers to gene flow. To preserve a maximum of genetic diversity, we recommend considering both genetic clusters as management units, and to increase gene flow among toad occurrences with the aim of restoring and protecting functional meta-populations within each of the clusters. Several isolated populations with especially low genetic diversity and signs of inbreeding need particular short-term conservation attention to avoid extinction. We also recommend to allow natural gene flow between both clusters but not to use individuals from one cluster for translocation or reintroduction into the other. Our results underscore the utility of molecular tools for species conservation, highlight outcomes of habitat fragmentation onto the genetic structure of an endangered amphibian and reveal particularly threatened populations in need for urgent conservation efforts.
Similar content being viewed by others


Introduction
Despite conservation efforts, biodiversity of vertebrates is declining worldwide with more than a fifth of species classified as threatened (Hoffmann et al. 2010). Since the trend of population decline is increasing in many vertebrates, urgent research is necessary to counteract this biodiversity loss, especially since most declines are due to anthropogenic activities (Allan et al. 2019). Amphibians appear particularly vulnerable to such anthropogenic activities. A higher percentage of amphibians are threatened compared to other vertebrates and conservation efforts have been neglected compared to efforts allocated to birds and mammals (Hoffmann et al. 2010). Amphibian populations suffer from land-use change leading to habitat fragmentation and loss, and associated contamination and spreading of infectious diseases (Stuart et al. 2004; Scheele et al. 2019). Thirty-two species of more than 6770 amphibian species are already extinct and many other species (> 2600) are affected by population declines (IUCN 2019). In Europe, amphibian populations mainly declined in areas where suitable habitat is decreasing and where a high number of alien species is present (Falaschi et al. 2018). An increasing body of research reveals reduction of population size and depletion of genetic variation because habitat fragmentation reduces gene flow among populations. For example, in the coastal dune meta-population of the natterjack toad (Epidalea calamita) connectivity was negatively affected by urbanization resulting in small isolated sub-populations with low genetic diversity (Cox et al. 2017). For amphibians, the reduction of genetic diversity significantly reduces fitness especially under stressful conditions (reviews: Cushman 2006; Allentoft and O’Brien, 2010; McCartney-Melstad and Shaffer 2015). Additionally, various landscape genetic studies have shown that genetic variation within and among amphibian populations depended on environmental heterogeneity of the surrounding landscape. Factors that can influence population genetics of amphibians include both natural (e.g., slope, vegetative land cover; Mims et al. 2015; Winiarski et al. 2020) as well as anthropogenic features (e.g., roads, urban areas; Goldberg and Waits 2010; McKee et al. 2017). Understanding such landscape impacts on genetic diversity and structure is important for identifying populations facing greatest risks of genetic depletion due to habitat destruction, and for developing optimal conservation strategies.
In addition to identifying landscape effects on genetic variation, it is also vital to consider the historical context of remaining populations. Therefore, defining (sub-) species and population boundaries for developing conservation programs (McCartney-Melstad and Shaffer 2015; Coates et al. 2018) is mandatory. Knowledge of historical lineages is necessary to maintain fixed genetic differences involved in processes for adaptive evolution (Storfer 1999). However, within species or ancient lineages the loss of genetic connectivity and diversity have been identified as two of the main reasons for population declines (Frankham 2010; Coates et al. 2018). Reconnecting demes that were originally linked via gene flow can help to maximize genetic diversity, counteract inbreeding and increase fitness (Madsen et al. 1999; Cushman 2006; Beauclerc et al. 2010). Thus, reintroductions or translocations can be conservation measures that maintain long-term population viability as well as mixing of gene pools of less diverged populations. Before developing such management designs for endangered species, studies of gene flow, population structure and evolutionary connections to infer historical associations are necessary.
To inform conservation managers about genetic population structure and how to use this knowledge for conservation measures two types of conservation units have been developed: Evolutionary significant units (ESUs) and management units (MUs) (Moritz 1994, 1999; Shaffer et al. 2015; Coates et al. 2018). ESUs are considered as historical, major genetic lineages within a species and are commonly identified by phylogenetic structure through mtDNA. Management units in turn are subunits within ESUs and often identified by divergence in allele frequencies at microsatellite loci but lacking deep evolutionary isolation. In the western spadefoot (Spea hammondi), for example, two major allopatric phylogenetic lineages have been defined based on nuclear and mtDNA and these two lineages together with ecological differences have been proposed to be considered as ESUs and to be managed separately (Neal et al. 2018). Indeed, some studies provided evidence that mixing gene pools of such lineages might lead to outbreeding depression (Burton 1990: copepods; Sagvik et al. 2005: frogs). Translocations among populations that historically did not exchange genes have not been recommended (Moritz 1999; Edmands 2007). In northern Spain, the agile frog Rana dalmatina is structured into three main genetic (microsatellite) clusters, each cluster harboring a meta-population in optimal habitat, with more gene flow within and little gene flow among them. In this case, each meta-population was recommended to be treated as a management unit (Sarasola-Puente et al. 2012).
Two Bombina species occur in Europe; the fire-bellied toad in the flat plains of Eastern and Northern Europe (Bombina bombina), and the yellow-bellied toad (Bombina variegata) in the uplands in Western, Eastern and Southern Europe (Carparthian, Balkans and Apennine mountains). Evidence of hybridization between both species exists in a narrow zone in Eastern Europe (Vörös et al. 2006). Several phylogeographic analyses have tried to disentangle the relationship among several Bombina lineages and identified refugial areas and expansion patterns back towards the north after the glacial periods (Fijarzyk et al. 2011; Pabijan et al. 2013). Using mtDNA, Firjarzyk et al. (2011) and Pabijan et al. (2013) recognized three major clades within the yellow-bellied toad: Balkano–Western B. variegata (toads from the Balkan and Western Europe including), B. variegata pachypus (geographically isolated on the Apennine peninsula) and Carpathian B. variegata, however with conflicting evidence for relatedness among these lineages.
European Bombina are declining and are threatened by extinction in parts of their distribution range (IUCN 2019). Bombina variegata is still abundant in southern and eastern parts of its distribution range and accordingly classified as Least Concern (Kuzmin et al. 2009). However, the species has undergone a steady decline since the 1990s (Nöllert 1996), and is categorized as endangered in Germany and threatened with extinction at the northern edge of its distribution range (Kühnel et al. 2009). Irrespective of inconclusive previous phylogenetic patterns, identifying the genetic affiliation within the species B. variegata managed in conservation projects is crucial. Unfortunately, the phylogeographic studies of European populations of B. variegata have included few (Hofman et al. 2007; Fijarczyk et al. 2011) or no samples from Germany (Pabijan et al. 2013). For this reason, one of the main goals of this study was the analysis of mt haplotype diversity within our study area and to verify whether our B. variegata populations belong to the Balkano-Western clade as earlier studies suggested.
Several endangered amphibian species have been protected by connectivity measures and translocation projects (e.g. European tree frogs, Hyla arborea: Angelone and Holdregger 2009; Broquet et al. 2010; fire-bellied toads, B. bombina: Schröder et al. 2012). The Nature and Biodiversity Conservation Union (NABU) started a project to improve habitat connectivity of yellow-bellied toad populations to counteract declines from northern to southwestern Germany (Nadjafzahdeh and Buschmann 2014). The species has lost its original habitat (floodplains) nearly completely and remaining toad groups rely on secondary habitats like stone quarries and military zones in mountainous areas where they find small, ephemeral ponds and puddles for survival and reproduction (Szymura 1993). The aim of the conservation project was to create new reproduction ponds, stepping-stone habitats and perform resettlements or reintroductions for reconnecting and strengthening the populations, which are often isolated and small, especially in the northern distribution area.
In support of the species conservation program, our main questions in this study were (1) Are the populations within the studied areas genetically distinguishable to a level of ESUs and MUs? (2) How genetically diverse is the whole population as well as local occurrences and are there occurrences which are highly isolated and genetically depleted? (3) Which landscape parameters influence the genetic diversity within and differentiation among local sub-populations?
To answer these questions, we combine phylogeographic information based on mt haplotypes with analyses of population structure based on microsatellites and Bayesian statistics, conventional population and landscape genetic analyses. Based on our results, we suggest short- and long-term conservation actions that are most important for ensuring the genetic integrity and long-term persistence of toad occurrences in our study area, including translocations, habitat restoration, reintroductions and defragmentation measures to increase connectivity.
Material and methods
Sampling
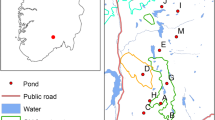
In spring and summer 2012 and 2013, we collected DNA from buccal swabs (Medical ware swabs MW100) of 7 to 45 yellow-bellied toad specimen from each of the 47 localities throughout the region of the NABU conservation project in Germany (Fig. 1; Table 1). Initially, toads were sampled in additional 13 localities but sample size was too small (< 7 individuals) so that they were discarded from population genetic analyses. The DNA was extracted from buccal swabs using the DNeasy Kit (QIAGEN, Hilden, Germany) following manufacture’s protocol.

Localities of buccal swab sampling of yellow-bellied toads in Germany of the NABU conservation project
Phylogeography
Mitochondrial DNA was amplified for three genes: Cytochrome B (Cyt b), Cytochrome oxidase I (COI) and nicotinamide adenine dinucleotide dehydrogenase subunit 4 (ND4) by polymerase chain reaction (PCR) following Brusa et al. (2013). The primers applied were MTAL forward (Hauswaldt et al. 2011) and Cytb–c reverse (Bossuyt and Milinkovitch, 2000) for Cyt b; LCO1490 forward (Folmer et al. 1994) and COI-H reverse (Marchodom et al. 2003) for COI; and ND4 forward and Leu reverse (Arévalo et al. 1994) for ND4. The sequences of the three genes were visually inspected, aligned, and edited using GENEIOUS 9.1 (Kearse et al. 2012). Haplotype network reconstructions were performed by combining the sequence data and maximum likelihood trees (MEGA X v10.0.5; Kumar et al. 2018) with Haplotype Viewer (http://www.cibiv.at/~greg/haploviewer). Further sequences of B. variegata (Fijarczyk et al. 2011) were downloaded from GenBank (http://www.ncbi.nlm.nih.gov/genbank/) to establish a molecular relationship of the toads from our localities with toads from other European populations.
Population structure
Ten previously published microsatellite markers (Stuckas and Tiedemann 2006; Hauswaldt et al. 2007) were applied in this study. In total, 885 individuals from 47 localities (with N ≥ 7) were genotyped for the ten markers (Auffarth et al. 2020). Genotyping was performed on an ABI-3100 sequencer. Allele sizes were scored with GeneMapper 5.0 by using the size standard ROX-350 (Applied Biosystems Inc.). Micro-Checker 2.2.3 (van Oosterhout et al. 2004) was used to check for genotyping errors due to null alleles, allelic dropout and stuttering. The program Fstat v. 2.9.3 (Goudet 1995) was used to test for genotypic disequilibrium of all pairs of loci in each sample.
The genetic diversity was assessed with the software ARLEQUIN v. 3.5 (Excoffier et al. 2005) by calculating expected and observed heterozygosities (Ho, He) as well as average allelic richness (AR). The number of private alleles (NU) was calculated by using GeneALEx v. 6.502 (Peakall and Smouse 2006, 2012). The inbreeding coefficient FIS was calculated by sample site using Genetix v. 4.05 (Belkhir et al. 2004). Significance was tested with a permutations test (1000 permutations).
To infer population structure, a Bayesian model-based clustering algorithm was conducted using the software Structure 2.3.3 (Pritchard et al. 2000). The program assigns individuals to K clusters on the basis of their multilocus genotypes, without prior knowledge of the sample origin. The admixture model was chosen with correlated allele frequencies between populations. We compared results of two estimation approaches, i.e., with and without sampling locations as prior. The run length was set to 105 MCMC iterations following a burn-in period of 105. Ten replicates were performed for every number of clusters (K), and the corresponding ΔK ad hoc statistics was calculated (Evanno et al. 2005). CLUMPP (Jakobsson and Rosenberg 2007) was applied to align replicates by using the full search algorithm and corresponding graphs were created with Distruct 1.1 (Rosenberg 2004). Hierarchical analyses were conducted with each of the clusters most supported by Evanno’s ΔK. Hereby, sample localities were restricted to those that showed at least 80% of cluster affiliation in the previous analysis (Dufresnes et al. 2013).
Our sample size was uneven among sample localities due to sometimes very small sub-population sizes. Structure might fail to recover the true population structure under this condition when calculating ΔK statistics (Puechemaille 2016). Therefore, we followed recommendations given in Puechemaille (2016): first we used a random subsample of 15–20 individuals per sample site and removed sample localities with less than 15 toads from the data set. Thus, we repeated the Structure analysis with 655 toads from 36 sample localities. Secondly, we calculated further K estimators (corrected ΔK, corrected posterior probabilities, MedMeaK, MaxMeaK, MedMedK and MaxMedK) with the R scripts available at http://batlab.ucd.ie/~spuechmaille/. All datasets were analyzed with parallel structure (Besnier and Glover 2013), an R-based implementations of Structure (Pritchard et al. 2000), at the CIPRES Gateway (Miller et al. 2010).
We calculated genetic distances between the sample localities as pairwise FST (Weir and Cockerham 1984) and Dest (Jost 2008) values as implemented in Arlequin (Excoffier et al. 2005) and DEMEtics (Gerlach et al. 2010). Mean FST were calculated for each locality with the PopGenReport in R available at http://www.popgenreport.org/. The relationship between genetic (FST, Dest) and geographic distance was analysed with a Mantel test implemented in IBDWS 3.23 (Jensen et al. 2005). As recommended by Rousset (1997), we performed logarithmic transformation of geographic distances and expressed genetic distance as FST/(1 − FST), respectively Dest/(1 − Dest). The significance for r ≤ 0 was estimated via 10,000 randomizations. The linear geographic distances were calculated in QGIS 2.12.2. We further conducted Mantel Tests for each of the major clusters defined by Structure for populations assigned with a probability of at least 80%.
Landscape genetics
To assess whether genetic variation of sampling localities was influenced by characteristics of the surrounding landscape, we conducted a multi-scale landscape genetic analysis, similar to McKee et al. (2017). Specifically, we measured landscape heterogeneity in circles of varying radii around sampling localities and used these data as covariates in spatial regression models of genetic diversity and differentiation. Radii were 625, 1250, 2500 and 5000 m, with the smallest radius approximating average reported dispersal distances, and the largest radius reflecting maximum reported dispersal distances of our study species (Jakob et al. 2009). We hypothesized that genetic variation would depend on land cover, anthropogenic development and topography. Hence, we measured the proportion of the land cover classes agriculture, pastures, forests, and urban areas within the different circles. Additionally, we measured road density within the circles and assessed both topographic steepness and ruggedness. For steepness, we calculated the mean, sum, and maximum values of topographic slopes in the circles, with higher values indicating steeper landscapes surrounding the sampling localities. For ruggedness, we used the standard deviation of elevation, which increases with increasing elevational variation. Land cover classes were derived from 2012 Corine Land Cover (CLC) data (EEA 2013), road densities from an open-access layer provided by ESRI (2019) and topography from a digital surface model provided by the Copernicus Land Monitoring Service (EU-DEM, EEA 2017).
Since our data showed a significant IBD pattern (see Results), we used a spatial-lag regression model (i.e., a conditional autoregressive model, CAR) with geographic distances among sampling localities as a spatial weighting matrix. Using this weighting matrix, the CAR adds the distance-weighted mean of the dependent variable at neighbouring locations as a predictor to the model. This additional predictor essentially accounts for IBD, because genetic variation at a sampling location is partially dependent on the distance to other sampling locations (Wagner and Fortin 2016). Furthermore, since we detected two main genetic clusters and IBD patterns were different for these two clusters, we conducted all analyses using all occurrences, and also separately for each cluster. Dependent variables for genetic differentiation were FST and Dest values, with pairwise values averaged for each sampling locality (Balkenhol et al. 2013). Genetic diversity was measured as allelic richness (AR) corrected for sampling size using rarefaction, calculated in R package diveRsity (Keenan et al. 2013). Since we suspected that local population sizes also influenced genetic variation, we included sample size as an additional covariate. This is a rather crude proxy of population size, but since sampling intensity was the same across all sampling localities, it is reasonable to assume that larger sample sizes stem from larger populations. We implemented the CAR models in software GeoDa1.12 (Anselin 2018) and compared models based on delta AIC values corrected for small sample sizes. Specifically, we first used delta AIC values to find the most meaningful buffer size to measure each individual variable, and then combined the best representation of each variable in multi-variate models, which were again compared based on delta AIC. Before constructing multi-variate models, we first checked for correlations among covariates and only included variables with |r|< 0.7 in the same model (Dormann et al. 2013).
Results
Phylogeography and genetic diversity for mt genes
Sequencing mitochondrial DNA genes provided polished fragment lengths of 780 bp for Cytb (73 sequences from 46 locations), 604 bp for COI (17 sequences from 17 locations), and 814 bp for ND4 (20 sequences from 19 locations). For our sampling area the mtDNA exhibited almost no variability in the three tested markers (Cytb, COI & ND4). The alternating haplotypes differed only by one mutation, which were silent in all cases. At Cytb three haplotypes were present in 73 sequences from 46 localities with Forbach and Heppenheim showing unique haplotypes (For12, Hes13) besides the main one; at COI two haplotypes were found in 17 sequences from 17 localities, where Brander Wald presented a unique haplotype (Bra34); ND4 exhibited only one haplotype in 20 sequences from 19 localities. When the Cytb haplotypes were compared with other European sequences of B. variagata and B. bombina, phylogenetic analysis (Neighbor joining and maximum likelihood trees) results showed that all analyzed samples belonging to Germany were included into the Balkano-Western clade (Fig. 2). Our haplotype network revealed that the B. bombina sequence JF898356 published in Fijarczyk et al. (2011) is actually a sequence from B. v. variegata. The genetic distances (p-distances) among the five main Bombina lineages (B. bombina, B.v. Carpathian, B.v. Rhodopean, B.v. Apennine, B.v. Balkano-Western) are high and vary among 3% to 12% (Fig. 2).

Haplotype network and p-distances based on mitochondrial DNA (Cytb) of yellow-bellied toads in the study area (green with yellow border) and additional European Bombina samples from Genbank
Population structure and genetic diversity
All ten microsatellite markers were polymorphic, and the total number of alleles per locus ranged from 9 (12F) to 19 (B13), with an average of 13.5. All loci were monomorphic at least in one population (F12, F2) to 31 (1A) and 36 (F22) populations, but showed more alleles in the other populations; no locus was monomorphic across the whole study area. Most of the monomorphic loci were in LBK (5 loci) and OCH (6 loci). There was no significant linkage disequilibrium at any pair of loci after applying Bonferroni corrections (Online Resource 1).
The test for null alleles (van Oosterhout et al. 2004) uncovered null alleles in 75 of 470 locus-sampling sites combinations (i.e. 10 microsatellites × 47 sites). Therefore, we reran Structure with two approaches, (i) discarding all loci with high load of null alleles, (ii) considering null alleles as missing data. In general, Bayesian analysis conducted with Structure uncovered two major genetic groups, separating northern and southern/central populations (K = 2; Fig. 3a, d, e). There was no considerable difference in the outcome when sampling locations were used as prior information or not. When loci with a high number of null alleles were excluded from the analysis, results from Structure remain unchanged (Online Resource 2). Both major genetic clusters were further subdivided in two sub-clusters (Fig. 3b, c). In the northern group BRM, and in the southern/central group OCH + DOB, LBK + BRA as well as HOC + KIR seem to be less connected by gene flow to the other localities.

Population structure of yellow-bellied toads as outcome of the Structure analysis, a for the total study area, b for the northern genetic cluster, c for the southern genetic cluster, d mean likelihood for each K and e ΔK for each K for the total area
The subsampled data set also resulted into the most likely population structure of K = 2 and no differences were found in the assignment of sample localities or individuals to the two clusters compared to the full data set (Online Resource 3). Also, the corrected ΔK indicated exactly the same population structure (Online Resource 4). However, the other estimators of K pointed to K = 1 (for PPKCor0.8) to K = 19 (for MaxMedK0.5). We conclude that the B. variegata population in our study area is indeed hierarchically structured which is best represented with the ΔK statistic (Puechemialle 2016), but that both main clusters can be further subdivided. However, neither of the clusters is genetically isolated, i.e. gene flow occurred across clusters and sub-clusters as indicated by the many admixtured individuals (Fig. 3).
Genetic distances (FST) among sample sites varied from 0.005 between OSB and NAT in the north to 0.666 between OCH and LBK. Among the 1081 pairwise comparisons most FST values are significant (Online Resource 5), with only four comparisons not being significant before Bonferroni correction and 90 not significant after Bonferroni correction. Fifty-four FST values are low (< 0.01) while 52 FST values are high (FST > 0.40). We found two areas which seem to be less fragmented, indicated by low (partly not significant) FST values: the area in the Weser Hills with AST, AWW, OSB, NAT, BOR, and SCH; and the same area in comparison to two populations in the south: VAT and LEF. Additionally, our analyses revealed that three localities are highly differentiated from other localities: OCH, LBK and LAN. Overall, the Dest values showed the same geographic pattern for genetic distances, even if in most cases they seem to be smaller than the corresponding FST values (Online Resource 6).
We found a weak but significant correlation between genetic differentiation (FST, Dest) and geographical distance (FST: Z = 756.27, r = 0.14, p = 0.003, Dest: Z = 545.59, r = 0.16, p < 0.0001) indicating that genetic differentiation among sites is only partially explained by geographical distance. When the two clusters were analyzed separately, the correlation within each was more significant (North-Cluster: Z = 10.0, r = 0.59, p = 0.017 (FST), Z = 11.0, r = 0.57, p = 0.032 (Dest) South/Central Cluster: Z = 303.48, r = 0.17, p = 0.036 (FST), Z = 172.28, r = 0.21, p = 0.005 (Dest)) (Online Resource 7).
Overall, genetic diversity was low to moderate (Table 1). The observed heterozygosity (Ho) ranged from 0.19 (OCH) to 0.59 (AWW), the expected heterozygosity (He) from 0.26 (OCH) to 0.68 (NAT), and the allelic richness (AR) from 1.51 (LBK) to 4.69 (OSB). At most sample sites, the Ho was lower than He, indicating low genetic diversity and possible inbreeding (Fig. 4). In eighteen sample sites, the toads had private alleles (NU), mostly one or two, but AST had eight, and OSB and IST each had five private alleles (Table 1).

Observed versus expected heterozygosity for 47 populations of yellow-bellied toads. In most populations the expected heterozygosity is higher than the observed indicating low genetic diversity and/or inbreeding
We detected ten localities in the north and 21 localities in the south with a high inbreeding index (FIS) significantly differing from zero (Table 1); in all these cases the observed heterozygosity was considerable lower than the expected one. Most severe inbreeding was detected in OSB in the northern cluster.
Landscape effects on genetic variation
Best CAR models explaining genetic diversity and differentiation of the toad occurrences involved the three variables urban areas, road density, and topographic ruggedness (Table 2). All of these three variables produced lowest delta AIC values when measured in a radius of 2500 m. Since the variables urban areas and road density were highly correlated (r = 0.748), we could not include them in the same model. Thus, we instead rescaled both landscape layers to range from 0 to 1 and then added them up, creating a new landscape layer that had highest values when both the amount of urban areas and road densities were high (‘Urban areas + Road density’ in Table 2). Results for genetic differentiation were congruent among both estimates of genetic differentiation (Mean FST and Dest), hence we here only report values obtained from mean FST values. The relative performance of the three explanatory variables differed depending on the occurrences used and also on whether genetic diversity (AR) or genetic differentiation (FST) were modelled.
When using all occurrences, best models involved the variables road density and urban areas, but R2 values were generally low with best models explaining about 14.5% of the variation in FST values and about 9.8% in AR values. Using only occurrences from the southern/central cluster improved R2 values to 0.170 and 0.151 for FST and AR, respectively. Furthermore, topographic ruggedness was suggested as a somewhat important predictor of genetic differentiation in this cluster (R2 = 0.127, delta AICc = 1.789). Highest R2 values were obtained for the northern cluster, where the combination of urban areas and road density explained 60.5% of the variation in FST values and 55.5% in the AR values. All of best models shown in Table 2 with delta AICc values < 2 were also highly significant (p = 0.001). All of the other variables we tested had delta AICc-values > 9, led to low R2 values (< 0.05) and were insignificant (p > 0.05). Sample size, as a proxy of population size, was almost significant (p = 0.066), and explained about 3% of the genetic diversity (R2 = 0.029) when considering all occurrences.
Discussion
In this study, our goals were to determine whether the occurrences of yellow-bellied toads in a project region in Germany were genetically structured and, if this was the case, whether different genetic units have to be taken into consideration for conservation measures. Moreover, we were interested to measure genetic diversity across the study region to identify genetically depleted populations and anthropogenic habitat alterations that might hamper dispersal and be associated with low genetic diversity. Our molecular analyses revealed that our B. variegata occurrences jointly fall into the previously described Balkano-Western mtlineage, while microsatellites in combination with Bayesian cluster analyses revealed the existence of two major genetic clusters whose geographic border runs from western Germany towards the Weser Hills in the north. We recommend considering both clusters as different management units even though some gene flow is or was present. The genetic diversity of the toads is low over the whole study area with significant inbreeding in 66% of the study localities (31 out of 47). Genetic depression can be primarily explained by habitat fragmentation due to the presence of roads and urban areas, which likely present important migration barriers for the toads.
Phylogeography
Within our study area B. variegata exhibited almost no variability at mitochondrial DNA genes (Cytb, COI, ND4). For each mitochondrial gene we found only one abundant haplotype in 96–100% of the analyzed samples. The few remaining unique haplotypes occurred within one mutation step from the main haplotype. This lack of polymorphisms indicates that populations of yellow-bellied toads in Germany are not structured at the level of mtDNA.
When the resulting mtDNA sequences were compared with other European sequences of B. variagata obtained from online databanks (GenBank and BOLD), phylogenetic analysis showed that all our samples were included into the Balkano-Western clade, which was previously described by other authors (Hoffman and Szymura 2007; Fijarczyk et al. 2011). This low level of variation at mitochondrial markers can be explained by the loss of genetic variation by repeated founder events and bottlenecks during the post-glaciation expansion northwestward out of the Balkan refugium (Hoffman et al. 2007; Fijarczyk et al. 2011). Our haplotype network also supports the earlier finding that four main lineages of B. variegata exist in Europe: one lineage in the Carpathian (with 2 sub-lineages: W and E), one lineage in the Rhodopean, one lineage at the Apennine peninsula (Italy) and one lineage at the Balkan extending into western Europe (with two sub-lineages “Western” and “scabra”) (see Fijarczyk et al. 2011). Our data do not support the idea that the lineage at the Apennine peninsula forms a separate species (Lanza and Vanni 1991), nor do they support the subspecies status “B.v. scabra” within the Balkano Western clade. Instead the four main lineages could be considered as subspecies. The genetic distances among B. bombina and the four lineages of B. variegata however show that the distance between B. v. Carpathian and Balkano-Western is as high as between B. bombina and the Carpathian variegata lineage (~ 8%). Further phylogenetic studies including more genetic and ecological information would be helpful discussing the taxonomic and conservation status of European Bombina clades. The 5% threshold in sequence divergence is sometimes considered to be enough for species delimitation in amphibians (e.g. Hagemann and Pröhl 2007; but see Vences and Wake 2007 for a critical discussion) and would justify species status to the B.v. Carpathian and Balkano-Western lineages. In terms of conservation issues, our results are important because they demonstrate that yellow-bellied toads all belong to one mt clade indicating the absence of highly diverged evolutionary significant units (Moritz 1994) within our study area.
Population structure
Our Bayesian cluster analyses revealed two major genetic clusters: a northern cluster and a southern/central cluster. The border between the two groups runs in the northern Weser Hills, particularly between POT at the easternmost range of the Weser hills and all other localities from LIE to FUL in the same forest (Fig. 1: top small panel; Fig. 3a: red cluster). Interestingly, the geographic position between the two clusters corresponds roughly with the course of an ancient trade route the “Hellweg” (Leidinger 1999). It might be possible that yellow-bellied toads used this route along water filled traffic lanes for migration and colonization of the northern area before and during the middle age. In the northern cluster, BRM is genetically different from the other localities. This isolation is probably due to limited gene flow to the other localities in the study area since BRM is located some km apart, outside of the Weser hills, where all other yellow-bellied toads of the northern cluster are located. The northern cluster is at the northernmost distribution of the yellow-bellied toad in Europe.
The Structure analysis also revealed that gene flow has been present between the two clusters and is not completely absent, indicated by the many individuals with mixed (red and blue) genotypes. It is noticeable that in particular some populations far in the south have higher proportions of red (JAE, DIE, FOR) than the populations of the southern/central cluster in close proximity to the northern cluster (DOB, OCH, POT). Pötzen (POT) belongs to the southern/central cluster but is located within the Weser hills at the south-eastern border and there is no obvious migration barrier to all other populations in the Weser hills affiliated with the northern cluster. Limited gene flow between both clusters in this area might indicate that local and unique selection pressures prevent pronounced admixture, though this warrants further investigations using genetic markers under selection. Indeed, nothing is known about local adaptation in yellow-bellied toads in this area and whether it could impede gene flow. Further, Structure and all other analyses also point to further fragmentation of both genetic clusters. Since original habitats of yellow-bellied toads are almost completely destroyed, the toads rely on secondary habitats (mining areas, stone quarries) which are largely isolated from each other (Gollmann and Gollmann 2012; Nadjafzahdeh and Buschmann 2014). The fragmentation of the toad’s gene pool reflects the geographic separation of their secondary habitats.
As mentioned above, defining conservation units is mainly based on molecular tools, e.g. the population structure inferred from mt DNA and microsatellites. The application of these tools is rather consistent across studies for different vertebrate groups: divergent, reciprocally monophyletic mt lineages have been interpreted as ESUs, which in some cases have been found to be ecologically divergent (frogs: Neal et al. 2018; fish: Hutama et al. 2017). The definition of management units mostly relies on the faster evolving microsatellites for which the population structure is calculated by using Bayesian cluster analyses, sometimes in combination with information on the distribution and frequencies of mt haplotypes (amphibian: Allentoft et al. 2009; Dolgener et al. 2012; Sarasola-Puente et al. 2012; Keely et al. 2015; mammals: Wiemann et al 2010; fish: Wetjen et al. 2019). In these cases, microsatellite clusters are considered as management units and individual management is recommended. We follow this practice and advise to consider the northern and southern/central genetic cluster of yellow-bellied toads as two different management units. For conservation measures like reintroductions of toads into suitable habitats or translocations, we recommend to not mix toads from both genetic clusters, but instead use toads from several geographically close and genetically related subpopulations to enhance genetic diversity for increasing fitness and the potential for adaptation (Allentoft and O’Brien 2010). However, we do not recommend against the establishment of connectivity measures as for example the creation of swallow ponds to enable migration between both genetic clusters. Both clusters are not genetically separated and evidence of limited gene flow is present; thus in earlier times the natural situation probably allowed some gene flow. The presence of only one mt haplotype (with only two exceptions) also indicates that the evolutionary divergence between both groups is minor. In case animals migrate into a new genetically distinct population four scenarios are possible: (1) immigrating animals enhance local diversity and thereby improve the adaptability of the whole local population or (2) genetically distinct animals cause outbreeding depression in the new habitat (Edmands 2007), or (3) in case habitat characteristics are very different from the original one, migrating animals might avoid new habitats which do not match their phenotype, and/or (4) they adapt to the new environment by phenotypic plasticity (e.g. Lowe and Addis 2019). The relationships between inbreeding, outbreeding, genetic distance and ecological differences between populations have not been tackled in yellow-bellied toads or its relatives, and thus need further scientific attention.
Genetic diversity
The genetic diversity of yellow-bellied toads in the study area is low: the mt haplotype diversity is nearly completely invariant, diversity measures for microsatellites point into the same direction (low He, Ho and AR) and the inbreeding coefficient (FIS) is significant in most occurrences. The sister species of B. variegata, the fire-bellied toad B. bombina is abundant in eastern Germany where its population genetics have been studied by Dolgener et al. (2012). A direct comparison reveals a much higher genetic diversity in this species: overall 19 mt haplotypes have been detected, and genetic diversity indices for microsatellites are considerably higher than in our study species (He: 0.59–0.78, Ho: 0.44–0.74 and AR: 3.5–5.3 for single localities). The low genetic diversity in B. variegata is alarming since clear genetic-fitness-correlations have been found in 34 studies on 17 amphibian species, especially in face of increasing environmental pollution and infection with pathogens (Allentoft and O’Brien 2010). For example, Johansson et al. (2007) compared genetic diversity and fitness in the frog Rana temporaria in continuous (forested) versus fragmented (agriculture) landscapes. The tadpoles in the fragmented habitats were smaller and genetically less diverse and had lower survival than tadpoles from the continuous landscape, indicating that fragmentation and loss of genetic diversity reduce fitness. Similar findings are known from natterjack toads and tree frogs (Rowe and Beebee 2003; Andersen et al. 2004). In tree frogs in France, low genetic diversity in isolated populations was associated with increased froglet mortality when tadpoles had been infected with an amphibian parasite, the chytrid fungus (Luquet et al. 2012). This finding is of concern since chytrid fungus infection is increasing during the last decade in B. variegata (Ohst et al. 2013) and first evidence exists that population isolation and low genetic diversity enhances prevalence (Oswald et al. 2020) while the infection reduces survival of juvenile yellow-bellied toads (Spitzen-van der Sluijs et al. 2017).
Spatial and landscape effects on genetic variation
Our landscape genetic analyses revealed that both genetic structure and diversity can be explained by landscape characteristics in a 2500-m radius around occurrences. These landscape variables explained genetic differentiation much better than IBD, which explained about 35% of the variation in FST values in the northern cluster, but only about 3% overall and in the south/central cluster. While topographic ruggedness seems to influence genetic differentiation in the southern/central cluster, best models were obtained with the anthropogenic variables urban areas and road density, regardless of which occurrences we used. Both variables have a significantly negative effect on genetic diversity and connectivity throughout our study area, and explain substantial amounts of genetic variation.
Even though other studies have shown that gene flow among amphibian populations can sometimes be predicted by natural landscape characteristics such as topography, vegetation or moisture indices (e.g., Murphy et al. 2010; Gutiérre-Rodríguez et al. 2017; Lenhardt et al. 2017) our results are congruent with other studies that have demonstrated negative effects of anthropogenic land-use on amphibians in human-dominated landscapes (e.g., Goldberg and Waits 2010; Safner et al. 2011; Cox et al 2017).
The fact that we obtained best models when measuring variables in a 2500 m radius might indicate that most multi-generational gene flow occurs across this distance. Generally, amphibians are poor and short-distance dispersers (Smith and Green 2005), but long distance dispersal has been reported in several species in the absence of substantial barriers to movement (reviewed in Fonte et al. 2019). For example, Smith and Green (2006) reported a dispersal distance of 34 km for a Fowler’s toad (Bufo fowleri) in Canada. Reports for yellow-bellied toads are variable, however according to most studies the observed dispersal distance was lower than 300 m within two years for most individuals (Beshkov and Jameson 1980; Barandun and Reyer 1998). A mark-recapture study in northern Rhineland-Palatinate (Germany) aimed to estimate migration among four yellow-bellied toad habitats in close proximity (0.5–2.4 km distance; Hantzschmann and Sinsch 2019). However, no single migration event from one to another sub-population was detected. The authors assumed earlier connection but recent isolation and ongoing genetic differentiation despite the short geographic distance. The overall longest migration distance reported was 5138 m for a yellow-bellied toad female recaptured after 3 years (Jacob et al. 2009). Thus, yellow-bellied toads might migrate distances as far as 5 km when migration barriers are absent. However, the presence of urban areas and associated roads surrounding toad habitats can present severe impediments to amphibian movement and gene flow (Cushman 2006), and jeopardize the genetic diversity and persistence of local populations (Shaffer et al. 2015). Based on our results, we conclude that the isolation of sampled toad occurrences is strongly dependent on the amount of urban areas and road densities in the surrounding landscape.
Implications for conservation
In our study area, yellow-bellied toad occurrences are divided into two genetic main clusters, which show further sub-structuring. Together with low genetic diversity at two molecular markers (microsatellites and mtDNA) and strong negative effects of urban areas and road density, our results point to considerable, human-caused fragmentation with associated loss of genetic diversity. We recommend that conservation measures should first aim at enhancing local and overall genetic diversity. While the restoration of the original habitats, i.e. course of rivers and floodplains should be a long term goal, a short term goal should be to protect occurrences with very low diversity and high inbreeding (Table 1: OCH, OSB, DOB, GUW, HOM, LAN, KIR), as well as those that are most isolated (high FST values: BRM, OCH, LBK, HOM, LAN, KIR) which are the same in four cases. These toad populations should receive genetic refreshment by introducing toads from other localities. Translocating individuals from genetically or environmental different source populations however might disrupt locally adapted gene complexes and lead to outbreeding depression. Frankham et al. (2011) developed a decision tree for the probability of outbreeding depression between two populations that we can directly apply to our situation. Outbreeding depression is expected when two populations – or two genetic clusters—have fixed chromosomal differences, gene flow was absent during the last 500 years, live in substantial different environments and have been separated by more than 20 generations. While it is unlikely, that toads from both clusters have fixed chromosomal differences and gene flow was absent during the last 500 years, environmental factors (climate, habitat type ect.) have not been assessed systematically in this study. Moreover, we do not know whether the two cluster have been separated by more or less than 20 generations. Many subpopulations are also extremely small (N < 50 toads; Buschmann pers. obs.). Because of the lack of genetic diversity, small populations are less likely to be locally adapted than larger populations. Therefore they should benefit from “genetic capture or genetic rescue” by the introduction of individuals from larger populations. It is further recommended, mixing individuals from several source occurrences while limiting the introduced proportion to 5–20% of the size of the recipient occurrence to guarantee genetic variation and minimize potential harmful consequences of outbreeding depression (Weeks et al. 2011; Aitken and Whitlock 2013). Probably the easiest way to enhance genetic diversity at critical yellow-bellied toad occurrences is to release tadpoles from other occurences that are geographically close, belong to the same genetic cluster and are ecologically similar (Dolgener et al. 2012). This latter point is important, because divergence in fitness-related traits as a consequence of local adaptation has been found in amphibians (e.g. Rogell et al. 2011) including yellow-bellied toads (Dittrich et al. 2016). Regular monitoring should accompany such tadpole translocations to ensure that occurrences benefit from this conservation action. Where possible, we further recommend establishing stepping stone habitats to improve connectivity and mitigate the negative impacts of roads and other urban infrastructures. However, this is only meaningful when occurrences are no more than 2–5 km apart from each other because of the short dispersal distances of the toads. Reintroductions might help to improve connectivity in case appropriate habitats are available. For this purpose, we advise mixing of tadpoles from several, genetically more diverse source populations to obtain relatively high numbers of alleles for improving the potential of adaptation to the new local environment. For translocations and reintroductions, the northern and southern/central genetic cluster should be considered as two management units, but connectivity between them via natural gene flow should not be restrained. Finally, efforts to reduce the isolating effects of urbanization and roads should be increased, for example via amphibian tunnels, modified drainage culverts or overpasses specifically designed for amphibians (Woltz et al. 2008; Smith and Sutherland 2014). In an ideal case, all of these conservation actions should continue until several meta-populations (network of a number of subpopulations connected via dispersal) harboring an effective population size (Ne) of > 1000 toads in each cluster are re-established and maintained for safe-guarding a maximum of genetic diversity and adaptive potential (Frankham et al. 2014). Finally, more research is necessary. We need to know how local adaptation, phenotypic plasticity and genetic diversity interact to increase or decrease fitness in yellow-bellied toads and other amphibians. For instance, toads from different genetic clusters and from different ecological habitats should be crossed and the fitness of offspring measured and compared to matching pairs. In future, adaptive components of genetic diversity should also be included in conservation genetic studies for efficiently protecting populations that are adapted to divergent local environments (Bonin et al. 2006).
Data availability
All data generated or analyzed during this study are available in Genbank (accession number of mt sequences: MW296267 - MW 296376) and Zenodo (microsatellite data: https://zenodo.org/record/4612584#.YFI24ecxlbU) or included in this published article and its supplementary information files (Online Resources 1–7).
References
Aitken SN, Whitlock MC (2013) Assisted gene flow to facilitate local adaptation to climate change. Annu Rev Ecol Evol Syst 44:367–388
Allan JR, Watson JEM, Marco MD et al (2019) Hotspots of human impact on threatened terrestrial vertebrates. PLoS Biol 17:e3000598
Allentoft ME, O’Brien J (2010) Global amphibian declines, loss of genetic diversity and fitness: a review. Diversity 2:47–71
Allentoft ME, Siegismund HR, Briggs L, Andersen LW (2009) Microsatellite analysis of the natterjack toad (Bufo calamita) in Denmark: populations are islands in a fragmented landscape. Conserv Genet 10:15–28
Anderson L, Kåre F, Damgaard C (2004) Habitat fragmentation causes bottlenecks and inbreeding in the European tree frog (Hyla arborea). Proc R Soc Lond B 271:1293–1302
Angelone S, Holderegger R (2009) Population genetics suggests effectiveness of habitat connectivity measures for the European tree frog in Switzerland. J Appl Ecol 46:879–887
Anselin L (2018) GeoDa 1.12.1.161 September 2018. Spatial Analysis Laboratory (SAL). Department of Agricultural and Consumer Economics, University of Illinois, Urbana-Champaign, IL, USA. Downloaded from https://geodacenter.github.io/
Arévalo E, Davis SK, Sites JW (1994) Mitochondrial DNA sequence divergence and phylogenetic relationships among eight chromosome races of the Sceloporus grammicuscomplex (Phrynosomatidae) in Central Mexico. Syst Biol 43:387–418
Auffarth J, Pollmann D, Buschmann H, Pröhl H (2020) Conservation genetics of yellow-bellied toads (Bombina variegata) in the Northern Rhineland. Mertensiella 29:50–58.
Balkenhol N, Pardini R, Cornelius C, Fernandes F, Sommer S (2013) Landscape-level comparison of genetic diversity and differentiation in a small mammal inhabiting different fragmented landscapes of the Brazilian Atlantic Forest. Cons Gen 14:355–367
Barandun J, Reyer H-U (1998) Reproductive ecology of Bombina variegata: habitat use. Copeia 2:497–500
Beauclerc K, White B (2010) Genetic rescue of an inbred captive population of the critically endangered Puerto Rican crested toad (Peltophryne lemur) by mixing lineages. Conserv Genet 11:21–32
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (2004) GENETIX4. 05, logiciel sous Windows TM pour la génétiquedes populations. Laboratoire génome, populations, interactions, CNRS UMR. 5000. 1996–2004
Beshkov VA, Jameson DL (1980) Movement and abundance of the yellow-bellied toad Bombina variegata. Herpetologica 36:365–370
Besnier F, Glover KA (2013) Parallel structure: A R package to distribute parallel runs of the population genetics program STRUCTURE on multi-core computers. PLoS ONE 8:e70651
Bonin A, Nicole F, Pompanon F, Miaud C, Taberlet P (2006) Population adaptive index: a new method to help measure intraspecific genetic diversity and prioritize populations for conservation. Conserv Biol 21:697–708
Bossuyt F, Milinkovitch MC (2000) Convergent adaptive radiations in Madagascan and Asian ranid frogs reveal covariation between larval and adult traits. Proc Nat Acad Sci 97:6585–6590
Broquet T, Angelone S, Jaquiery J et al (2010) Genetic bottlenecks driven by population disconnection. Conserv Biol 24:1596–1605
Brusa O, Bellati A, Meuche I, Mundy N, Pröhl H (2013) Divergent evolution in the polymorphic granular poison-dart frog, Oophaga granulifera: genetics, coloration, advertisement calls and morphology. J Biogeogr 40:394–408
Burton RS (1990) Hybrid breakdown in development time in the copepod Tigriopus californicus. Evolution 44:1814–1822
Coates DJ, Byrne M, Moritz C (2018) Genetic diversity and conservation units: dealing with the species-population continuum in the age of genomics. Front Ecol Evol 6:165
Cox K, Maes J, van Claster H, Mergeay J (2017) Effect of the landscape matrix on gene flow in a coastal amphibian metapopulation. Conserv Genet 18:1359–1375
Cushman SA (2006) Effects of habitat loss and fragmentation on amphibians: a review and prospectus. Biol Conserv 128:231–240
Dittrich C, Drakulić S, Schellenberg M, Thein J, Rödel M-O (2016) Some like it hot? Developmental differences in Yellow-bellied toad (Bombina variegata) tadpoles from geographically close but different habitats. Can J Zool 94:69–77
Dormann CF, Elith J, Bacher S, Buchmann C, Carl G, Carré G, Marquéz JR, Gruber B, Lafourcade B, Leitao PJ, Münkemüller T, McClean C, Osborne PE, Reineking B, Schröder B, Skidmore AK, Zurell D, Lautenbach S (2013) Collinearity: a review of methods to deal with it and a simulation study evaluating their performance. Ecography 36(1):27–46
Dolgener N, Schröder C, Schneeweiss N, Tiedemann R (2012) Genetic population structure of the Fire-bellied toad Bombina bombina in an area of high population density: implications for conservation. Hydrobiologia 689:111–120
Dufresnes C, Wassef J, Ghali K et al (2013) Conservation phylogeography: does historical diversity contribute to regional vulnerability in European tree frogs (Hyla arborea)? Mol Ecol 22:5669–5684
EEA (2013) Corine Land Cover (CLC) data 2012, Version 20. European Environment Agency. https://land.copernicus.eu/pan-european/corine-land-cover/clc-2012
EEA (2017) Digital elevation model over Europe, EU-DEM v1.1. European Environment Agency. https://land.copernicus.eu/imagery-in-situ/eu-dem/eu-dem-v1.1
ESRI (2019) Strassenachsen Deutschland. https://opendata-esri-de.opendata.arcgis.com/
Edmands S (2007) Between a rock and a hard place: evaluating the relative risks of inbreeding and outbreeding for conservation and management. Mol Ecol 16:463–475
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Laval G, Schneider S (2005) ARLEQUIN. Ver 3.0. An integrated software package for population genetic data analysis. Evol Bioinform 1:47–50
Falaschi M, Manenti R, Thuiller W, Ficetola GF (2018) Continental-scale determinants of population trends in European amphibians and reptiles. Glob Change Biol 2019:1–12
Fijarczyk A, Nadachowska K, Hofman S (2011) Nuclear and mitochondrial phylogeography of the European fire-bellied toads Bombina bombina and Bombina variegata supports their independent histories. Mol Ecol 20:3381–3398
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for ampliWcation of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotech 3:294–299
Fonte LF, Mayer M, Lötters S (2019) Long-distance dispersal in amphibians. Front BioGeogr 11. https://doi.org/10.21425/F5FBG44577
Frankham R (2010) Challenges and opportunities of genetic approaches to biological conservation. Biol Conserv 143:1919–1927
Frankham R, Ballou JD, Eldridge MDB et al (2011) Predicting the probability of outbreeding depression. Conserv Biol 25:465–475
Frankham R, Bradshaw CJA, Brook BW (2014) Genetics in conservation management: revised recommendations for the 50/500 rules, Red list criteria and population viability analyses. Biol Conserv 170:56–63
Gerlach G, Jueterbock A, Kraemer P, Deppermann J, Harmand P (2010) Calculations of population differentiation based on GST and D: Forget GST but not all of statistics. Mol Ecol 19:3845–3852
Goldberg CS, Waits LP (2010) Comparative landscape genetics of two pond-breeding amphibian species in a highly modified agricultural landscape. Mol Ecol 19:3650–3663
Gollmann B, Gollmann G (2012) Die Gelbbauchunke. Von der Suhle bis zur Radspur. Beiheft der Zeitschrift für Feldherpetologie 4. Laurenti Verlag, Bielefeld
Gutiérre-Rodríguez J, Gonçalves J, Civantos E, Martínez‐Solano I (2017) Comparative landscape genetics of pond‐breeding amphibians in Mediterranean temporal wetlands: The positive role of structural heterogeneity in promoting gene flow. Mol Ecol 26: 5407– 5420
Hagemann S, Pröhl H (2007) Mitochondrial paraphyly in a polymorphic poison frog species (Dendrobatidae; D. pumilio). Mol Phylogenet Evol 45:740–747
Hantzschmann A, Sinsch U (2019) Struktur und dynamik von Gelbbauchunken-populationen (Bombina variegata) im Westerwald–Konsequenzen für das regionale Artenschutz-management. Z Feldherpetol 26:218–235
Hauswaldt JS, Schröder C, Tiedemann R (2007) Nine new tetranucleotide microsatellite markers for the fire-bellied toad (Bombina bombina). Mol Ecol Notes 7:49–52
Hauswaldt JS, Ludewig A-K, Vences M, Pröhl H (2011) Widespread co-occurrence of divergent mitochondrial haplo-type lineages in a Central American species of poison frog (Oophaga pumilio). J Biogeogr 38:711–726
Hoffmann M, Hilton-Taylor C, Angulo A et al (2010) The impact of conservation on the status of the world’s vertebrates. Science 330:1503–1509
Hofman S, Spolsky C, Uzzell T et al (2007) Phylogeography of the fire-bellied toads Bombina: independent Pleistocene histories inferred from mitochondrial genomes. Mol Ecol 16:2301–2316
Hutama A, Dahruddin H, Busson F et al (2017) Identifying spatially concordant evolutionary significant units across multiple species through DNA barcodes: application to the conservation genetics of the freshwater fishes of Java and Bali. Glob Ecol Conserv 12:170–187
IUCN (2019) IUCN red list of threatened species. https://www.iucnredlist.org/. Accessed 2 Sept 2019
Jacob A, Scheel B, Buschmann H (2009) Raumnutzung in einer Metapopulation der Gelbbauchunke (Bombina variegata) an ihrer nördlichen Verbreitungsgrenze. Z Feldherpetol 16:1–18
Johansson M, Primmer CR, Merilä J (2007) Does habitat fragmentation reduce fitness and adaptability? A case study of the common frog (Rana temporaria). Mol Ecol 16:2693–2700
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jensen J, Bohonak A, Scott K (2005) Isolation by distance, web service. BMC Genet 6:13
Jost L (2008) G(ST) and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Kearse M, Moir R, Wilson A et al (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–2164
Keely CC, Hale JM, Heard GW et al (2015) Genetic structure and diversity of the endangered growling grass frog in a rapidly urbanizing region. R Soc Open Sci 2:140255
Keenan K, McGinnity P, CrossTF, Crozier WW, Prodöhl PA (2013) diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Meth Ecol Evol 4:782–788
Kühnel K-D, Geiger A, Laufer H, Podloucky R, Schlüpmann M (2009) Rote Liste und Gesamtartenliste der Lurche (Amphibia) und Kriechtiere (Reptilia) Deutschlands. In: Haupt H et al (eds) Rote Liste gefährdeter Tiere, Pflanzen und Pilze Deutschlands. Band 1: Wirbeltiere. Bundesamt für Naturschutz: Naturschutz und biologische Vielfalt 70:259–288
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Kuzmin S, Denoël M, Anthony B et al (2009) Bombina variegata. The IUCN Red List of Threatened Species 2009: e.T54451A11148290. Accessed 23 Feb 2016
Lanza B, Vanni S (1991) Notes on the biogeography of the Mediterranean Island amphibians. International symposium on the biogeographical aspects of insularity. Atti dei Convegni Lincei, Accademia Nazionale dei Lincei 85:335–344
Leidinger P (1999) Der Westfälische Hellweg als frühmittelalterliche Etappenstraße zwischen Rhein und Weser. Westfälische Zeitschrift 149:9–34
Lenhardt PP, Brühl CA, Leeb C, Theissinger K (2017) Amphibian population genetics in agricultural landscapes: does viniculture drive the population structuring of the European common frog (Rana temporaria)? PeerJ 5: e3520
Lowe WH, Addis BR (2019) Matching habitat choice and plasticity contribute to phenotype-environmental covariation in a stream salamander. Ecology 100:e02661
Luquet E, Garner TWJ, Léna J-P et al (2012) Genetic erosion in wild populations makes resistance to a pathogen more costly. Evolution 66:1942–1952
Machordom A, Araújo R, Erpenbeck D, Ramos MA (2003) Phylogeography and conservation genetics of endangered European margaritiferidae (Bivalvia: Unionoidea). Biol J Linn Soc 78:235–252
Madsen T, Shine R, Olsson M, Wittzell H (1999) Restoration of an inbred adder population. Nature 402:34–35
McCartney E, Shaffer HB (2015) Amphibian molecular ecology and how it has informed conservation. Mol Ecol 24:5084–5109
McKee AM, Maerz JC, Smith LL, Glenn TC (2017) Habitat predictors of genetic diversity for two sympatric wetland-breeding amphibian species. Ecol Evol 7: 6271–6283
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Proceedings of the Gateway Computing Environments Workshop (GCE), 14 Nov. 2010, New Orleans, LA 1–8
Mims MC, Phillipsen IC, Lytle DA, Hartfield Kirk EE, Olden JD (2015) Ecological strategies predict associations between aquatic and genetic connectivity for dryland amphibians. Ecology 96:1371–1382
Moritz C (1994) Defining evolutionary significant units for conservation. Trends Ecol Evol 9:373–375
Moritz C (1999) Conservation units and translocations: strategies for conserving evolutionary processes. Hereditas 130:217–228
Murphy MA, Dezzani R, Pilliod DS, Storfer A (2010) Landscape genetics of high mountain frog metapopulations. Mol Ecol 19:3634–3649
Nadjafzahdeh M, Buschmann H (2014) Stärkung und Vernetzung von Gelbbauchunken-Vorkommen in Deutschland. Rana 15:67–74
Neal KM, Johnson BB, Shaffer HB (2018) Genetic structure and environmental niche modelling confirm two evolutionary and conservation units within the western spadefoot (Spea hammondii). Conserv Genet 19:937–946
Nöllert A (1996) Verbreitung, Ökologie und Schutz der Gelbbauchunke. Naturschutzreport 11:1–260
Ohst T, Gräser Y, Plötner J (2013) Batrachochytrium dendrobatidis in Germany: distribution, prevalences, and prediction of high risk areas. Dis Aquat Org 107:49–59
Oswald P, Rodríguez A, Bourke J, Wagner N, de Buhr N, Buschmann H, Köckritz-Blickwede M, Pröhl H (2020) Locality, time and heterozygosity affect chytrid infection in yellow-bellied toads. Dis Aquat Org 142:225–237
Pabijan M, Wandycz A, Hofman S et al (2013) Complete mitochondrial genomes resolve phylogenetic relationships within Bombina (Anura: Bombinatoridae). Mol Phylogenet Evol 69:63–74
Peakall R, Smouse PE (2006) genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Puechmaille SJ (2016) The program structure does not reliably recover the correct population structure when sampling is uneven: subsampling and new estimators alleviate the problem. Mol Ecol Resour 16:608–627
Rogell B, Berglund A, Laurila A, Höglund J (2011) Population divergence of life history traits in the endangered green toad: implications for a support release programme. J Zool 285:46–55
Rosenberg N (2004) Distruct: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Rowe G, Beebee T (2003) Population on the verge of a mutational meltdown? Fitness costs of genetic load for an amphibian in the wild. Evolution 57:177–181
Safner T, Miaud C, Gaggiotti O, Decout S, Rioux D, Zundel S, Manel S (2011) Combining demography and genetic analysis to assess the population structure of an amphibian in a human-dominated landscape. Cons Gen 12:161–173
Sagvik J, Uller T, Olsson M (2005) Outbreeding depression in the common frog, Rana temporaria. Conserv Genet 6:205–211
Sarasola-Puente V, Madeira MJ, Gosá A et al (2012) Population structure and genetic diversity of Rana dalmatina in the Iberian Peninsula. Conserv Genet 13:197–209
Scheele BC, Pasman SLF et al (2019) Amphibian fungal panzootic causes catastrophic and ongoing loss of biodiversity. Science 363:1459–1463
Schröder C, Pokorny I, Dolgener N et al (2012) Allochthonous individuals in managed populations of the fire-bellied toad Bombina bombina. Genetic detection and conservation implications. Limnologica 42:291–298
Shaffer H, Gidis M, McCartney-Melstad E et al (2015) Conservation genetics and genomics of amphibians and reptiles. Annu Rev Anim Biosci 3:113–138
Smith M, Green D (2005) Dispersal and the metapopulation paradigm in amphibian ecology and conservation: are all amphibian populations metapopulations? Ecography 28:110–128
Smith R, Sutherland W (2014) Amphibian conservation: global evidence for the effects of interventions. Pelagic Publishing, Exeter
Spitzen-van der Sluijs A, Canessa S, Martel A, Pasmans F (2017) Fragile coexistence of a global chytrid pathogen with amphibian populations is mediated by environment and demography. Proc R Soc Lond B 284:20171444
Storfer A (1999) Gene flow and endangered species translocations. Top Revisit Biol Conserv 87:173–180
Stuart S, Chnason JS, Cox NA et al (2004) Status and trends of amphibian declines and extinctions worldwide. Science 306:1783–1786
Stuckas H, Tiedemann R (2006) Eight new microsatellite loci for the critically endangered fire-bellied toad Bombina bombina (Linnaeus, 1761) and their cross-species applicability among anurans. Mol Ecol Notes 6:150–152
Szymura JM (1993) Analysis of hybrid zone with Bombina. In: Harisson RG (ed) Hybrid zones and evolutionary process. Oxford Univ Press, New York, pp 261–289
Van Oosterhout C, Hutchinson W, Wills D, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Vences M, Wake DB (2007) Speciation, species boundaries and phylogeography of amphibians. In: Heatwole H, Tyler M (eds) Amphibian biology. Surrey Beatty Sons, Chipping Norton, pp 2613–2671
Vörös J, Alcobendas M, Martínez-Solano I, García-París M (2006) Evolution of Bombina bombina and Bombina variegata (Anura: Discoglossidae) in the Carpathian Basin: a history of repeated mt-DNA introgression across species. Mol Phylogenet Evol 38:705–718
Wagner HH, Fortin M-J (2016) Basics of spatial data analysis: linking landscape and genetic data for landscape genetic studies. In: Balkenhol N, Cushman S, Storfer A & Waits LP (eds) Landscape genetics: Concepts, methods, applications. Wiley-Blackwell, Hoboken, pp 77–98
Week AR, Sgro CM, Young AG et al (2011) Assessing the benefits and risks of translocations in changing enviroments: a genetic perspective. Evol Appl 4:709–725
Weir BS, Cockerham CC (1984) Estimating Fstatistics for the analysis of population structure. Evolution 38:1358–1370
Wetjen M, Schmidt T, Schrimpf A, Schulz R (2019) Genetic diversity and population structure of burbot Lota lota in Germany: implications for conservation and management. Fish Manag Ecol 00:1–15
Wiemann A, Andersen LW, Berggren P et al (2010) Mitochondrial Control Region and microsatellite analyses on harbour porpoise (Phocoena phocoena) unravel population differentiation in the Baltic Sea and adjacent waters. Conserv Genet 11:195–211
Winiarski KJ, Peterman WE, Whiteley AR, McGarigal K (2020) Multiscale resistant kernel surfaces derived from inferred gene flow: an application with vernal pool breeding salamanders. Mol Ecol Resour 20:97–113
Woltz H, Gibbs J, Ducey P (2008) Road crossing structures for amphibians and reptiles: informing design through behavioral analysis. Biol Conserv 141:2745–2750
Acknowledgements
We thank the Nature and Biodiversity Conservation Union (NABU) of Lower Saxony for financial and logistic support. Field work was conducted by Christian Höppner, Tobias Mika, Mirjam Nadjafzahdeh, Bruno Scheel, Matthias Schindler and students. (Land-)Kreis Schaumburg (A2:443304), Minden-Lübbecke (687607 Mi), Hildesheim (3245/1901), Region Hannover (36.04-1416/20.14), Kreis Warendorf (613901), Rhein-Siegkreis (67.1-2.03.20-24/13-M), Stadt Hamm (31.1/2.2), Stadt Karlsruhe (55-8852.15), Stadt Freiburg (55-8852.44/010) granted research permits. Sabine Sippel conducted microsatellite analyses and Johara Bourke mtDNA analyses in the lab. Sönke von der Berg prepared several figures for the manuscript and Ariel Rodriguez and Andreas Zarling advised on statistical analyses.
Funding
Open Access funding enabled and organized by Projekt DEAL. Funding was provided by the Nature and Biodiversity Conservation Union (NABU) of Lower Saxony.
Author information
Authors and Affiliations
Contributions
HP, HB and NB contributed to the study conception and design. Field work was supervised by HP and HB, lab work was supervised by JA and HP. JA, TB, NB and HP analyzed the data. The first draft of the manuscript was written by HP and NB, and was reviewed and edited by JA, TB, and HB.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Informed consent
All authors agree to publish the produced data.
Research involving human and animal rights
The welfare of the animals examined was not affected. The research was approved by the corresponding conservation offices.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pröhl, H., Auffarth, J., Bergmann, T. et al. Conservation genetics of the yellow-bellied toad (Bombina variegata): population structure, genetic diversity and landscape effects in an endangered amphibian. Conserv Genet 22, 513–529 (2021). https://doi.org/10.1007/s10592-021-01350-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-021-01350-5